Introduction
Neuroblastoma is the most common childhood
malignancy derived from the primitive cells of the sympathetic
nervous system (1). It accounts for
~15% of all pediatric oncology deaths (2). The treatment of patients with advanced
neuroblastoma or high-risk neuroblastoma is still a challenge due
to several factors including, dose-limiting toxicity to standard
chemotherapeutic agents, disease heterogeneity and tumor regression
(3). Moreover, these patients have a
very poor prognosis with the overall ten-year survival rates of
less than 10% (4). Therefore, the
development of innovate alternate treatment strategies is necessary
to increase the treatment effectiveness and lower toxicity.
MicroRNAs (miRNAs/miRs) are a large family of small
(~22–25 nucleotides), endogenous, non-coding RNAs, which binds the
partial or perfect complementary sequences in the 3′untranslated
region (3′UTR) of target messenger RNAs (mRNAs) leading to
translational repression or mRNA degradation (5). They regulate the expression of genes
involved in proliferation, apoptosis, development and stress
response. Thus, miRNAs have shown to play an important role in the
initiation and progression of cancer (5). Depending on their respective target,
miRNAs can act as oncogenes and or tumor suppressors (6). miRNAs are differentially expressed
across cancer types and microRNA-profiling studies have revealed a
general downregulation in tumors as compared with normal tissues
(7). Interestingly, growing evidence
have now shown the potential of aberrant expression of miRNAs to
use as the prognostic and diagnostic biomarkers of human
malignancies (6).
miR-376 family of miRNAs comprises of miR-376a2,
miR-376b, miR-376c (earlier known as miR-368),
miR-376b1 and miR-376b2 and their sequence identity
is highly similar to mouse miR-376a-c miRNAs (8). miR-376 family members are located on
chromosome 14 in humans and at the distal end of mouse chromosome
12 (9). They are expressed in
placenta, developing embryos, and adult tissues (8). The expression of miR-376c is
downregulated in many human malignancies including cervical cancer
(10), prostate cancer (11), oral squamous cell carcinoma (12), intrahepatic cholangiocarcinoma
(13), melanoma (14), osteosarcoma (15) and gliomas (16). However, it has been shown that
miR-376c is upregulated and it act as oncogenic in acute
myeloid leukemia (17) and gastric
cancer (18). In addition, forced
expression of miR-376c enhance ovarian cancer cell survival
by targeting activing-receptor like kinase 7 (19). Whereas, forced overexpression of
miR-376c suppressed growth and invasion of non-small cell
lung cancer (20). According to our
knowledge, there is no report regarding the role of miR-376c
in neuroblastoma. Thus uncovering the mechanisms of miR-376c
function is critical to both the fundamental understanding of
neuroblastoma pathogenesis and novel therapeutic treatments.
The Cyclin D1 (CCND1) is one of the
extensively documented oncogene in human cancers. Functionally,
CCND1 binds with cyclin-dependent kinases (CDK 4/6) which
phosphorylate pRB family proteins, which in turn transactivates
genes necessary for cell cycle progression (21). Dysregulated expression of CCND1
is a common feature in various human cancers (22,23).
Inhibitors targeting CCND1 are thoroughly studied but no
results have yet been proven effective (22,23). The
CCND1 gene has one of the longest 3′UTR (~3.1 kb),
suggesting a strong functional relevance (24). To date, many experimentally validated
miRNAs targeting CCND1 in different cancers are identified.
For example, let-7e, miR-9-5p, miR-15a-5p, miR-16,
miR-17, miR-20a, miR-106b, miR-34a and miR-206
(25–31). However, no miRNA directly targeting
CCND1 3′UTR is yet identified in neuroblastoma.
In this study, we examined the relationship between
miR-376c-3p expression and neuroblastoma tumorigenesis. In
our previous deep sequencing study, we have analyzed
miR-376c-3p expression in neuroblastoma cell lines with
different genetic characteristics (32). miR-376c-3p was downregulated in
most of the cell lines tested and therefore we overexpressed
miR-376c-3p, which have had significant effects on cell
growth and survival of neuroblastoma cells.
In addition, we demonstrated that CCND1 is a
direct target of miR-376c-3p in neuroblastoma and
overexpression of miR-376c-3p might have a significant
influence on inhibition of neuroblastoma tumorigenesis.
Materials and methods
Cell culture
Human neuroblastoma cell lines, CHLA-15 and CHLA-20
were cultivated in Iscove's Modified Dulbecco's Medium
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented with
20% fetal bovine serum, 4 mM L-Glutamine and 1X ITS (5 µg/ml
insulin, 5 µg/ml transferrin, and 5 ng/ml selenous acid). CHLA-15
and CHLA-20 were obtained from the same neuroblastoma patient prior
to and following treatment with combination chemotherapy regimens,
respectively. SKNAS, BE(2)-C, Kelly and SHSY-5Y cells were grown in
RPMI-1640 medium (Sigma-Aldrich; Merck KGaA) supplemented with 10%
fetal bovine serum and 2 mM L-Glutamine (final concentrations). All
cells were split before confluence and maintained at 37°C in a
humidified incubator with 4.5 to 5% CO2 atmosphere. The
cell lines were authenticated by short tandem repeat profiling at
the Center of Forensic Genetics, The Arctic University of
Norway-UiT, Norway and tested negative for mycoplasma
contamination. CHLA-15 and CHLA-20 cell lines were kindly provided
by Children's Oncology Group, Cell Culture and Xenograft
Repository, Texas Tech University Health Science Centre (Lubbock,
TX, USA). BE(2)-C, SKNAS, Kelly and SHSY-5Y were provided by Dr.
John Inge Johnsen (Childhood Cancer Research Unit, Department of
Women's and Children's Health, Karolinska Institutet, Stockholm,
Sweden).
Transfections
MicroRNA miR-376c-3p mimics or negative
control (NC) mimics were purchased from GenePharma Co., Ltd.
(Shanghai, China) and Ambion (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Transfections of miRNA and NC mimics (25–40 nM)
were carried out using Lipofectamine® 2000 reagent
(Thermo Fisher Scientific, Inc.) according to manufacturer's
instructions.
Cell viability assay
CHLA-15, CHLA-20, BE(2)-C, Kelly and SHSY-5Y cells
were seeded in 24-well plates and reverse transfected with 25 nM
miR-376c-3p or NC mimics using Lipofectamine®
2000 reagent (Thermo Fisher Scientific, Inc.). Every 24 h
post-transfection, alamarBlue® (Thermo Fisher
Scientific, Inc.) cell viability assay was performed according to
manufacturer's instructions. Ten percent of the alamarBlue reagent
was added to the cultured cells, mixed gently and incubated at 37°C
for three hours. 100 µl of medium was then transferred to
black-walled 96-well plate and fluorescence was monitored at 540 nm
excitation wavelength and 590 nm emission wavelength in a
microplate reader (CLARIOstar; BMG Labtech GmbH, Offenburg,
Germany). Cell viability was calculated as the percentage of NC
transfected cells set to 100 percent.
Flow cytometric analysis of cell cycle
distribution
The BE(2)-C, Kelly and SHSY-5Y cells were seeded in
25 cm2 culture flasks and reverse transfected with 25 nM
mimics as described previously. 24 h post-transfection cells were
trypsinized and washed with 1X phosphate-buffered saline (PBS). The
cells were then fixed in ice-cold 70% ethanol and incubated
overnight at −20°C. Next day, the ethanol fixed cells were
centrifuged for 10 min at 0.8 × g and washed twice with 1X PBS and
resuspended in the propidium iodide (PI) (Thermo Fisher Scientific,
Inc.) staining solution (PBS with 100 µg/ml RNase, 50 µg/ml PI).
The cells were then incubated for 30 min protected from light and
stored on ice until analyzed. Fluorescence emitted from the PI-DNA
complex was analyzed by flow cytometry using BD
LSRFortessa™ cell analyzer (BD Biosciences, Franklin
Lakes, NJ, USA). FlowJo 7.6.5 software was used to analyze the cell
cycle data using the Dean-Jett-Fox model for cell cycle
evaluation.
Bioinformatics target prediction
TargetScan (version 6.2; www.targetscan.org/) target prediction software was
used to predict miR-376c-3p targets related to cell cycle.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The Kelly, SHSY-5Y and BE(2)-C cells were seeded in
6-well plates and transfected with 40 nM of miR-376c-3p or
NC mimics. Cells were harvested 48 h post-transfection and total
RNA was isolated with QIAzol®Lysis reagent (Qiagen
Sciences, Inc., Gaithersburg, MD, USA) according to manufacturer's
instructions and quantified by NanoDrop™ 1000
spectrophotometer (Thermo Fisher Scientific, Inc.).
For miRNA expression analysis, complementary DNA
(cDNA) was synthesized from isolated total RNAs using the miScript
II RT kit (Qiagen Sciences, Inc.) according to manufacturer's
instructions. The reaction mixture (1X) (20 µl reaction volume) for
reverse transcription was as follows: Total RNA, 1 µg (diluted in
RNase Free Water up to 12 µl); 5X miScript HiSpec Buffer, 4 µl; 10X
miScript Nucleics Mix, 2 µl; miScript Reverse Transcriptase Mix, 2
µl; The cycling conditions were 37°C for 60 min followed by 95°C
for 5 min. The cDNA obtained was diluted with 80 µl RNase Free
Water to achieve 10 ng/µl concentration and stored at −20°C until
use. To quantitate miR-376c-3p levels, quantitative
polymerase chain reaction was performed with miScript primer assay
for miR-376c (cat. no MS00004046) using miScript
SYBR®Green PCR kit (Qiagen Sciences, Inc.,). The
miScript primer assay for miR-4286 (cat. no MS00021371) was used as
an internal control for normalization. The reaction mixture (1X)
(20 µl reaction volume) for real time PCR was as follows: cDNA (1
ng/5 µl), 5 µl; QuantiTect SYBR-Green PCR Master Mix, 10 µl;
Specific miScript primer assay, 2 µl; 10X miScript Universal
Primer, 2 µl; RNase Free Water, 1 µl.
For basic miRNA expression levels in neuroblastoma
cell lines, the following method was used for calculations. Raw
fluorescence values (non-baseline corrected) generated from RT-qPCR
reactions were used to calculate mean PCR efficiencies in the
LinRegPCR software (Version 11.0; http://LinRegPCR.HFRC.nl.). N0 values (starting
concentrations calculated by LinRegPCR software, N0=threshold/(mean
amplicon efficiencyCq)) were used to calculate the
expression of miR-376c-3p relative to the stably expressed
miR-4286 (32). qPCR reactions
were performed in triplicates on 2 independent biological
replicates. Standard deviations were calculated taking into account
the principle of error propagation (including technical and
biological replicates).
For mRNA expression analysis, complementary DNA
(cDNA) was synthesized from isolated total RNAs using the maxima
reverse transcriptase (Thermo Fisher Scientific, Inc.) according to
manufacturer's instructions. The reaction mixture (1X) (20 µl
reaction volume) for reverse transcription of mRNAs was as follows:
Oligo DT primer (20 µM), 1 µl; dNTP (10 mM each), 1 µl; Total RNA,
1 µg (diluted in RNase Free Water up to 13.75 µl); Incubate 65°C
for 5 min followed by addition of 5× RT Buffer, 4 µl; Maxima
Reverse Transcriptase, 0.25 µl; The cycling conditions were 60°C
for 30 min followed by 85°C for 5 min. The cDNA obtained was
diluted with 80 µl RNase Free Water to achieve 10 ng/µl
concentration and stored at −20°C until use. To quantitate
CCND1 levels, quantitative polymerase chain reaction was
performed with Power SYBR-Green PCR Master Mix (Thermo
Fisher Scientific, Inc.). SDHA housekeeping gene was used as an
internal control for normalization. The reaction mixture (1X) (20
µl reaction volume) for real time PCR was as follows: cDNA (1 ng/1
µl), 10 µl; Power SYBR-Green PCR Master Mix, 5 µl; Forward Primer
(10 µM), 0.4 µl; Reverse Primer (10 µM), 0.4 µl; RNase Free Water
4.2 µl.
Amplifications were carried out using Light Cycler
96 SW 1.1 (Roche Diagnostics GmbH, Mannheim, Germany) and
expression levels of miRNAs and mRNAs were evaluated using the
comparative ΔΔCq comparative cycle threshold method (33). The primers used were CCND1
(forward: 5′-CCGTCCATGCGGAAGATC-3′; reverse:
5′-ATGGCCAGCGGGAAGAC-3′) and SDHA (forward:
5′-CTGATGAGACAAGATGTGGTG-3′; reverse:
5′-CAATCTCCCTTCAATGTACTCC-3′).
Western blot analysis
Cells were trypsinized and lysed in 40 µl RIPA
buffer (50 mM Tris-HCL pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium
deoxycholate, 0.1% SDS) supplemented with 1X Protein Inhibitor
Cocktail (Roche Diagnostics GmbH) and 1 mM dithiothreitol (DTT).
Lysate was cleared with centrifugation (21.1 × g) and the total
protein concentrations were determined using DC™Protein Assay kit
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) according to
manufacturer's instructions. 40 µg protein was then loaded in each
well and separated on a NuPAGE® Novex 4–12% Bis-Tris
precast polyacrylamide gel (Thermo Fisher Scientific, Inc.). The
separated proteins were transferred on Immobilon-FL PVDF membrane
(EMD Millipore, Billerica, MA, USA) and blocked for 1 h at room
temperature in 5 ml Odyssey Blocking Buffer (LI-COR Biosciences,
Lincoln, NE, USA). The PVDF membrane was then incubated overnight
at 4°C with primary antibodies anti-Cyclin D1-(H-295)-Human Cyclin
D1 Rabbit, polyclonal; (1:1,000; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) and anti-actin-(ab3280)-Human Actin Mouse,
monoclonal (1:1,000; Abcam, Cambridge, UK). The secondary
antibodies used were goat anti-rabbit-IRDye800CW,
(1:5,000) (Rockland Immunochemicals, Inc., Gilbertsville, PA, USA)
and goat anti-mouse-Alexa Fluor 680, (1:5,000; Thermo Fisher
Scientific, Inc.). Antibody binding was detected using the Odyssey
CLx Infrared Imaging System (LI-COR Biosciences). ImageJ software
was used to quantify western blot results.
Luciferase reporter assay
The pMIR-Report-Cyclin D1-UTR-WT construct was a
generous gift from Dr. Laura Barkley (30) and pMIR-Report-Cyclin D1-UTR-MUT
construct with a mutation in the putative miR-376c-3p seed
sequence (mutant) was generated using QuikChange® Multi
Site-Directed Mutagenesis kit (Agilent Technologies, Inc., Santa
Clara, CA, USA). The primers used for mutagenesis were
CCND1_3′UTR_miR-376c-3p (forward: 5′-CACATCTTGGCATACTAATTCTTG-3′;
reverse: 5′-CAAGAATTAGTATGCCAAGATGTG-3′). To validate for mutation
in seed sequence the mutant plasmid was sequenced using sequencing
primer 5′-CATCTGATTGGACAGGCATG-3′. The cells seeded at a density of
5×104cells/well in a 12-well plate were co-transfected
with 40 nM NC or miR-376c-3p mimics, 20 ng pRL-SV40
construct (Promega Corporation, Madison, WI, USA) and 100 ng wild
type or mutant luciferase constructs using
Lipofectamine® 2000 reagent (Thermo Fisher Scientific,
Inc.). 24 h post-transfection, firefly and renilla luciferase
activities analyzed using the Dual-Luciferase Reporter Assay
(Promega Corporation), according to manufacturer's instructions.
Firefly luciferase activity was normalized against renilla
luciferase activity and luciferase activities of miR-376c-3p
transfected cells were calculated relative to NC transfected cells
set to 100 %.
MicroRNA expression data from
neuroblastoma tumors
miRNA expression data from 226 primary neuroblastoma
tumors were obtained through the Neuroblastoma Research Consortium
(NRC). Differential miRNA expression was analyzed using a
Kruskal-Wallis test.
Statistical analysis
The data was expressed as mean ± standard deviation
(SD). Unless otherwise stated, all experiments were performed at
least three times independently. Statistical analysis was performed
using the software GraphPad Prism version 5.00 for Windows
(GraphPad Software, Inc., La Jolla, CA, USA; available at
www.graphpad.com). Statistical differences
between means were determined using Student's t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
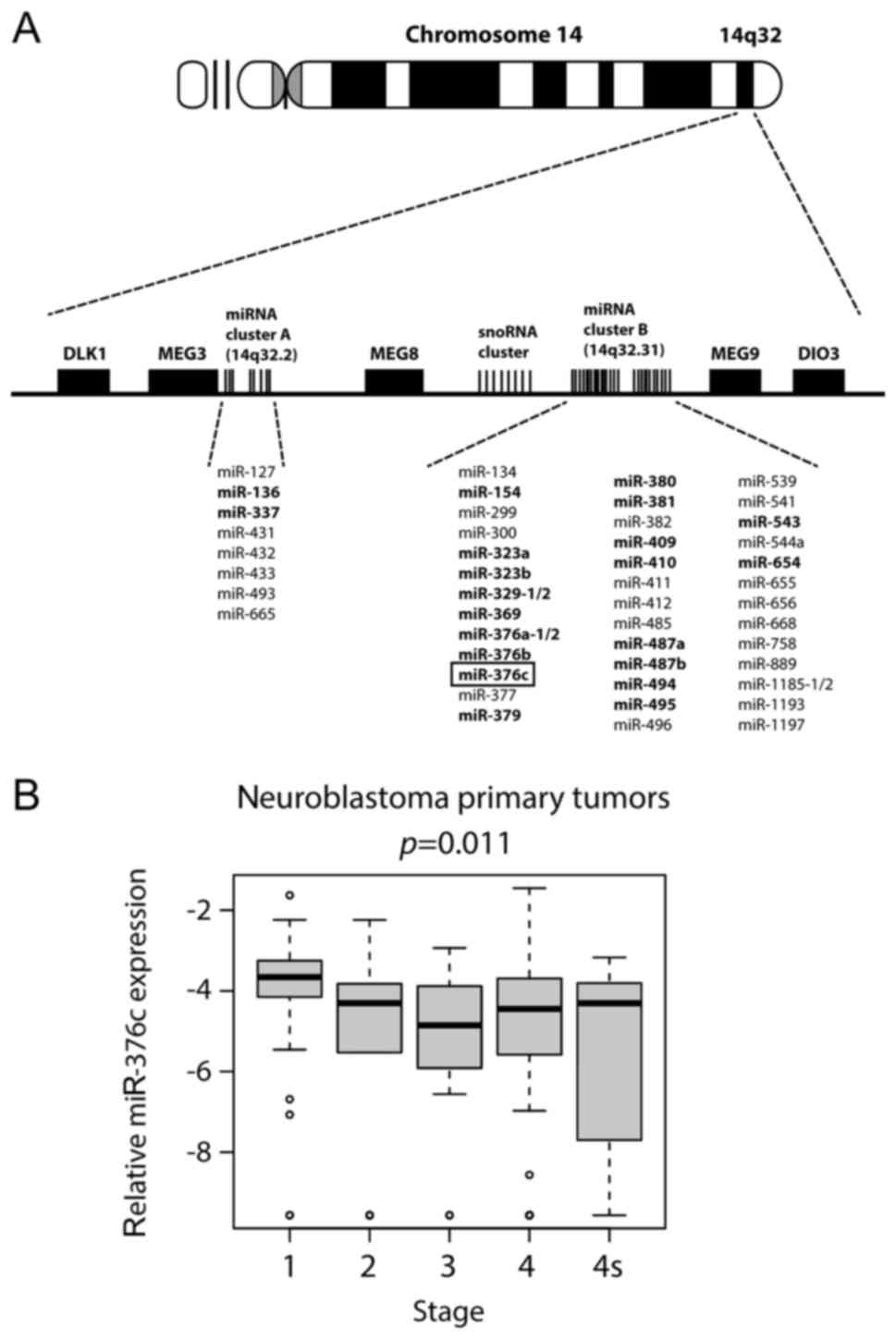
Multiple miRNAs located at 14q32 chromosomal region
are downregulated and their lower levels are associated with poor
prognosis factors in neuroblastoma. Results from our previous
study, where we used next generation sequencing technology (SOLiD
ligation sequencing) to determine miRNA expression profiles in
neuroblastoma cell lines established from patients at diagnosis and
at relapse after treatment, identified 22 downregulated microRNAs
from 14q32 miRNA cluster. The expression of these downregulated
miRNAs was confirmed in a cohort consisting of 226 primary
neuroblastomas (32).
miR-376c-3p, was one of the 22 miRNAs that was downregulated
in most of the cell lines isolated from patients after the
treatment (Fig. 1A). When
miR-376c expression was compared in neuroblastoma primary
tumors of different stages from the 226-cohort, we observed a trend
towards lower expression in advanced stage disease compared to
tumors in stage 1 (Fig. 1B). Thus, we
sought out to focus on the functional role of miR-376c-3p in
neuroblastoma.
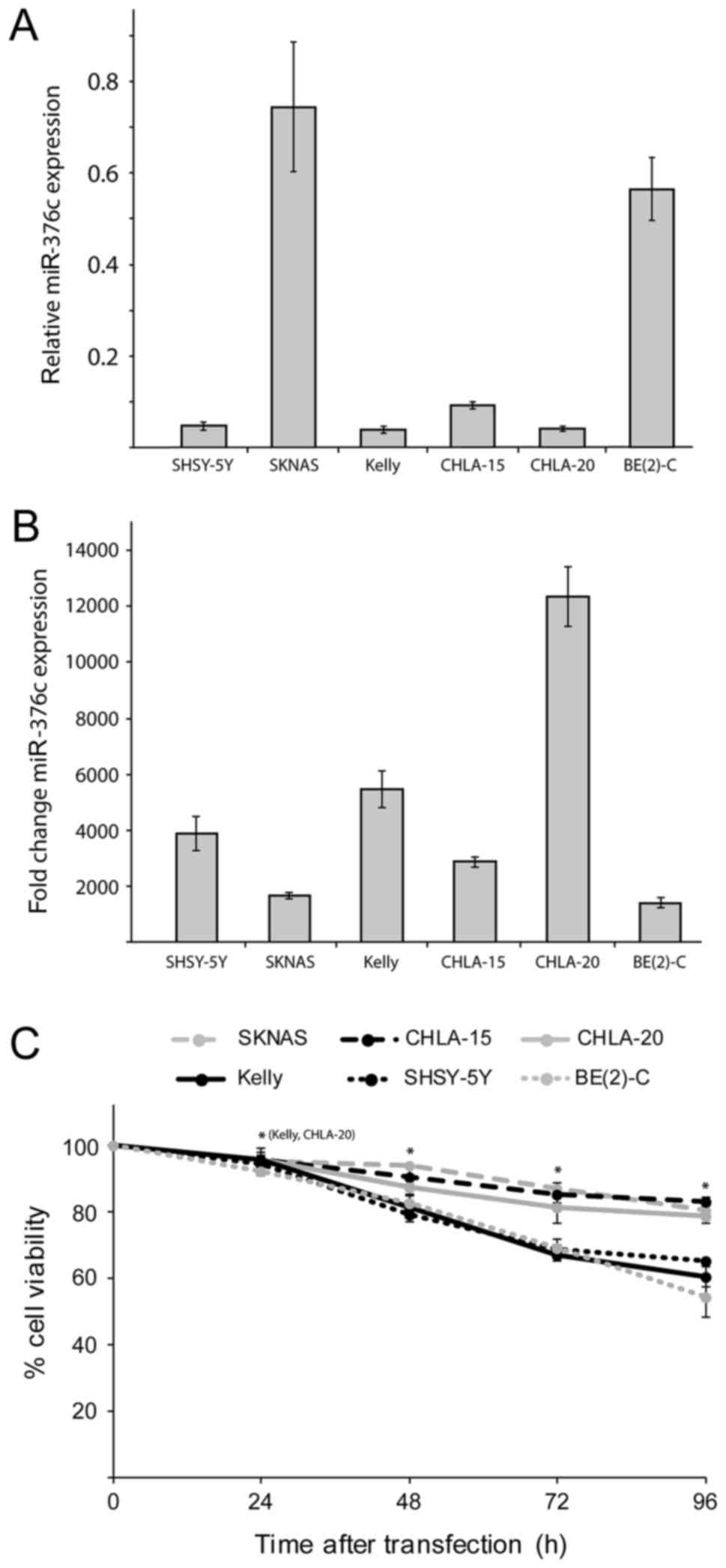
miR-376c-3p reduces cell viability in
neuroblastoma cell lines
Even though miR-376c-3p has been shown to
play either the oncogenic or the tumor suppressive role in
different cancers (10–20) the functional role of
miR-376c-3p is not yet determined in neuroblastoma.
Therefore, we first used RT-qPCR to measure miR-376c-3p
basic expression levels relative to miR-4286 in 6
neuroblastoma cell lines. miR-4286 was previously showed to
be stably expressed in neuroblastoma cell lines (32). Among the cell lines, SKNAS and BE(2)-C
cells have the highest expression level of miR-376c-3p,
whereas SHSY-5Y, Kelly, CHLA-15 and CHLA-20 cells showed barely
detectable levels of expression (Fig.
2A). In order to find out the potential role of
miR-376c-3p in neuroblastoma, cell viability assay was
performed on several neuroblastoma cell lines by overexpressing NC
or miR-376c-3p mimics. The expression of miR-376c-3p
was significantly increased in miR-376c-3p transfected cell
lines compared with NC transfected cells, as validated and
confirmed by RT-qPCR (Fig. 2B). Cell
viability alamarBlue assay was performed at 24, 48, 72 and 96 h
post-transfection, which showed that neuroblastoma cell lines
transfected with miR-376c-3p mimics, had significantly
reduced cell viability as compared to NC transfected cells. Thus,
ectopic expression of miR-376c-3p reduced the growth of
SKNAS, CHLA-15, CHLA-20, SHSY-5Y, Kelly and BE(2)-C cells as
compared to NC transfected cells (Fig.
2C). These results indicate that cell growth of neuroblastoma
cell lines is significantly affected by overexpression of
miR-376c-3p.
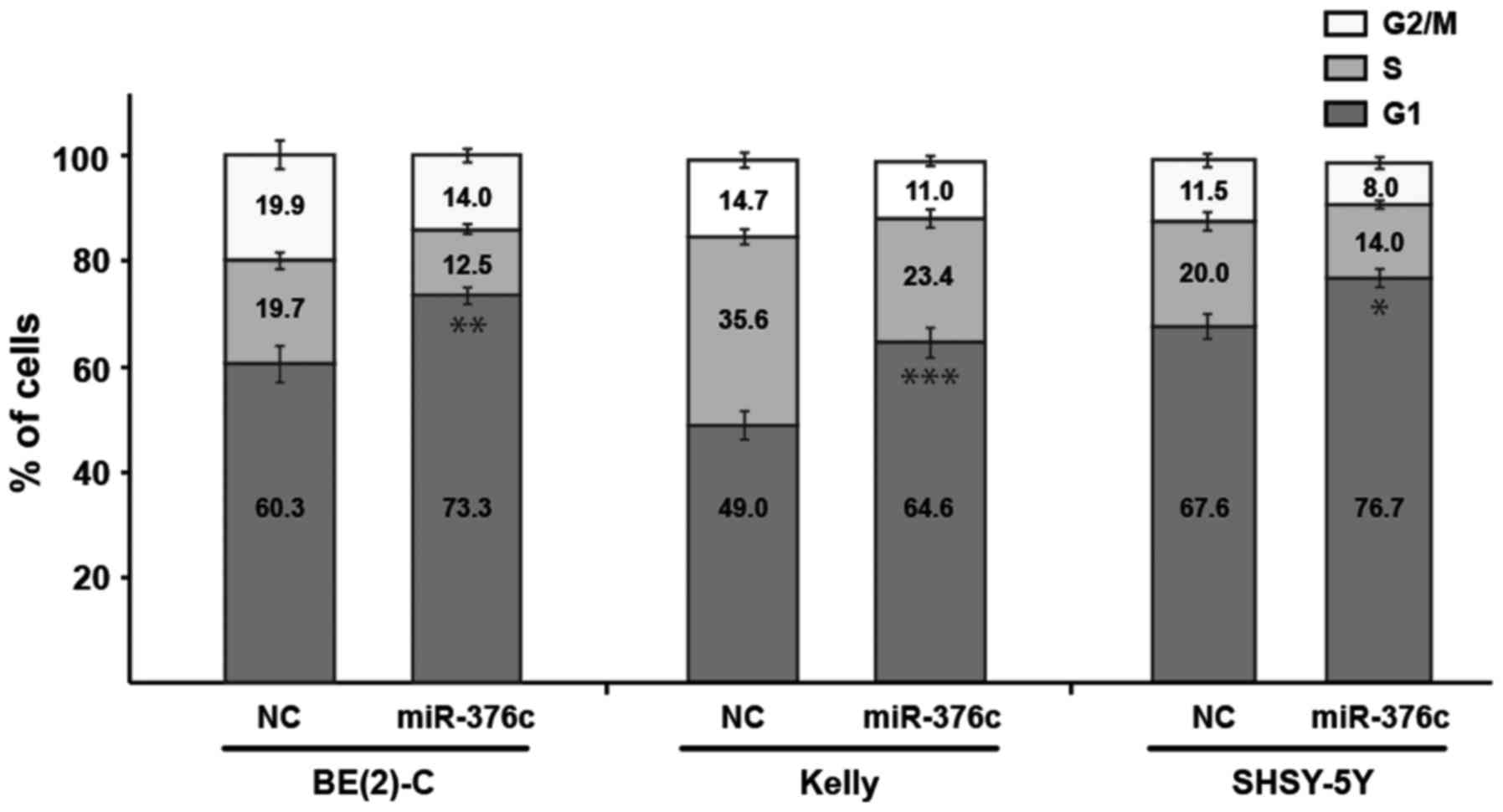
miR-376c-3p induces a G1-cell cycle
arrest in neuroblastoma cells
We did not detect significant apoptosis in
miR-376c-3p transfected neuroblastoma cells using Annexin V
and PARP cleavage assay (data not shown). Thus, we investigated the
effects of miR-376c-3p overexpression on cell cycle
distribution of representative neuroblastoma cell lines BE(2)-C,
Kelly and SHSY-5Y by flow cytometric assay. Ectopic expression of
miR-376c-3p in BE(2)-C, Kelly and SHSY-5Y resulted in
increased percentage of cells in G1-phase of cell cycle as compared
to NC transfected cells by 13% (**P=0.0023), 16% (***P=0.0001) and
9% (*P=0.0106), respectively with a corresponding reduction in the
percentage of cells in the S and G2/M-phase (Fig. 3). Thus, this observation led us to the
conclusion that decrease in cell viability might be due to
induction of G1-cell cycle arrest in neuroblastoma cells and not
apoptosis.
CCND1 is a direct target of
miR-376c-3p in neuroblastoma
In order to investigate the underlying molecular
mechanisms of miR-376c-3p induced suppression of the cell
viability and G1-cell cycle arrest, a bioinformatics analysis was
performed using miRNA target prediction algorithm TargetScan
(Release 6.2; www.targetscan.org/) to predict the target genes of
miR-376c-3p mainly associated with the cell cycle
progression. TargetScan revealed 254 potential downstream targets
with the conserved sites for miR-376c-3p (data not shown).
It was theoretically demonstrated that miR-376c-3p had
single binding site in the 3′UTR of CCND1 oncogene. Thus, to
determine whether miR-376c-3p could directly target the
predicted 3′UTR of CCND1, a dual-luciferase reporter assay
was performed in BE(2)-C and SHSY-5Y cells. Here, we used a
luciferase reporter containing full-length (~3.1 kb), wild type
CCND1 3′UTR (wt) or mutant CCND1 3′UTR (MUT)
construct having a mutation of the putative miR-376c-3p
target site shown in bold and italics (Fig. 4A).
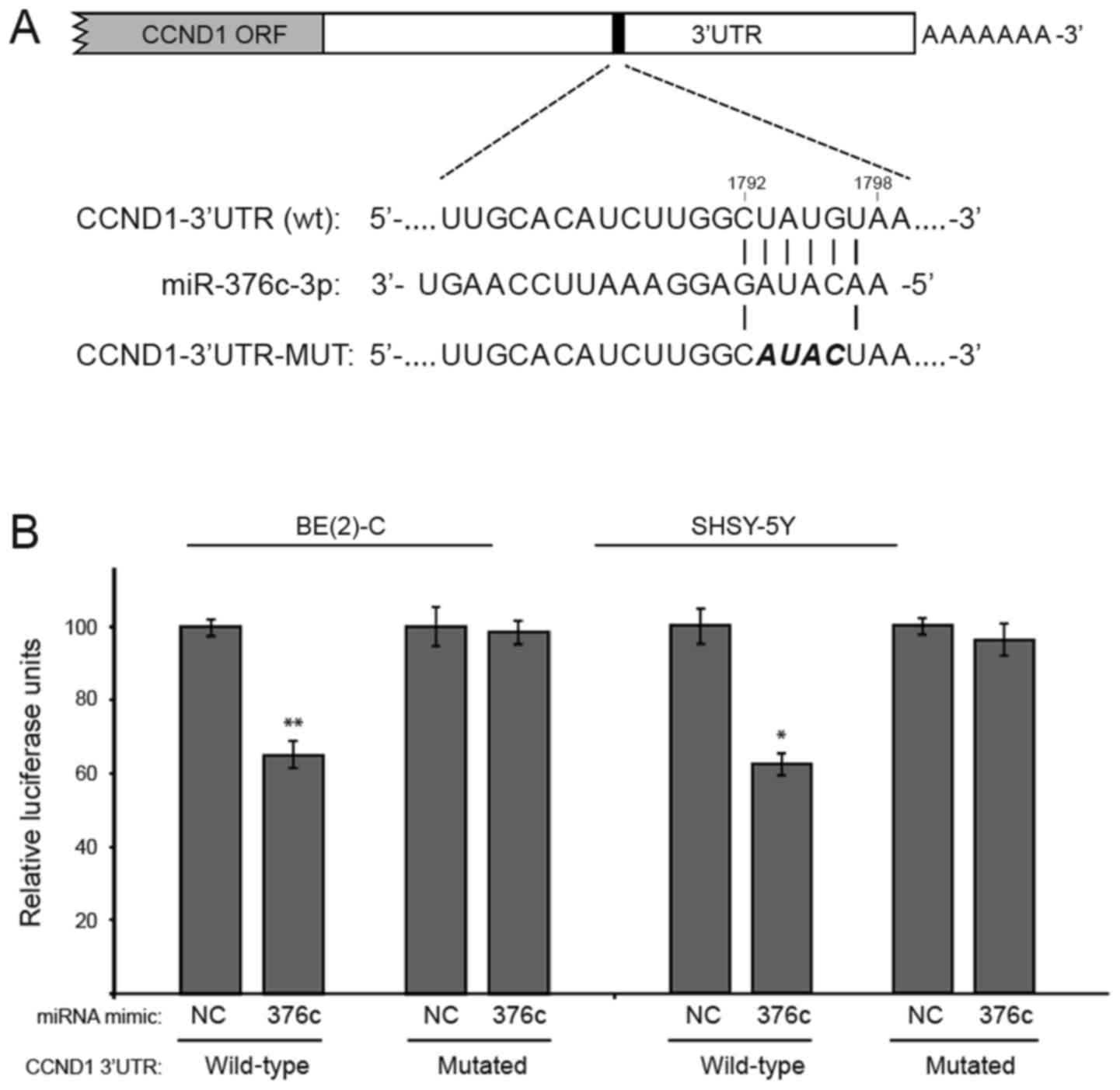 | Figure 4.CCND1 is a direct target of
miR-376c-3p in neuroblastoma. (A) The CCND1 3′UTR
contain one predicted miR-376c-3p binding site (nucleotides
1,792 to 1,798). In the figure, the alignment of the seed region of
miR-376c-3p with CCND1 and the site of target
mutagenesis are shown in bold and italics. (B) pMIR-Report-CCND1
luciferase constructs containing a full-length wt or mutated
CCND1 3′UTR and miR-376c-3p or NC mimics, were
co-transfected into BE(2)-C and SHSY-5Y cells. Luciferase activity
of wt construct was significantly reduced compared with mutated
constructs. Relative repression of firefly luciferase activity is
normalized with renilla luciferase activity. Error bars
indicate mean ± standard devaition of three independent experiments
repeated in triplicates. *P<0.05, **P<0.01 vs. the adjacent
NC. miR, microRNA; NC, negative control; CCND1, cyclin D1; ORF,
open reading frame; UTR, untranslated region; wt, wild type; MUT,
mutated. |
Transient co-transfection of BE(2)-C and SHSY-5Y
cells with miR-376c-3p mimics and the wild type CCND1
3′UTR (wt) reporter construct suppressed the luciferase activity as
compared to NC transfected cells by 35% (**P=0.0091) and 38%
(*P=0.0135), respectively. However, the activity of the reporter
construct mutated at the specific miR-376c-3p target site is
unaffected (Fig. 4B). These data
indicated that miR-376c-3p represses CCND1 expression
by directly binding to the 3′UTR sequence of CCND1 mRNA.
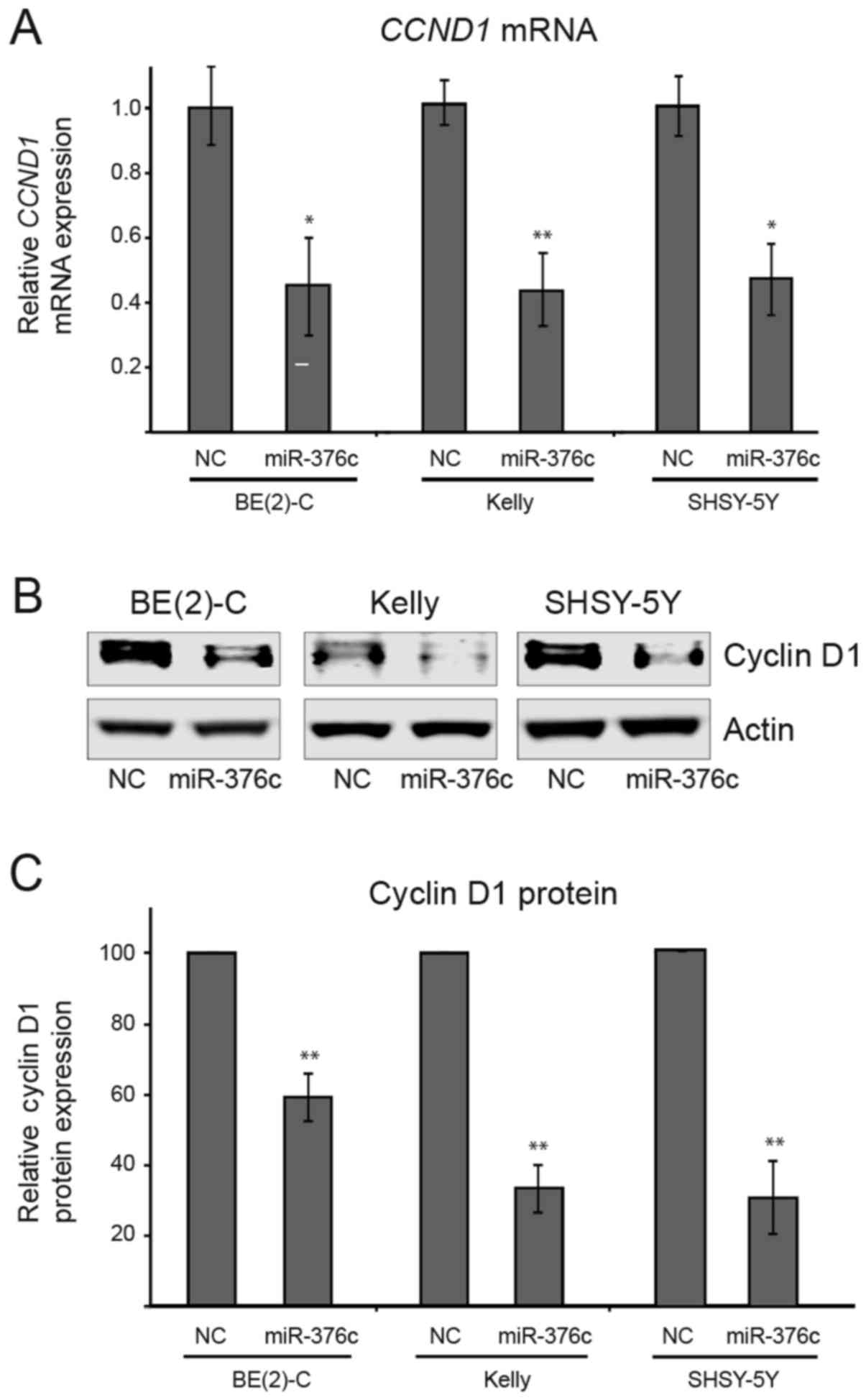
miR-376c-3p reduces mRNA and protein
levels of CCND1 in neuroblastoma cells
To further confirm whether miR-376c-3p
directly targets CCND1 gene, neuroblastoma cells were
transfected with miR-376c-3p or NC mimics and the expression
levels of CCND1 was analyzed by quantitative RT-qPCR
analysis. The levels of CCND1 mRNA was markedly decreased by
miR-376c-3p overexpression in BE2-(C), Kelly and SHSY-5Y
cells by 57% (*P=0.0110), 57% (**P=0.0017), and 53% (*P=0.0134),
respectively as compared to NC transfected cells (Fig. 5A). Moreover, we also performed western
blot analysis and observed significant decrease in levels of cyclin
D1 proteins upon miR-376c-3p overexpression in BE2-(C),
Kelly and SHSY-5Y cells by 41% (**P=0.0089), 67% (**P=0.0032), and
69% (**P=0.0069), respectively as compared to NC transfected cells
(Fig. 5B and C). Taken together,
these results demonstrates that endogenous expression of
CCND1 gene is directly regulated by miR-376c-3p and
suggest that overexpression of CCND1 gene could be reduced
by enforced expression of miR-376c-3p in neuroblastoma
cells.
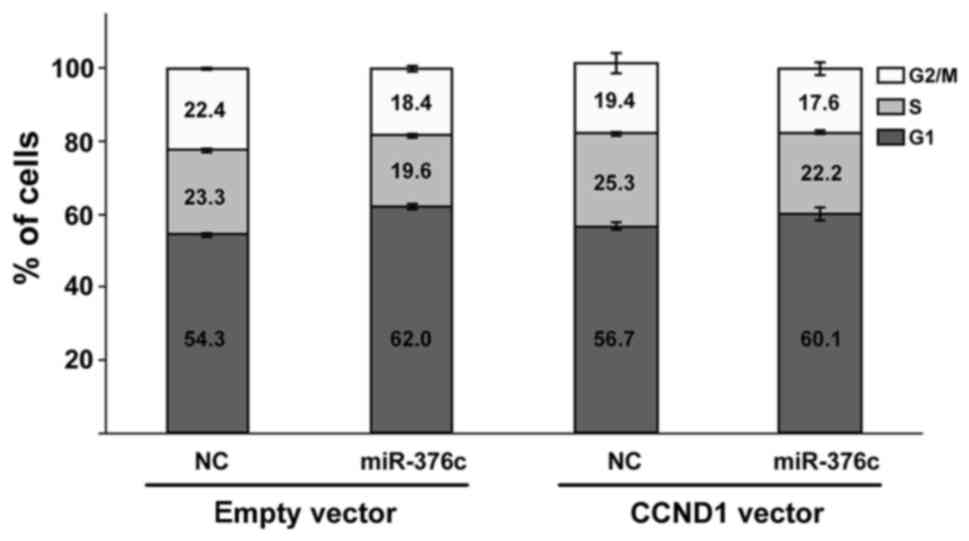
In addition, to test whether CCND1 could also
counteract the effect of miR-376c-3p induced G1 cell cycle
arrest, we overexpressed CCND1 in SHSY-5Y cells. However, we
only saw a modest and statistically insignificant effect of
CCND1 overexpression on reducing the effect of miR-376c-3p
(Fig. 6).
Discussion
Current treatment strategies for high-risk
neuroblastoma patients have limitations due to the refractory
nature of the disease (2–4). Hence, alternative strategies are
necessary for diagnosis and treatment of this disease. Mounting
evidence have shown the potential of miRNAs as key regulators of
cancer pathogenesis by acting as oncogenes or tumor suppressors
(6). Researchers have thus exploited
these properties for the development of the novel anticancer
therapies. miRNAs act by inhibiting the expression of its one or
more target genes. Hence, identification of specific miRNAs and
their targets is very important for the diagnosis and therapy of
cancer (34).
Results from our previous deep sequencing analysis
study identified 22 downregulated microRNAs from 14q32 miRNA
cluster differentially expressed in neuroblastoma cell lines
isolated from six patients at diagnosis and at relapse after
intensive treatments. miR-376c-3p, is one of the 22 miRNAs
that was downregulated in most of the cell lines isolated from
patients after treatment (32).
Moreover, the expression of miR-376c-3p was reduced in
International Neuroblastoma Staging System (INSS) stage 4 compared
to stage 1–2 in a cohort of 226 primary neuroblastoma tumors
(32). However, the functional role
of miR-376c-3p in neuroblastoma is not yet established.
Earlier reports from various cancers have
demonstrated the dual nature of miR-376c-3p to act as either
the oncogenic or the tumor suppressive miRNA depending on the
cellular contexts (10–20). miR-376c-3p is downregulated in
multiple human cancers, including prostate cancer (11), cervical cancer (10), oral squamous cell carcinoma (12), intrahepatic cholangiocarcinoma
(13), melanoma (14), osteosarcoma (15) and gliomas (16). In contrast, miR-376c-3p was
upregulated in acute myeloid leukemia (17) and gastric cancer (18). In these cancers, miR-376-3p has
been shown to target a set of genes including B-cell-specific
moloney murine leukemia virus insertion site 1 (BMI1), homeobox B7
(HOXB7), growth factor receptor-bound protein 2 (GRB2),
transforming growth factor-alpha (TGFA), liver receptor homolog-1
(LRH-1), insulin growth factor 1 receptor (IGF1R) and activin
receptor-like kinase 7 (ALK7). Other than affecting cell growth and
proliferation, these genes act as important mediators of cell
invasion (10,12,15, 20),
migration (11–14), cell cycle arrest (10), apoptosis (12) and chemoresistance (19).
In order to find out how miR-376c-3p affects
the growth and viability of neuroblastoma cells we first performed
alamarBlue cell viability assay and demonstrated that transient
overexpression of miR-376c-3p resulted in inhibition of cell
viability in neuroblastoma cell lines. Consistent with this
finding, we also observed that miR-376c-3p induced G1-cell
cycle arrest suggesting that cell cycle genes may be affected and
could serve as miR-376c-3p targets. To test this hypothesis,
we used miRNA bioinformatics algorithms to predict
miR-376c-3p targets related to the cell cycle regulation.
TargetScan algorithm predicted the CCND1 gene as a probable
target of miR-376c-3p. CCND1 is an important cell cycle
regulator whose mRNA contains a conserved miR-376c-3p
binding site on the 3′UTR. By dual-luciferase reporter assay, we
further demonstrated that miR-376c-3p could significantly
reduce luciferase activity of wild type constructs but not mutated
CCND1 3′UTR construct confirming the direct regulation on
CCND1 by miR-376c-3p. Our experimental data further
showed that the expression of CCND1 mRNA and protein levels
were significantly reduced after transfection with
miR-376c-3p mimics as compared to NC mimics transfected
cells. Additionally, we also performed flow cytometric rescue
experiment by overexpression of CCND1 in neuroblastoma cells
to see if CCND1 counteracts the effect of miR-376c-3p
induced G1 cell cycle arrest. However, we only saw a modest and
statistically insignificant effect of CCND1 overexpression
on reducing the effect of miR-376c-3p overexpression.
Cyclin D1, Cyclin D2 and Cyclin D3 belongs to the
class of cyclins, which activates the cyclin-dependent kinases
(CDKs). These cyclins through the phosphorylation of the substrates
at specific cell stages co-ordinates the sequential completion of
DNA replication and cell division (23). Unlike other cyclins, CCND1 is
induced by extracellular signals, including growth factor receptor
activation and integrin-derived adhesion signaling (23,35).
Functionally, CCND1 activates CDK4/6, which
phosphorylates pRB family proteins causing the release of E2F
transcription factors, which are essential for transcription of
genes necessary for G1-S transition of cell cycle. Hence, the
expression of cyclins are tightly regulated by variety of signaling
pathways at the transcriptional as well as post-transcriptional
levels (21).
The CCND1 gene has been proposed as an
important oncogene in various cancers. Numerous studies have
documented the relationship between deregulation of CCND1
and onset of tumorigenesis in wide variety of cancers. For
instance, Molenaar et al (36,37) found
that CCND1 levels were increased in neuroblastoma tumors and
high expression of CCND1 led to tumorigenesis in
neuroblastoma. In addition, Rihani et al (22) found that knockdown of CCND1
reduced cell proliferation, induced G1-cell cycle arrest and
inhibited the cyclin D1-Rb pathway in neuroblastoma cells.
Similarly, Sun and colleagues also found that inhibition of
CCND1 and CDK6 by miR-34a resulted in G1-cell
cycle arrest in non-small cell lung cancer (38). In line with these reports, our data
indicate that ectopic expression of miR-376c-3p leads to the
suppression of endogenous CCND1 gene resulting in the
reduced cell growth and G1-cell cycle arrest in the neuroblastoma
cells.
Taken together, our study for the first time
demonstrates that miR−376c-3p directly target CCND1
gene leading to reduced cell growth and G1-cell cycle arrest in
neuroblastoma. Therefore, we suggest that ‘miR-376c-CCND1’
axis could be a potential molecular target for preventing
neuroblastoma tumorigenesis.
Acknowledgements
The authors would like to thank the Children's
Oncology Group Cell Culture and Xenograft Repository (Texas Tech
University Health Science Center, Lubbock, TX, USA) for providing
the BE(2)-C, CHLA-15 and CHLA-20 cell lines, and Dr. Laura Barkley
(National Centre for Biomedical Engineering Science, National
University of Ireland Galway, Galway, Ireland) for providing the
full-length CCND1 3′UTR construct (pMIR-Report-Cyclin
D1-UTR-WT). The authors would also like to thank Professor Pieter
Mestdagh (Ghent University, Ghent, Belgium) for providing the miRNA
expression data from primary neuroblastoma tumors obtained through
the Neuroblastoma Research Consortium initiative.
Funding
This work was supported by grants from the
Northern-Norway Health Authorities (grant no. SFP 1278-16).
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Author's contributions
SPB, TF and CE designed the research. SPB performed
the experiments. CL assisted with the experiments. CE, CL and TF
supervised the experimental work. SPB wrote the manuscript. CE, CL
and TF critically amended the manuscript. The final manuscript was
read and approved by all authors.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
miRNA
|
microRNA
|
|
NC
|
negative control
|
|
miR-376c-3p
|
microRNA-376c-3p
|
|
CCND1
|
Cyclin D1
|
References
|
1
|
Brodeur GM: Neuroblastoma: Biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garaventa A: High risk neuroblastoma:
Small steps towards cure. Pediatr Blood Cancer. 61:964–965. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garaventa A, Parodi S, De Bernardi B, Dau
D, Manzitti C, Conte M, Casale F, Viscardi E, Bianchi M, D'Angelo
P, et al: Outcome of children with neuroblastoma after progression
or relapse. A retrospective study of the Italian neuroblastoma
registry. Eur J Cancer. 45:2835–2842. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Croce CM and Calin GA: miRNAs, cancer, and
stem cell division. Cell. 122:6–7. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Berindan-Neagoe I, Pdel Monroig C,
Pasculli B and Calin GA: MicroRNAome genome: A treasure for cancer
diagnosis and therapy. CA Cancer J Clin. 64:311–336. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kawahara Y, Zinshteyn B, Sethupathy P,
Iizasa H, Hatzigeorgiou AG and Nishikura K: Redirection of
silencing targets by adenosine-to-inosine editing of miRNAs.
Science. 315:1137–1140. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seitz H, Royo H, Bortolin ML, Lin SP,
Ferguson-Smith AC and Cavaillé J: A large imprinted microRNA gene
cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 14:1741–1748.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Deng Y, Xiong Y and Liu Y: miR-376c
inhibits cervical cancer cell proliferation and invasion by
targeting BMI1. Int J Exp Pathol. 97:257–265. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Formosa A, Markert EK, Lena AM, Italiano
D, Finazzi-Agro' E, Levine AJ, Bernardini S, Garabadgiu AV, Melino
G and Candi E: MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c,
miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p,
mapped to the 14q32.31 locus, regulate proliferation, apoptosis,
migration and invasion in metastatic prostate cancer cells.
Oncogene. 33:5173–5182. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang K, Jin J, Ma T and Zhai H:
miR-376c-3p regulates the proliferation, invasion, migration, cell
cycle and apoptosis of human oral squamous cancer cells by
suppressing HOXB7. Biomed Pharmacother. 91:517–525. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iwaki J, Kikuchi K, Mizuguchi Y,
Kawahigashi Y, Yoshida H, Uchida E and Takizawa T: miR-376c
down-regulation accelerates EGF-dependent migration by targeting
GRB2 in the HuCCT1 human intrahepatic cholangiocarcinoma cell line.
PLoS One. 8:e694962013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zehavi L, Avraham R, Barzilai A, Bar-Ilan
D, Navon R, Sidi Y, Avni D and Leibowitz-Amit R: Silencing of a
large microRNA cluster on human chromosome 14q32 in melanoma:
Biological effects of mir-376a and mir-376c on insulin growth
factor 1 receptor. Mol Cancer. 11:442012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jin Y, Peng D, Shen Y, Xu M, Liang Y, Xiao
B and Lu J: MicroRNA-376c inhibits cell proliferation and invasion
in osteosarcoma by targeting to transforming growth factor-alpha.
DNA Cell Biol. 32:302–309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang Q, Wang C, Hou Z, Wang G, Lv J, Wang
H, Yang J, Zhang Z and Zhang H: Serum microRNA-376 family as
diagnostic and prognostic markers in human gliomas. Cancer Biomark.
19:137–144. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dixon-McIver A, East P, Mein CA, Cazier
JB, Molloy G, Chaplin T, Lister Andrew T, Young BD and Debernardi
S: Distinctive patterns of microRNA expression associated with
karyotype in acute myeloid leukaemia. PLoS One. 3:e21412008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shiotani A, Murao T, Kimura Y, Matsumoto
H, Kamada T, Kusunoki H, Inoue K, Uedo N, Iishi H and Haruma K:
Identification of serum miRNAs as novel non-invasive biomarkers for
detection of high risk for early gastric cancer. Br J Cancer.
109:2323–2330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye G, Fu G, Cui S, Zhao S, Bernaudo S, Bai
Y, Ding Y, Zhang Y, Yang BB and Peng C: MicroRNA 376c enhances
ovarian cancer cell survival by targeting activin receptor-like
kinase 7: Implications for chemoresistance. J Cell Sci.
124:359–368. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang W, Tian Y, Jiang S, Liu S, Zhao X
and Tian D: MicroRNA-376c suppresses non-small-cell lung cancer
cell growth and invasion by targeting LRH-1-mediated Wnt signaling
pathway. Biochem Biophys Res Commun. 473:980–986. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arand J and Sage J: G1 cyclins protect
pluripotency. Nat Cell Biol. 19:149–150. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rihani A, Vandesompele J, Speleman F and
Van Maerken T: Inhibition of CDK4/6 as a novel therapeutic option
for neuroblastoma. Cancer Cell Int. 15:762015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deshpande A, Pastore A, Deshpande AJ,
Zimmermann Y, Hutter G, Weinkauf M, Buske C, Hiddemann W and
Dreyling M: 3′UTR mediated regulation of the cyclin D1
proto-oncogene. Cell Cycle. 8:3592–3600. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mitra D, Das PM, Huynh FC and Jones FE:
Jumonji/ARID1 B (JARID1B) protein promotes breast tumor cell cycle
progression through epigenetic repression of microRNA let-7e. J
Biol Chem. 286:40531–40535. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zheng L, Qi T, Yang D, Qi M, Li D, Xiang
X, Huang K and Tong Q: microRNA-9 suppresses the proliferation,
invasion and metastasis of gastric cancer cells through targeting
cyclin D1 and Ets1. PLoS One. 8:e557192013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bonci D, Coppola V, Musumeci M, Addario A,
Giuffrida R, Memeo L, D'Urso L, Pagliuca A, Biffoni M, Labbaye C,
et al: The miR-15a-miR-16-1 cluster controls prostate cancer by
targeting multiple oncogenic activities. Nat Med. 14:1271–1277.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Trompeter HI, Abbad H, Iwaniuk KM, Hafner
M, Renwick N, Tuschl T, Schira J, Müller HW and Wernet P: MicroRNAs
miR-17, miR-20a, and miR-106b act in concert to modulate E2F
activity on cell cycle arrest during neuronal lineage
differentiation of USSC. PLoS One. 6:e161382011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hermeking H: The miR-34 family in cancer
and apoptosis. Cell Death Differ. 17:193–199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Elliman SJ, Howley BV, Mehta DS, Fearnhead
HO, Kemp DM and Barkley LR: Selective repression of the oncogene
cyclin D1 by the tumor suppressor miR-206 in cancers. Oncogenesis.
3:e1132014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Buechner J, Tømte E, Haug BH, Henriksen
JR, Løkke C, Flægstad T and Einvik C: Tumour-suppressor microRNAs
let-7 and mir-101 target the proto-oncogene MYCN and inhibit cell
proliferation in MYCN-amplified neuroblastoma. Br J Cancer.
105:296–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Roth SA, Knutsen E, Fiskaa T, Utnes P,
Bhavsar S, Hald ØH, Løkke C, Mestdagh P, Johansen SD, Flægstad T
and Einvik C: Next generation sequencing of microRNAs from isogenic
neuroblastoma cell lines isolated before and after treatment.
Cancer Lett. 372:128–136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thorsen SB, Obad S, Jensen NF, Stenvang J
and Kauppinen S: The therapeutic potential of microRNAs in cancer.
Cancer J. 18:275–284. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Witzel II, Koh LF and Perkins ND:
Regulation of cyclin D1 gene expression. Biochem Soc Trans.
38:217–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Molenaar JJ, Ebus ME, Koster J, van Sluis
P, van Noesel CJ, Versteeg R and Caron HN: Cyclin D1 and CDK4
activity contribute to the undifferentiated phenotype in
neuroblastoma. Cancer Res. 68:2599–2609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Molenaar JJ, van Sluis P, Boon K, Versteeg
R and Caron HN: Rearrangements and increased expression of cyclin
D1 (CCND1) in neuroblastoma. Genes Chromosomes Cancer. 36:242–249.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R,
Sun Z and Zheng X: Downregulation of CCND1 and CDK6 by miR-34a
induces cell cycle arrest. FEBS Lett. 582:1564–1568. 2008.
View Article : Google Scholar : PubMed/NCBI
|