Introduction
Lung cancer is a major cause of cancer-associated
mortality, accounting for about 25% of all cancer mortalities
globally in 2015 (1). Small cell lung
cancer (SCLC), 15% of all lung cancer cases, is an extremely
aggressive malignancy with a tendency of fast growing rates, early
distant metastases and poor prognosis (1). The 5-year survival rate of all patients
with SCLC is <10% (2). There are
three kinds of SCLC treatment: The first is treatment with
chemotherapeutic drugs (3). The
second type involves targeted chemotherapy, which utilizes
compounds that function in tumor cells with specific mutations
prevent their proliferation, including gefitinib and imatinib
(3). The third is immunotherapy
(3). Although, SCLC is sensitive to
chemotherapy and radiotherapy, there remains no effective way to
fully treat SCLC (4). The majority of
patients relapsed following first-line treatment (4).
The effect of available molecular targeted drug is
not satisfactory (3,5), therefore, it is of great significance to
develop the molecule-targeted drugs with a good therapeutic effect
but limited side effects.
Marine is rich in biological resources and has a
complicated ecological environment (6,7). Marine
nature products with diverse structures are an important source of
drug discovery (8). For example,
Wentilactone A (WA), as a small molecule marine-derived endophytic
fungus, can inhibit the proliferation of SCLC cell line NCI-H446,
as indicated by previous research (9); however, this previous study did not
investigate the mechanism of WA inhibiting the proliferation of
SCLC cells. The present study aimed to examine the anti-SCLC
mechanism underlying WA in vitro and in vivo.
Materials and methods
Chemical compounds and reagents
The molecular weight of WA was 304 g/mol. The
density of WA was 1.53 g/cm3. The boiling point was
651.2°C at 760 mmHg. The WA used was supplied by the Institute of
Oceanology of the Chinese Academy of Sciences, Qingdao, China,
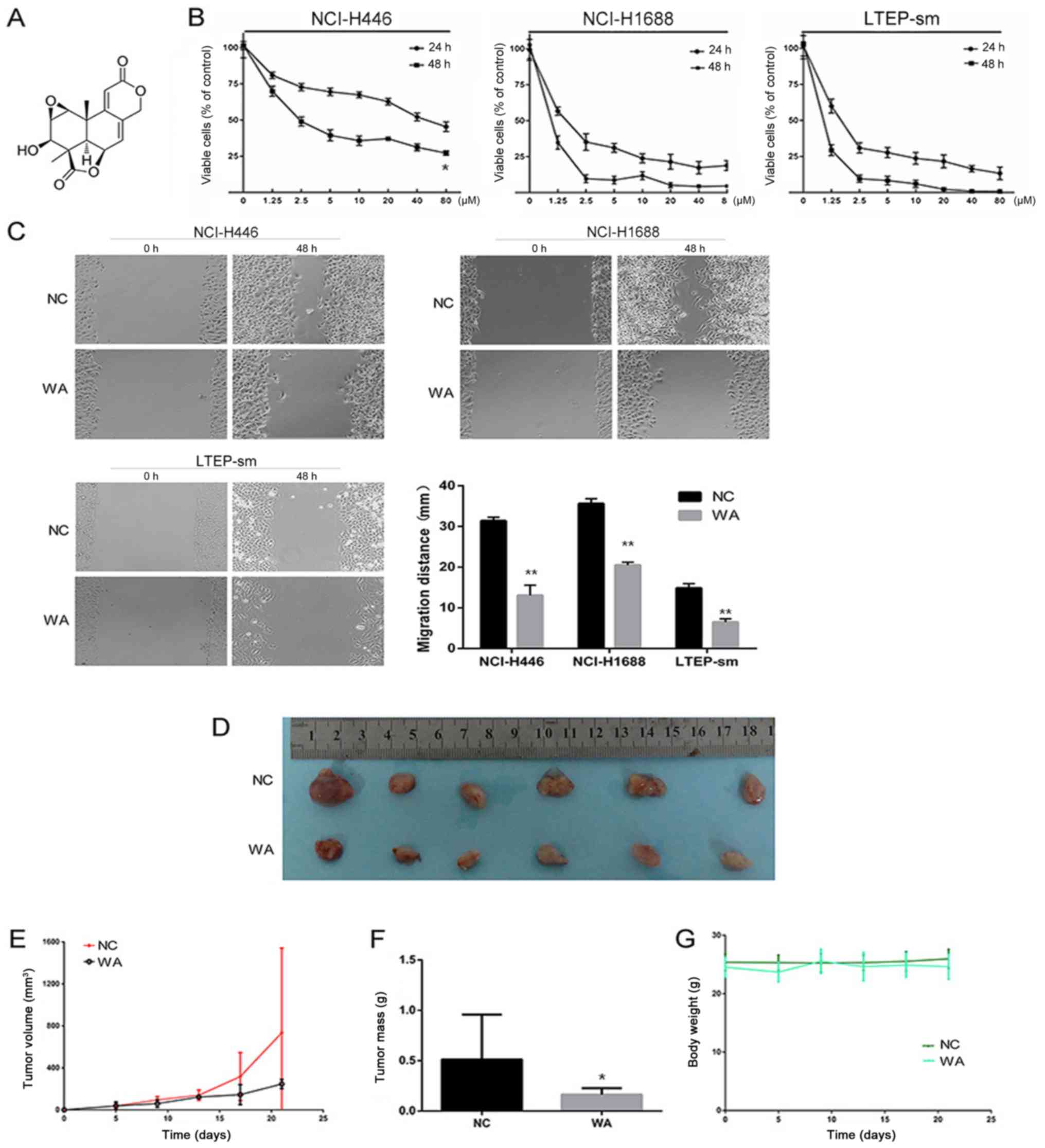
(chemical structure depicted in Fig.
1A, was dissolved in 100% dimethyl sulfoxide (DMSO) (final
concentration of WA was 1 mg/ml), stored at −20°C and diluted with
culture medium (final concentration of WA was 10 µM) (RPMI-1640
with 10% fetal bovine serum (FBS) and penicillin and streptomycin
(10 µl/ml). RPMI-1640, FBS, trypsin-EDTA, penicillin and
streptomycin were purchased from Biowest (Nuaillé, France). DMSO
was purchased from Bio-Light Biotech (Shanghai, China). The BCA
protein assay kit, Annexin V-fluorescein isothiocyanate/propidium
iodide (PI) apoptosis detection kit and Annexin V-APC Cell
Apoptosis Analysis kit (with PI) were purchased from Tianjin
Sungene Biotech Co., Ltd. (Beijing, China). Recombinant human
insulin like growth factor-1 (IGF-1; 100-11) was purchased from
PeproTech China (Suzhou, China). Anti-IGF-1 receptor (R) (ab182408;
1:1,000), anti-phosphorylated (p)IGF-1R (ab39398; 1:1,000), insulin
receptor substrate 1 (IRS1) (ab40777; 1:1,000), anti-p-IRS1
(ab46800; 1:1,000), anti-Caspase-3 (ab13847; 1:1,000),
anti-cleaved(c)-Caspase-3 (ab2302; 1:1,000), anti-aldo-keto
reductase family 1 member C1 (AKR1C1)/AKR1C2 (ab96087; 1:1,000),
anti-p-PI3K (ab182651; 1:1,000), anti-PI3K (ab1678; 1:1,000),
anti-AKT (ab179463; 1:1,000) and anti-p-AKT (ab81283; 1:1,000) were
purchased from Abcam (Cambridge, UK). Anti-Fas-associated death
domain-like interleukin-1-converting enzyme-like inhibitory protein
(FLIP) (ARG54328; 1:1,000) was purchased from Arigo Biolaboratories
(Taiwan, R.O.C. China). Anti-cleaved (c)-FLIP (sc-8347, 1:1,000)
was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). The chemiluminescence reagent was obtained from EMD Millipore
(Billerica, MA, US). AKR1C1 gene expression lentivirus and AKR1C1
shRNA lentivirus were purchased from Shanghai Genepharma Technology
Co., Ltd. (Shanghai, China). RevertAid First Stand cDNA Synthesis
Kit was obtained from Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). TRIzol® reagent was purchased from Thermo Fisher
Scientific, Inc.
Cell lines and culture
Human SCLC cell lines NCI-H446, NCI-H1688 and
LTEP-sm cells were purchased from Tongpai Biological Technology
Co., Ltd (Shanghai, China). NCI-H446, NCI-H1688 and LTEP-sm cells
were cultured in RPMI-1640 containing 10% FBS (Biological
Industries, Cromwell, CT, USA), supplemented with 100 U/ml
penicillin and 125 µg/ml streptomycin at 37°C in a 5%
CO2 incubator.
Cell proliferation assay
NCI-H446, NCI-H1688 and LTEP-sm cells were treated
with 10 µM WA at 0, 24 and 48 h at 37°C. Cell growth was measured
by Cell Counting Kit-8 assay (CCK-8; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan), according to the
manufacturer's protocols. Experiments were repeated in
triplicate.
Scratch assay
NCI-H446, NCI-H1688, LTEP-sm cells were placed on
6-well plates (Costar; Corning Incorporated, Corning, NY, USA) at a
cell density of 5×105 cells/well in RPMI-1640 medium at
37°C. At time 0 h, a 200 µl pipette tip was used to perform
scratches in the confluent monolayer. At 48 h, images were captured
of scratches, using the marked plate bottom for orientation, with
an Olympus 1X71 fluorescence microscope (×200), Olympus DP72 camera
and DPController Software (Olympus Corporation, Tokyo, Japan).
Measurements of scratch distance were completed by measuring the
difference between the length of the initial scratch and the length
of the scratch at 48 h.
Cell apoptosis analysis
Cell apoptosis was determined by Annexin V-APC/PI
apoptosis detection kit. Following treatment with WA (10 µM) at
37°C for 0, 24 or 48 h, cells were collected and washed once with
PBS and then suspended in 400 µl binding buffer (supplied with the
kit) for 30 sec. Following the addition of 4 µl Annexin V-APC at
room temperature, mixed and incubated in darkness for 10–15 min at
room temperature, then resuspended cells were centrifuged at room
temperature (600 × g for 3 min) to remove clear supernatant
extract. Additionally, 4 µl PI was added to every sample. Cells
were re-suspended in PBS for flow cytometry analysis.
cDNA microarray analysis
Total RNA of NCI-H446 cell was extracted prior to or
following a 48 h 10 µM WA treatment at 37°C using the RNeasy mini
kit (Qiagen GmbH, Hilden, Germany). The microarray experiment was
performed by Shanghai Genminix Informatics, Co., Ltd. (Shanghai,
China) using Affymetrix Human Transcriptome Array chip (version
2.0), AGCC Flution Control software, GeneChip System 3000Dx version
2.0 microarray scanner (Affymetrix; Thermo Fisher Scientific, Inc.)
and Genminix-Cloud Biotechnology Information platform (Genminix
Informatics Co., Ltd.), according to the manufacturer's
protocols.
Gene chip data analysis
GeneSifter program was used to analyze gene chip
data, which extracts patterns of gene expression from Affymetrix
gene expression data and uses Kyoto Encyclopedia of Genes and
Genomes (KEGG) (http://www.kegg.jp/), Gene Ontology
(GO) and Z-score reports to summarize the biological significance
of a gene list (10,11). All analyses were based on the
GeneSifter software (version 5.0; Geospiza, Inc., Seattle, WA,
USA). This program uses t-test and false discovery rate analysis to
identify differentially expressed genes (12,13).
Standard selection criteria to further identify differentially
expressed genes were as follows: |log2 ratios|≥1 and P<0.05
(log2 ratios≥1.0 represented up-regulated and log2 ratios≤−1.0
represented downregulated). The association of expression profiles
between the experimental and control groups was demonstrated by
unsupervised hierarchical clustering analysis tree. Using the KEGG
database, differently changed pathways were identified. Functional
differences of the expressed genes were analyzed by GO and assigned
into hierarchical categories.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) (9,14)
RT-qPCR was performed to determine the expressio of
AKR1C1 mRNA in each cell sample, using GAPDH as an endogenous
control for calibration. Total RNA was isolated using RNA Reagent
(Tiangen Biotech Co., Ltd., Beijing, China), and genomic DNA was
removed using Recombinant DNase I (Takara Biotechnology Inc.,
Dalian, Liaoning, China). RNA concentration and quality were
measured by NanoDrop 2000 (Thermo Fisher Scientific, Inc.,
Wilmington, DE, USA). Following analysis, samples with an OD
260/280 ratio from 1.9 to 2.1 were selected for further analysis.
The primer sequences were designed using an online tool (http://bioinfo.ut.ee/primer3/), and the primers were
purchased from BGI-Tech Solutions (Shanghai, China). The gene
primer sequences were: AKR1C1, forward, CGTTGCCAGCTCATTGCTCTT, and
reverse, TATGGCGGAAGCCAGCTTCAAT; GAPDH forward,
GAGTCAACGGATTTGGTCGT, and reverse, GACAAGCTTCCCGTTCTCAG. ABI 7500
real-time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) was used for the RT-qPCR reaction. The PCR solution (20 µl)
contained 10 µl SYBR green (RR091A, Takara Biotechnology Ltd.,
Dalian, China), 0.5 µl primer, 7 µl ddH2O and 2.5 µl cDNA. Thermal
cycler conditions for RT-qPCR were as follows: Pre-denaturation at
95°C for 180 sec. Denaturation at 95°C for 10 sec. Annealing for 20
sec at 60°C. Extension at 72°C for 20 sec. Melting curve analysis
was at 60°C for 6 sec. Using relative quantitative method and
taking the control group as blank control, the relative
transcriptional levels of mRNA of target genes were calculated
using the 2−ΔΔCq method (15). This method was used for subsequent
cell experiments. The experiment was repeated 3 times.
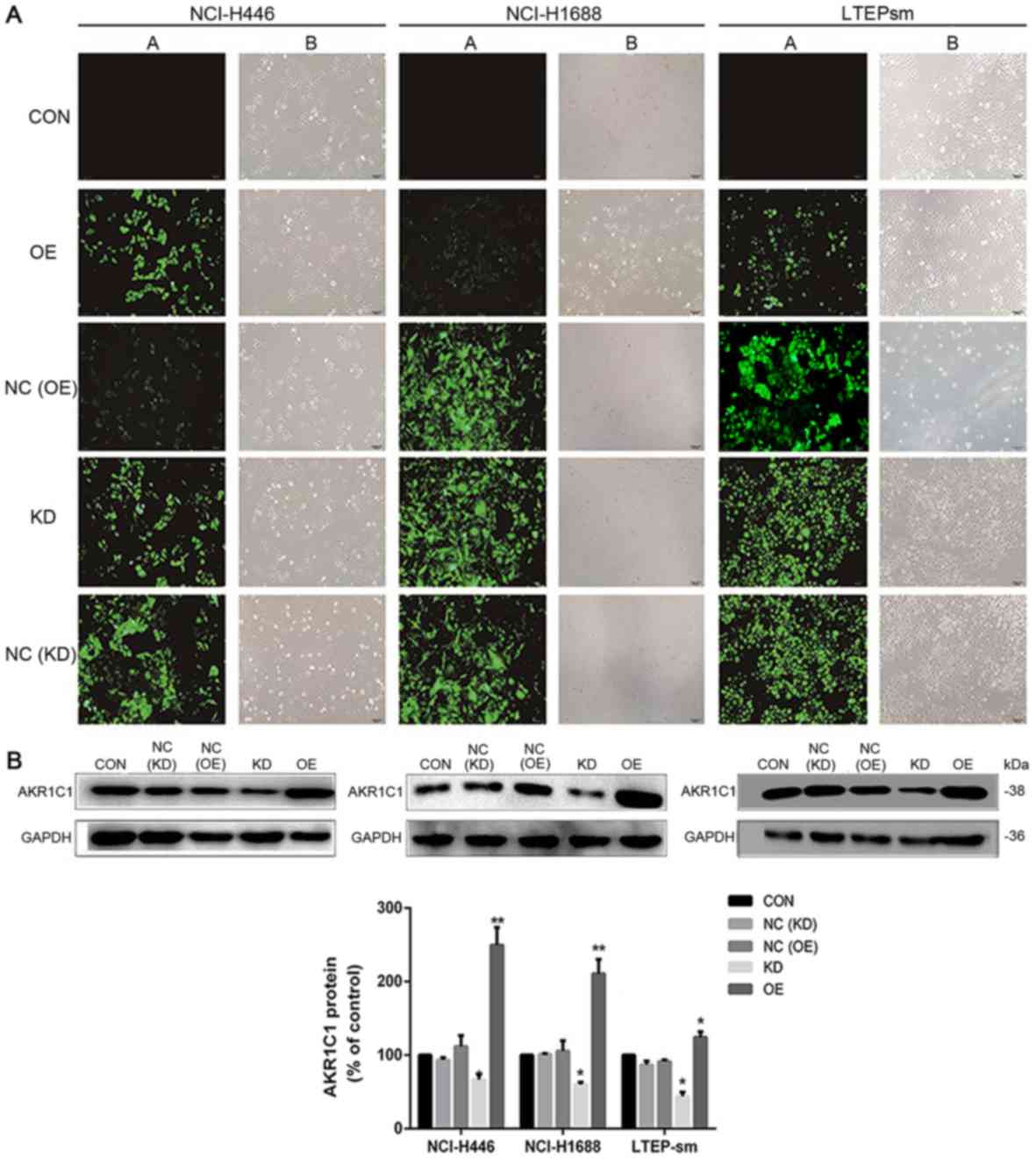
Transfection procedure
NCI-H446, NCI-H1688 and LTEP-sm cells were
respectively transfected with AKR1C1 expressing vector (OE group),
shRNA vector specific for AKR1C1 (KD group) and control green
fluorescent protein (GFP) vectors (NC group). The negative control
vector contained a GFP marker for cell tracking. The AKR1C1
expressing vector (pLenti-EF1α-GFP-puromycin-AKR1C1-Amp cDNA
expression lentiviral vector) contained a green fluorescent protein
marker. The shRNA vector specific for AKR1C1 vector was
pGLV-H1-GFP-puromycin-shRNA-AKR1C1 vector. All were purchased from
Shanghai GenePharma Co., Ltd. (Shanghai, China). Empty lentiviral
vector was used as control. To further investigate the biological
significance of the AKR1C1 gene in SCLC cells, NCI-H446, NCI-H1688
and LTEP-sm cells were transfected with AKR1C1 gene retroviral
vector plasmid (OE group) or lentiviral vector carrying sh-AKR1C1
plasmid (KD group). Following 48 h transfection, fluorescence
microscopy (×200) demonstrated the observed transfection
efficiency. Following 72 h infection, the efficiency was validated
by western blot analysis.
Cell proliferation assay following
transfection
AKR1C1 overexpression or knockdown carrier
transfected NCI-H446 cells were respectively plated in 96-well
plates at 4×103 cells/well. Following adherence
overnight, cell viability was measured at 0, 24, 48 and 72 h
following WA treatment at 37°C. Cell viability was determined by
CCK-8 assay (Dojindo Molecular Technologies, Inc.), according to
the manufacturer's protocols. The procedure also included adding
reconstituted CCK-8 in an amount equal to 10% of the RPMI-1640
medium volume and returning to the incubator at 37°C for 1 h.
Absorbance was measured at wavelength 450 nm. Experiments were
performed in triplicate, and three different experiments were
performed under the same experimental conditions.
Western blot analysis
Prior to WA treatment for the indicated time perods
(0, 12, 24 and 48 h), NCI-H446 and NCI-H1688 and LTEP-sm cells were
seeded to six-well plates, and split by radio immunoprecipitation
assay buffer containing phosphatase inhibitor, protease inhibitor
and phenylmethanesulfonyl fluoride (1:1,000). Then the protein
concentration was quantified by BCA protein assay kit. Using 10%
SDS-PAGE, the same amount of protein (10 µg) was separated,
transferred to nitrocellulose membranes and incubated with the
aforementioned corresponding primary and secondary antibodies.
Using the chemiluminescence reagent, the immunocomplexes were
visualized and detected on images.
Nude mice xenograft model
The nude mice xenograft models were established by
injection of 2×106 cells into the right armpit of 5-week
old BALB/c male athymic mice (body weight, 18–22 g; National Rodent
Laboratory Animal Resource, Shanghai, China). The mice were kept at
room temperature, with a 12 h light/dark cycle with ad libitum
access to food and water for 20 days. The mice were randomized into
6 groups (6 mice per group; total=36): Vehicle control (1% DMSO);
10 mg/kg WA; cells transfected with AKR1C1 overexpression carriers,
and 1% DMSO; cells transfected with AKR1C1 overexpression carriers,
and 10 mg/kg WA; cells transfected with AKR1C1 knockdown carriers,
and 1% DMSO; and cells transfected with AKR1C1 knockdown carriers,
and 10 mg/kg WA when xenografts were palpable. Vehicle or drugs
were administered intravenously every four days until sacrifice;
where body weight and tumor size were measured and recorded
simultaneously. Tumor size was measured using an electronic
caliper, and the tumor volumes were calculated using the formula:
(Length × width2)/2. Following 20 days, mice were
sacrificed, and the tumors were collected, weighed and imaged.
Tumor inhibition effect was calculated using the following
equation: Tumor suppression (%)=(1-T/C) ×100, where T is the
average tumor weight of the treated group and C is the average
tumor weight of the control group. All protocols were approved by
the University Animal Ethics Committee of the Second Military
Medicine University.
Immunohistochemistry
All tumor xenograft tissues were fixed with 10%
formalin for 24 h at room temperature and embedded in paraffin.
Liquid paraffin wax was added to an iron mold and cooled slightly,
then the tissue was placed in the paraffin wax and orientated
correctly. Additional liquid paraffin was added, and the wax was
frozen. The wax block was cut into 5 µm sections each for 3
consecutive slices, slides were incubated with 8% goat serum (Dako;
Agilent Technologies, Inc., Santa Clara, CA, USA; 250 µl normal
goat serum added to 5 ml 50 mM Tris-Cl) at 4°C for 2 h and
incubated with 50 µl the primary anti-AKR1C1 antibody at 4°C
overnight. Sections were washed with PBS three times, each time for
5 min. Subsequent to removing the PBS liquid, 50 µl biotin-labeled
secondary antibody (ab6788; Abcam; 1:500) was dropped onto each
slice, which was then incubated at room temperature for 10 min. All
stained sections were examined under a light microscope
(magnification, ×200). The sections were stained and analyzed
histologically according to the operating manual.
Statistical analysis
Statistical analysis was performed using SPSS 17.0
statistical software (SPSS, Inc., Chicago, IL, USA). All numerical
results are expressed as the mean ± standard deviation. Significant
differences among groups were determined with a one-way ANOVA. When
the differences were significant, a Student-Newman-Keuls test at a
5% probability was conducted. P<0.05 was considered to indicate
a statistically significant difference.
Results
WA significantly inhibits the
proliferation of SCLC cells
As depicted in Fig.
1B, WA inhibited the proliferation of SCLC cells in a dose- and
time-dependent manner. The 48 h IC50 value of WA was
3.44±0.38 µmol/l for NCI-H446 cells. The value was 0.41±0.18 µmol/l
for NCI-H1688 cells and 0.57±0.10 µmol/l for LTEP-sm cells. The
result of the scratch assay demonstrated that the migration ability
of NCI-H446 cells was significantly decreased following 48 h WA
treatment (P<0.01). Compared with the 48 h WA treatment group,
the migration distance of NCI-H1688 cells was significantly
decreased (P<0.01). Compared with the 48 h WA treatment group,
the migration distance of LTEP-sm cells was decreased (P<0.01)
(Fig. 1C). Further analysis on the
inhibitory effect of WA in SCLC cells indicated that WA suppressed
the growth of NCI-H446 cell xenograft tumor and no mice succumbed
to causes other than interventional sacrifice (Fig. 1D-G). WA inhibited the growth of SCLC
in vivo and in vitro.
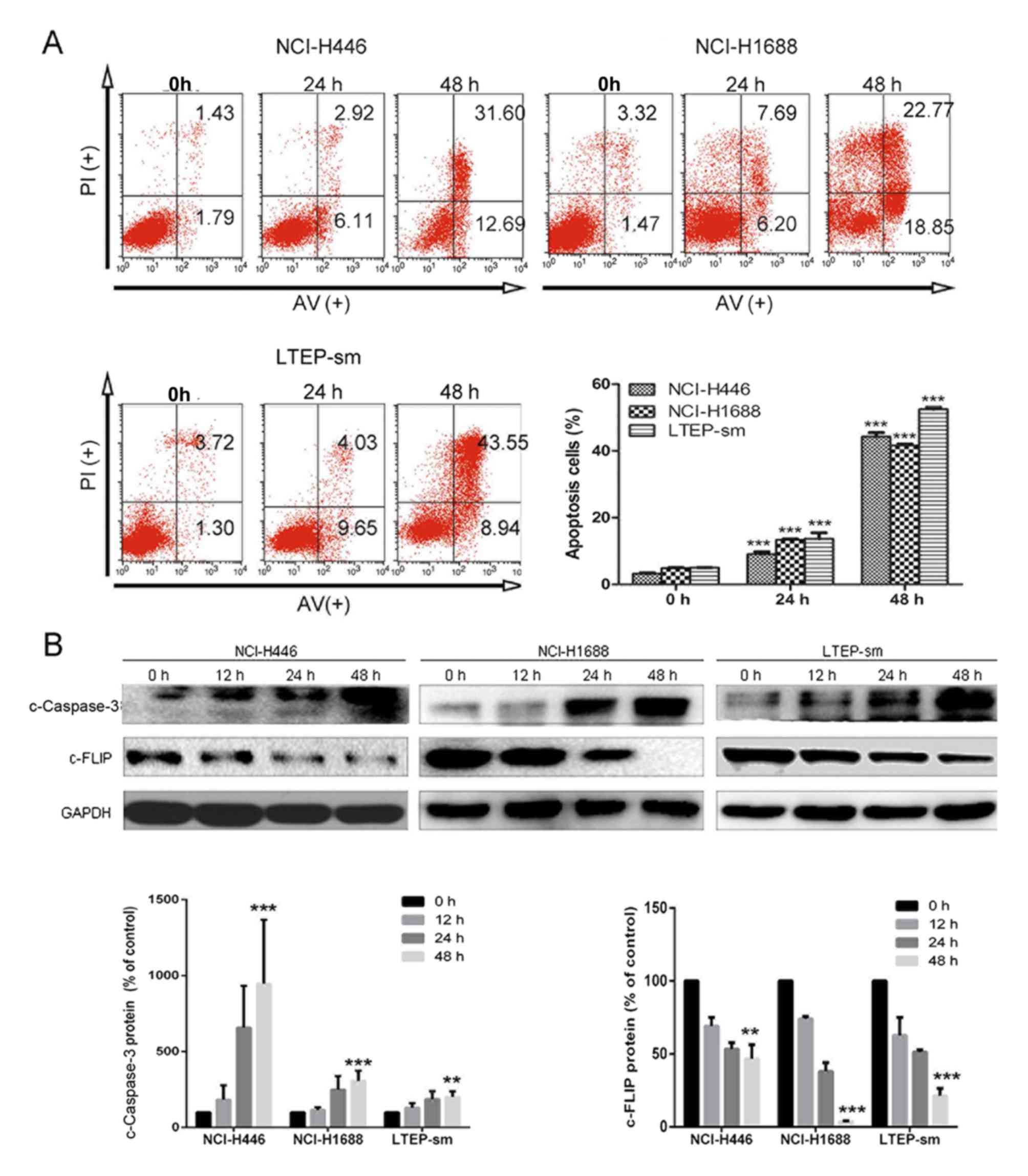
WA inhibits the growth of SCLC via
inducing apoptosis of tumor cells
The rates of cellular apoptosis were assessed in
three SCLC cell lines NCI-H446, NCI-H1688, LTEP-sm by flow
cytometry. The results demonstrated that the numbers of early and
late apoptotic cells at 24–48 h post-treatment of WA in SCLC cells
(Fig. 2A). Compared with the NC group
of NCI-H446 cells, the numbers of early and late apoptotic cells in
the WA treatment group was significantly increased (P<0.01).
Compared with the NC group of NCI-H1688 cells, the numbers of early
and late apoptotic cells following treatment of WA also
significantly increased (P<0.01). Following treatment with WA,
the numbers of early and late apoptotic LTEP-sm cells was
significantly increased (P<0.01). Western blot results indicated
that the apoptosis-associated protein levels of c-FLIP were
decreased in WA treatment groups in NCI-H446 (P<0.01), NCI-H1688
(P<0.001) and LTEP-sm (P<0.001) cells, compared with the NC
group; and the protein levels of c-Caspase-3 were increased in the
WA treatment groups compared with the NC group (P<0.001 for
NCI-H446, P<0.001 for NCI-H168 and P<0.01 for LTEP-sm). These
results indicated that WA induced cell apoptosis by regulating the
FLIP-dependent apoptosis pathway. The apoptosis must be
contributing factors leading to the growth attenuation in SCLC
cells.
AKR1C1 as a target gene of WA
Bioinformatics analyses demonstrated that AKR1C1 may
be a target gene of WA. According to the results of bioinformatics
analysis, there are 17,873 differentially expressed genes during
the progression of WA-mediated NCI-H446 cells. The association of
expression profiles between the two groups was demonstrated by
unsupervised hierarchical clustering analysis tree (Fig. 3A). The logarithm of the fluorescence
intensity ratio was represented by fold change, and a log2
ratio≥1.0 or a log2 ratios≤-1.0 means a two-fold change (Fig. 3B). These data indicated WA upregulated
10,560 genes and downregulated 7,313 genes. The AKR1C1 gene
underwent the most significant change following WA treatment
(P<0.001) (Fig. 3B). Pathway
analysis demonstrated that the differentially expressed genes were
mainly involved in 159 KEGG classical pathways, particularly in the
p53 signaling pathway (23 genes), mitogen-activated protein kinase
signaling pathway (44 genes), apoptosis (23 genes), PI3K-AKT
signaling pathway (40 genes), nuclear factor-κB signaling pathway
(17 genes), Janus kinase-signal transducer and activator of
transcription signaling pathway (15 genes), vascular endothelial
growth factor signaling pathway (16 genes) and insulin signaling
pathway (18 genes; Fig. 3D). RT-qPCR
analysis indicated that the mRNA expression level of gene AKR1C1
was notably decreased in the WA treatment NCI-H446 cells (Fig. 3E). Additionally, western blot analysis
demonstrated that the protein expression level of AKR1C1 was
notably decreased in the WA treatment NCI-H446 cells (Fig. 3F).
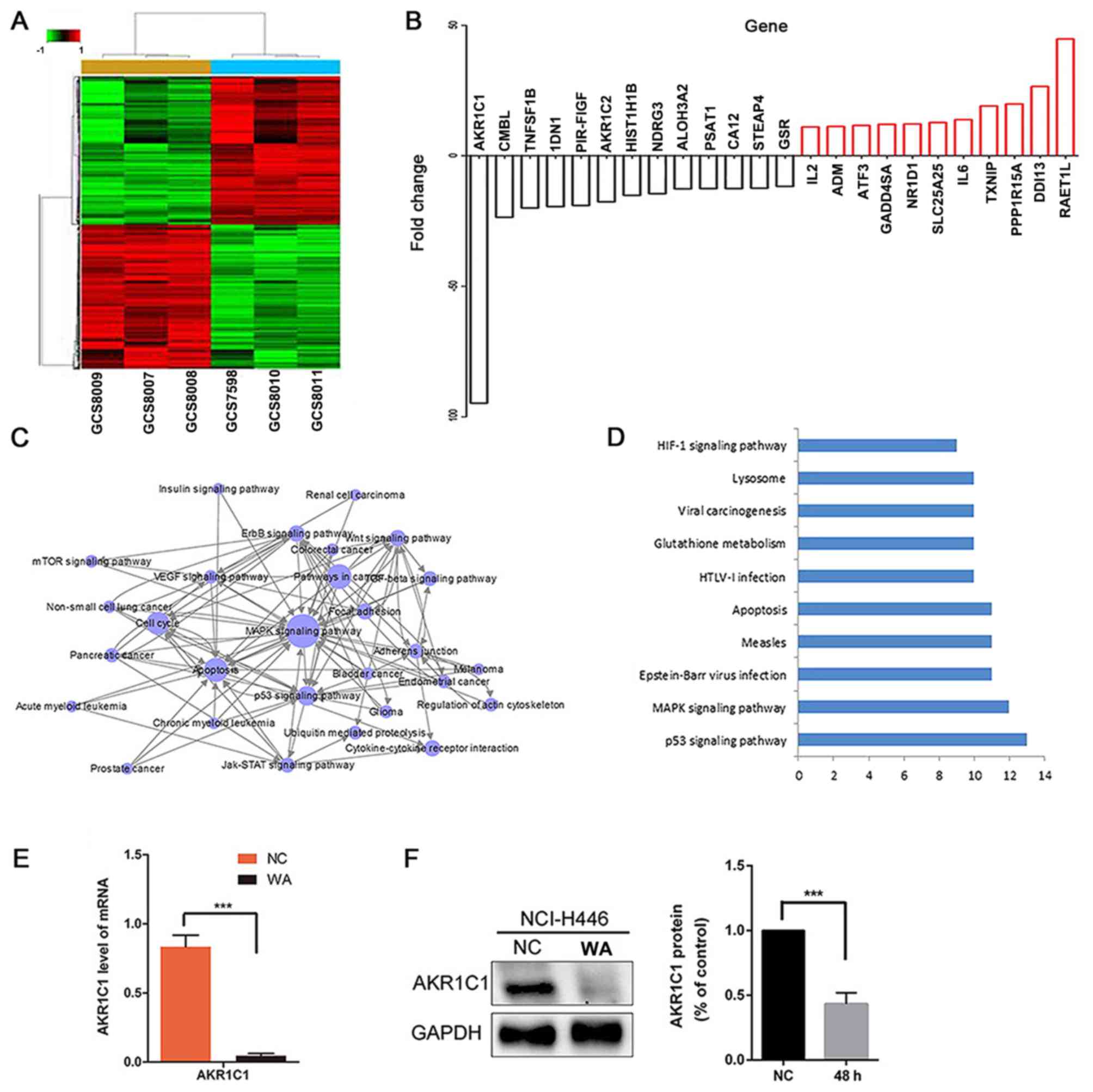 | Figure 3.(A) Genesifter program heat map
analysis. 17,873 genes were identified from the 60,000 transcripts
that significantly changed over the experimental time course at 0
and 48 h. (B) Top 24 significantly changed genes. (C) Gene Ontology
assay. (D) Pathway assay. (E) Reverse-transcription quantitative
polymerase chain reaction changes in AKR1C1 gene expression,
***P<0.001 with a 95% confidential level. (F) Protein level was
detected by western blot analysis. AKR1C1 was significantly
reduced, ***P<0.001 with a 95% confidential level. AKR1C1,
aldo-keto reductase family 1 member C1; MAPK, mitogen-activated
protein kinase. HIF-1, hypoxia inducible factor 1; HTLV, human
T-lymphotropic virus; WA, Wentilactone A; NC, control group. |
AKR1C1 attenuates SCLC cells apoptosis
in vitro
Following 48 h transfection, fluorescence microscopy
demonstrated that the observed transfection efficiency for
NCI-H446, NCI-H1688 and LTEP-sm SCLC cell lines were all ~99%
(Fig. 4A). Western blot analysis
demonstrated the transfection efficiency (Fig. 4B). Additionally, the anti-apoptotic
effect of AKR1C1 gene and pro-apoptotic effects of WA on SCLC cells
were investigated by flow cytometry analysis. As depicted in
Fig. 5A, compared with the NC group,
the overexpression of AKR1C1 gene decreased the apoptosis of SCLC
cells. Compared with NC group, WA treatment significantly induced
SCLC cell apoptosis (P<0.001). Compared with the apoptosis rate
41.81±0.60% in 48 h WA-treated NC group NCI-H446 cells, the
apoptosis rate was 18.86±0.23% in 48 h WA-treated OE group NCI-H446
cells and 53.59±1.05% in 48 h WA-treated KD group NCI-H446 cells.
The aforementioned results indicated that WA enhanced cell
apoptosis; however, overexpression of AKR1C1 reversed the effect of
WA, and knockdown of AKR1C1 promoted the effect of WA in SCLC
cells. As depicted in Fig. 5B, the
CCK-8 array demonstrated that the knockdown of AKR1C1 gene
attenuated cell proliferation. This attenuation is more notable in
NCI-H446 cells following WA treatment. Overexpression of the AKR1C1
gene improved cell proliferation, and WA treatment partially
reversed the anti-apoptotic effect of the AKR1C1 gene. Notably, the
flow cytometry analysis and colony formation assay obtained the
same results, that the knockdown of AKR1C1 gene and WA treatment
attenuated SCLC cells proliferation.
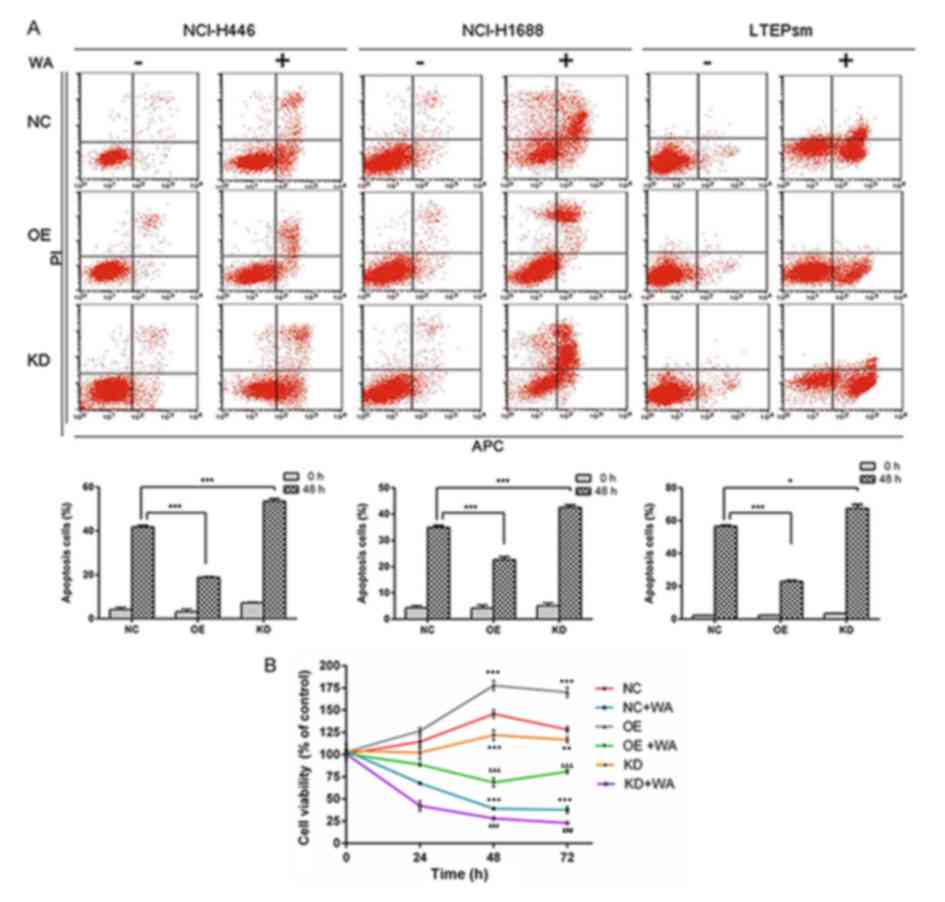 | Figure 5.(A) WA treatment significantly
induced SCLC cell apoptosis and overexpression of AKR1C1 gene
reversed the pro-apoptotic effect of WA. There were six groups. OE
group cells were infected with lentiviral vector LV5-AKR1C1; OE+WA
group cells were infected with lentiviral vector LV5-AKR1C1 and WA
treatment; NC group cells were non-WA-treatment; NC+WA group cells
were WA treatment; KD group cells were infected with lentivirus
particles carrying sh-AKR1C1; KD+ WA group cells were infected with
lentivirus particles carrying sh-AKR1C1 and WA treatment. (B) Cell
Counting Kit-8 assay was utilized to evaluate cell proliferative
capacity following transfection for 48 h, NC+WA group cells
revealed a significant slower proliferation compared with the NC
group. *P<0.05, **P<0.01 and ***P<0.001. KD, lentiviral
vector carrying sh-AKR1C1 plasmid compared with the KD group at the
same time point, ###P<0.001; OE, transfected with
AKR1C1 gene retroviral vector plasmid, compared with the OE group
at same time point, &&&P<0.001; AKR1C1,
aldo-keto reductase family 1 member C1; NC, control group; WA,
Wentilactone A; PI, propidium iodide; APC, allophycocyanin. |
AKR1C1 attenuates SCLC tumor growth in
vivo
To further confirm the growth-attenuating effect of
WA and the growth-promotion effect of AKR1C1 on SCLC cells, a
xenograft tumor growth assay was performed. Following 20 days, the
tumors were harvested. The total weight of the WA treatment tumors
was significantly lower in nude mice, compared with the control
mice (P<0.05, Fig. 6A). The
subcutaneous tumor volume growth curve of NCI-H446 stably expressed
in vivo was depicted in Fig.
6B. The tumor volume was significantly larger in the OE group
nude mice, compared with the NC group mice at 5, 10, 15 and 20 days
(P<0.05, Fig. 6B); however, the
tumor volume was reduced following WA treatment. Compared with the
NC group, there was no significant weight loss observed in the WA
treatment group animals (P>0.05) (Fig.
6C). Immunohistochemistry of AKR1C1 antigen was detected, and
results demonstrated expression of AKR1C1 was lower in the WA
treatment group (Fig. 6E). The
results revealed that WA could attenuate the expression of AKR1C1
and the proliferation of SCLC in vivo. These results provide
further evidence that WA serves a tumor-attenuated role in
SCLC.
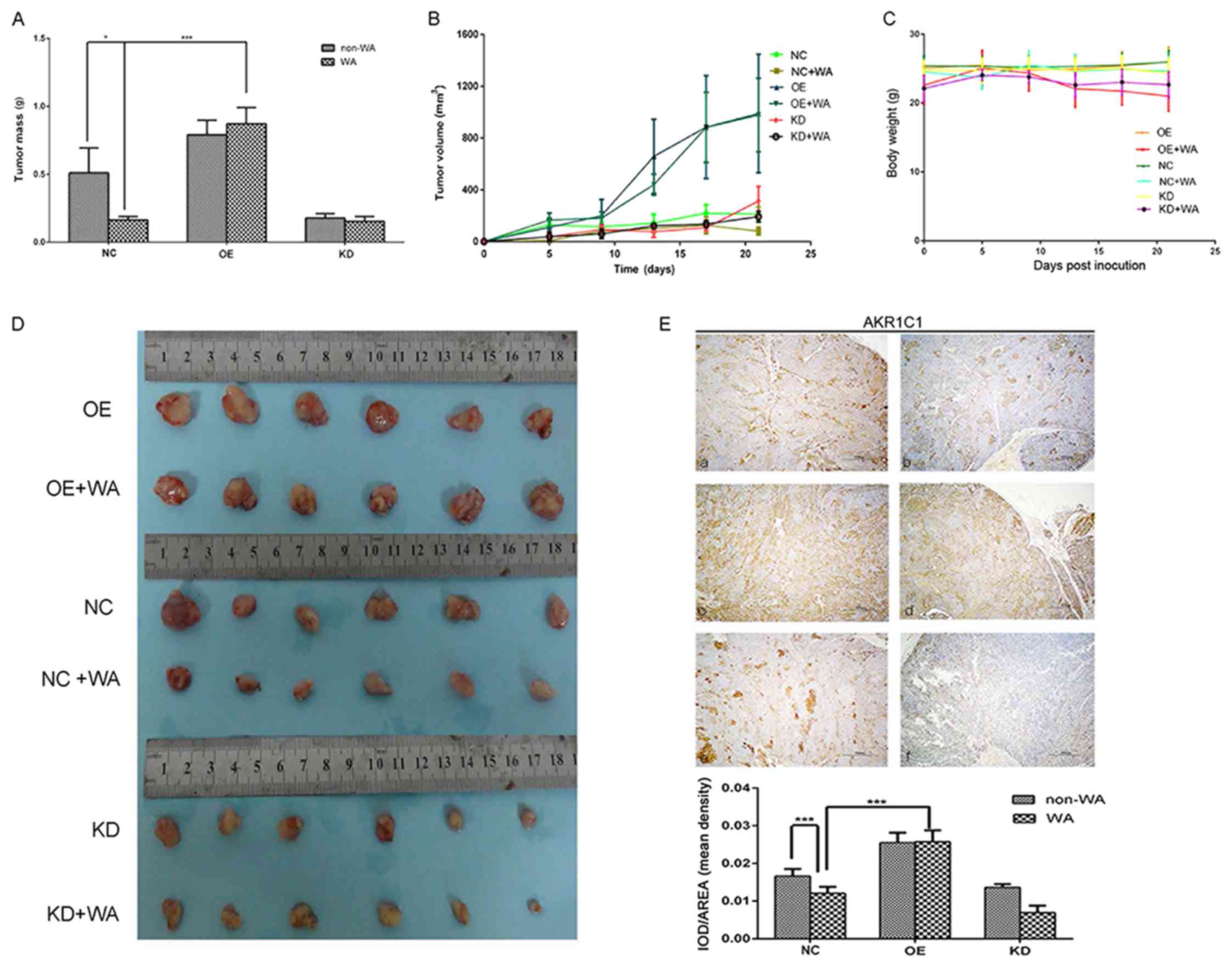 | Figure 6.WA inhibited expression of AKR1C1
gene and tumor growth. There were six groups. OE group cells were
infected with lentiviral vector LV5-AKR1C1; OE+WA group cells were
infected with lentiviral vector LV5-AKR1C1 and WA treatment; NC
group cells were non-WA-treatment; NC+WA group cells were WA
treatment; KD group cells were infected with lentivirus particles
carrying sh-AKR1C1; KD+WA group cells were infected with lentivirus
particles carrying sh-AKR1C1 and WA treatment. (A) The weight of
the metastatic tumors in each group. *P<0.05, ***P<0.001. (B)
The subcutaneous tumor volume growth curve of WA treatment was
depicted. (C) The body weight of the nude mice in each group. (D)
Tumors were harvested following 20 days. (E) Immunohistochemical
staining of (a) NC group, (b) NC+WA group, (c) OE group, (d) OE+WA
group, (e) KD group and (f) KD+WA group. ***P<0.001. KD,
lentiviral vector carrying sh-AKR1C1 plasmid; OE, transfected with
AKR1C1 gene retroviral vector plasmid; AKR1C1, aldo-keto reductase
family 1 member C1; NC, control group; WA, Wentilactone A;
IOD/AREA, Integrated optical density per stained area. |
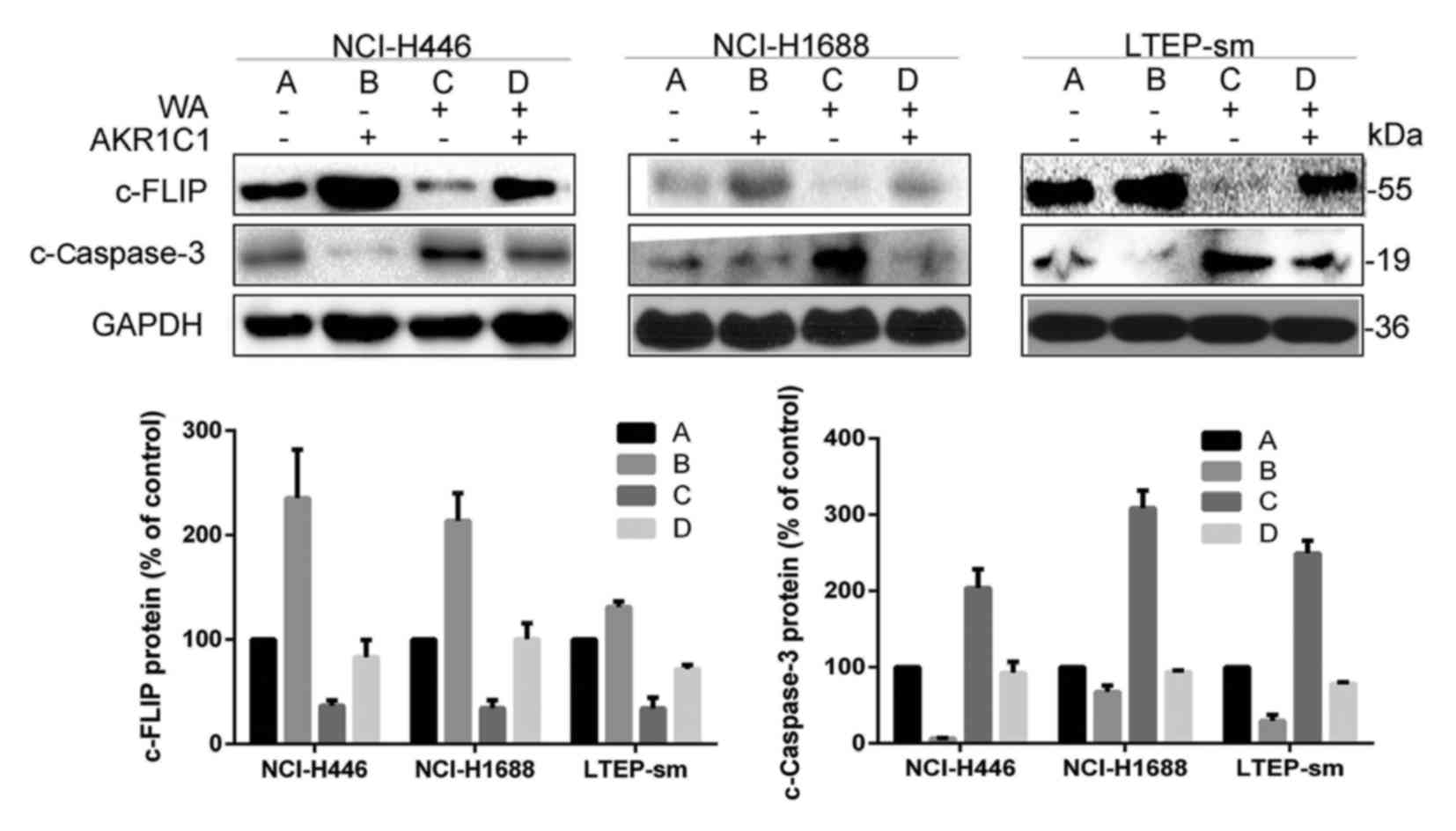
Overexpression of AKR1C1 gene may
partially reverse the pro-apoptotic function of WA via c-FLIP and
c-Caspase-3
In the present study, it was demonstrated that
overexpression of AKR1C1 gene promoted the expression of c-FLIP,
whilst WA treatment inhibited the expression of the c-FLIP protein,
activated c-Caspase-3 and induced apoptosis in SCLC cell lines
(Fig. 7). Palackal et al
(16) demonstrated that in patients
with SCLC, differential display demonstrated that AKR1C transcripts
are notably overexpressed. Wang et al (17) indicated that the downregulation of
c-FLIP drove lung cancer cells into cellular apoptosis. c-FLIP had
been demonstrated to induce apoptosis thus increasing Caspase-8 and
then Caspase-3 activation (18).
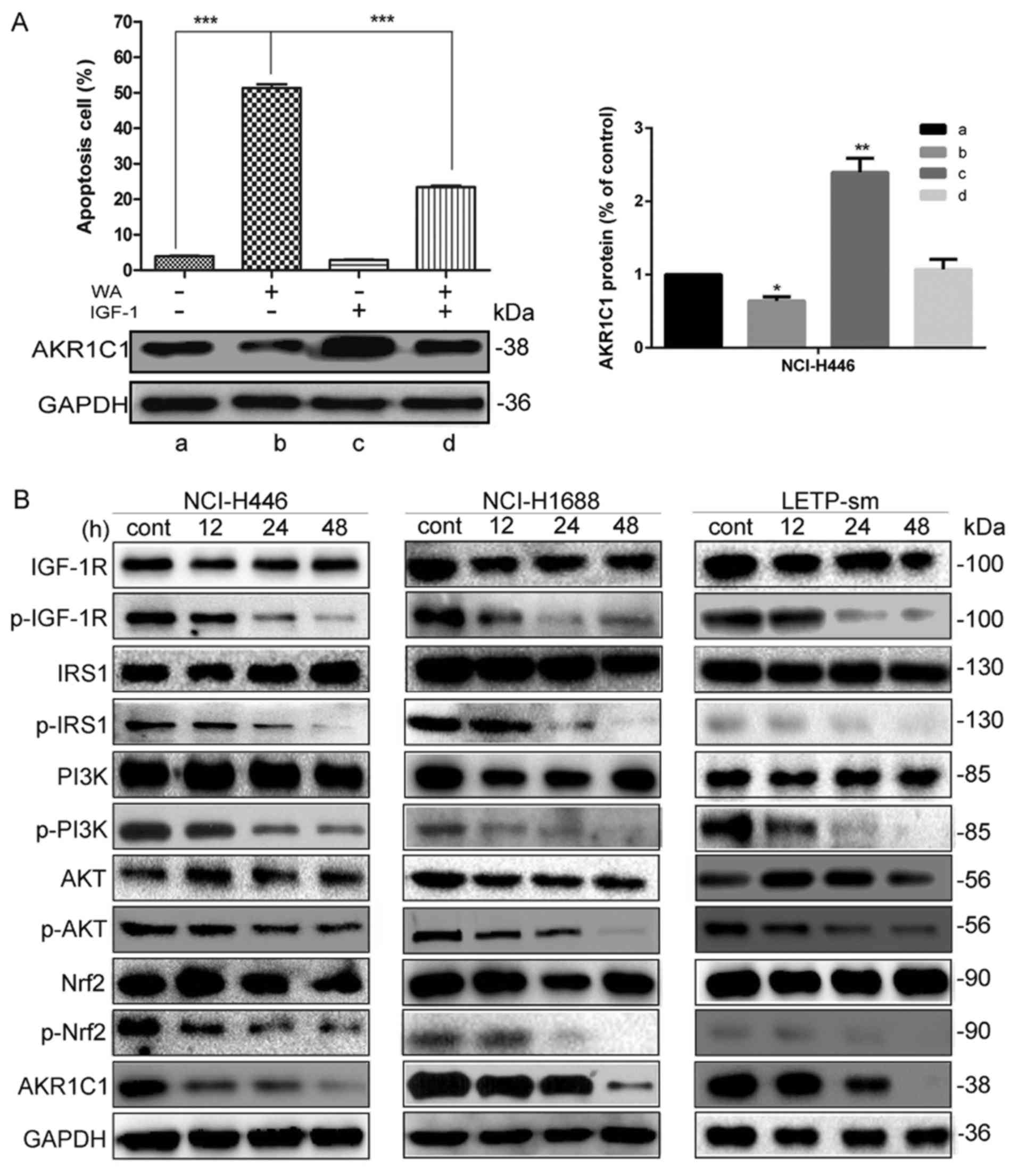
WA regulates the expression of AKR1C1
protein via the IGF-1R/IRS1/PI3K/AKT/nuclear factor-erythroid 2-
associated factor 2 (Nrf2) signaling pathway
To further determine the mechanisms underlying the
expression of AKR1C1 gene attenuation by WA, the investigation was
focused on whether IGF-1R, IRS-1, PI3K, AKT and Nrf2 were
associated with the WA-induced attenuated expression of AKR1C1 and
pro-apoptotic factors. Together with their protein abundances,
AKR1C1 was significantly reduced in the NCI-H446 cells following WA
treatment (P<0.001), compared with control cells. AKR1C1 were
significantly increased in the NCI-H446 cells following IGF-1
treatment. IGF-I, an activator of the IGF-IR pathway, promoted the
expression of AKR1C1 and altered the cell apoptosis following WA
treatment (Fig. 8A). It was
determined that the IGF-1 reverses the biological effect of WA on
SCLC cells. As depicted in Fig. 8B,
together with their protein abundances, p-IGF-1R (P<0.001),
p-IRS-1(P<0.001), p-PI3K (P<0.001), p-AKT (P<0.001),
p-Nrf2 (P<0.001) and AKR1C1 (P<0.001) were significantly
reduced in SCLC cells following WA treatment, compared with the
control. The result revealed that the attenuation effect of WA on
SCLC occurs at least partially by the targeting of AKR1C1 by the
IGF-1R/IRS1/PI3K/AKT/Nrf2 signaling pathway. The study demonstrated
that AKR1C1 may act as a potential target for the treatment of SCLC
in the future.
Discussion
SCLC is characterized by rapid growth and early
metastasis, which indicates strikingly high malignity and
metastatic potential (1). The
therapeutic potential of WA in SCLC treatment was highlighted
(9).
A recent study demonstrated that WA significantly
attenuated the growth of NCI-H446 cells, however the study did not
fully alleviate the notable inhibitory effect of WA on other SCLC
cell lines (9). In the present study,
the notable inhibitory effect of WA was investigated in three SCLC
cell lines NCI-H446, NCI-H1688 and LTEP-sm cells. Results of the
CCK-8 assay, flow cytometry, scratch assay and western blot
analysis indicated that WA had a notable effect on SCLC in
vitro and in vivo. It was determined in the present
study that WA inhibited c-FLIP in the three SCLC cell lines in a
time-dependent manner, inducing apoptosis by upregulating
c-Caspase-3, which was consistent with previous studies (19–23).
However, the target gene of WA is not clear. The focus of the
present study is the anti-SCLC mechanism of WA.
In the present study, bioinformatics analyses
indicated that AKR1C1 was possibly the target gene in SCLC. Results
of RT-qPCR and western blot confirmed AKR1C1 mRNA and protein
levels were lower following WA treatment. Tian et al
(24) demonstrated that high
expression of AKR1C1 gene was associated with progression and poor
prognosis of lung cancer. AKR1C1 is associated with numerous
important biological processes, including the oxidation reduction
and carcinogenesis of a number of vertebrate species (25). The effect and mechanism of AKR1C1 on
SCLC remains unclear. According to previous studies, Nrf2 was a
regulator of the AKR1C family (26–32). Yu
et al (33) demonstrated that
the PI3K-Akt pathway mediated the activation of NF-E2 associated
factor-2 (Nrf2). Tao et al (34) demonstrated that IGF1R-mediated
protection cells from apoptosis stopped upon the activation of
PI3K. IGF1-R pathway was essential to mediate tumor cell survival
and proliferation (35). IGF-1R
overexpression increased cell survival and suppressed cell
apoptosis (36). Further analysis on
the association of WA treatment with AKR1C1 expression demonstrated
that WA inhibited the expression of AKR1C1 via the
IGF-1R/IRS-1/PI3K/AKT/Nrf2 signaling pathway by western blot
analysis.
Further mechanism analysis indicated that WA
inhibited the expression of AKR1C1 and then induced the apoptosis
of SCLC cells by the FLIP/Caspase-3 pathway. According to previous
studies (21–23), apoptosis was known as a significant
terminal pathway to remove infected cells. There are two central
apoptosis pathways: The extrinsic pathway; and the intrinsic
pathway (22). The key regulatory
proteins in both pathways are Caspases (23). In addition, Caspases are classified as
the initiator (Caspase-8 and −9) and effector of Caspase-3
(37). Wang et al (17) and Safa et al (18) demonstrated that c-FLIP was involved in
chemotherapeutic drug resistance in numerous cancer cell types and
anti-apoptotic in human NSCLC; however, c-FLIP has generally been
demonstrated to act as a key negative regulator of apoptosis in
human cancer cells, despite the identified dual functionality of
c-FLIP as a pro- or anti-apoptotic factor in normal tissues.
Enhanced expression of c-FLIP (long or short isoform) has been
determined in various cancer types.
Apoptosis of SCLC cells were investigated by flow
cytometry prior to and following WA treatment. Flow cytometry assay
and western blot analysis both confirmed WA inhibited the
expression of AKR1C1 via the IGF-1R/IRS-1/PI3K/AKT/Nrf2 signaling
pathway and then inhibited c-FLIP and activated c-Caspase-3, which
induced the apoptosis of SCLC cells. This research provided the
experimental basis for the development of SCLC targeted drugs.
In summary, the present study revealed that WA
induced the apoptosis by
IGF-1R/IRS-1/PI3K/AKT/Nrf2/AKR1C1/c-FLIP/c-Caspase-3. Most
importantly, overexpression of AKR1C1 gene has pro-growth and
anti-apoptosis effects on SCLC cells, which can be reversed by WA
treatment. WA holds good promise as a novel potential AKR1C1
targeted drug candidate for future treatments of SCLC.
Acknowledgements
This authors thank the Laboratory of Marine Biology
and Biotechnology, Qingdao National Laboratory for Marine Science
and Technology, Key Laboratory of Experimental Marine Biology,
Institute of Oceanology of the Chinese Academy of Sciences for
their general support provided.
Funding
This present study was supported by the National
Natural Science Foundation of China (grant nos., 41776140, 81473239
and 41576160).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WJ conceived and designed the experiments and was
involved in the analysis and interpretation of data. BW performed
the experiments. BJ contributed to the conception of the study and
drafting the manuscript. CH contributed to the interpretation of
results obtained. HW perform data analysis with constructive
discussions and performed part of the experiments. LM contributed
to reagents and performed part of the experiments. GX contributed
to reagents and materials, and performed part of the experiments.
CL contributed to analytical tools and performed part of the
experiments. HT approved the final version and performed part of
the experiments. RC was involved in drafting the manuscript and
performed part of the experiments. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All samples were obtained with the informed consent
of the participants prior to their inclusion in the study,
according to Helsinki Declaration principles and after approval of
the ethics committee of Second Military Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stinchcombe TE: Current treatments for
surgically resectable, limited-stage, and extensive-stage small
cell lung cancer. Oncologist. 22:1510–1517. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mamdani H, Induru R and Jalal SI: Novel
therapies in small cell lung cancer. Transl Lung Cancer Res.
4:533–544. 2015.PubMed/NCBI
|
|
4
|
Liu B, Qin J and Zhou J: Advances in the
treatment of relapsed small cell lung cancer. Zhongguo Fei Ai Za
Zhi. 20:192–198. 2017.(In Chinese). PubMed/NCBI
|
|
5
|
George J, Jing SL, Jang SJ, Cun Y, Ozretić
L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, et al:
Comprehensive genomic profiles of small cell lung cancer. Nature.
524:47–53. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calcabrini C, Catanzaro E, Bishayee A,
Turrini E and Fimognari C: Marine sponge natural products with
anticancer potential: An updated review. Mar Drugs. 15:pii:
E3102017. View Article : Google Scholar
|
|
7
|
Hussain H and Green IR: A patent review of
the therapeutic potential of isoflavones (2012–2016). Expert Opin
Ther Pat. 27:1135–1146. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Russo P, Nastrucci C and Cesario A: From
the sea to anticancer therapy. Curr Med Chem. 18:3551–3562. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lv C, Hong Y, Miao L, Li C, Xu G, Wei S,
Wang B, Huang C and Jiao B: Wentilactone A as a novel potential
antitumor agent induces apoptosis and G2/M arrest of human lung
carcinoma cells, and is mediated by HRas-GTP accumulation to
excessively activate the Ras/Raf/ERK/p53-p21 pathway. Cell Death
Dis. 4:e9522013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sherlock G: Gene ontology: Tool for the
unification of biology. Can Instit Food Sci Technol J.
22:4152009.
|
|
11
|
Ge QM, Huang CM, Zhu XY, Bian F and Pan
SM: Differentially expressed miRNAs in sepsis-induced acute kidney
injury target oxidative stress and mitochondrial dysfunction
pathways. PLoS One. 12:e01732922017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wright GW and Simon RM: A random variance
model for detection of differential gene expression in small
microarray experiments. Bioinformatics. 19:2448–2455. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang H, Crawford N, Lukes L, Finney R,
Lancaster M and Hunter KW: Metastasis predictive signature profiles
pre-exist in normal tissues. Clin Exp Metastasis. 22:593–603. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yin J, Shackel N, Zekry A, McGuinness PH,
Richards C, Putten KV, McCaughan GW, Eris JM and Bishop GA:
Real-time reverse transcriptase-polymerase chain reaction (RT-PCR)
for measurement of cytokine and growth factor mRNA expression with
fluorogenic probes or SYBR Green I. Immunol Cell Biol. 79:213–221.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Palackal NT, Lee SH, Harvey RG, Blair IA
and Penning TM: Activation of polycyclic aromatic hydrocarbon
trans-dihydrodiol proximate carcinogens by human aldo-keto
reductase (AKR1C) enzymes and their functional overexpression in
human lung carcinoma (A549) cells. J Biol Chem. 277:24799–24808.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang X, Chen W, Zeng W, Bai L, Tesfaigzi
Y, Belinsky SA and Lin Y: Akt-mediated eminent expression of c-FLIP
and Mcl-1 confers acquired resistance to TRAIL-induced cytotoxicity
to lung cancer cells. Mol Cancer Ther. 7:1156–1163. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Safa AR and Pollok KE: Targeting the
anti-apoptotic protein c-FLIP for cancer therapy.
|
|
19
|
Goldar S, Khaniani MS, Derakhshan SM and
Baradaran B: Molecular mechanisms of apoptosis and roles in cancer
development and treatment. Asian Pac J Cancer Prev. 16:2129–2144.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sankari SL, Masthan KM, Babu NA,
Bhattacharjee T and Elumalai M: Apoptosis in cancer-an update.
Asian Pac J Cancer Prev. 13:4873–4878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Savitskaya MA and Onishchenko GE:
Mechanisms of apoptosis. Biochemistry (Mosc). 80:1393–1405. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li J and Yuan J: Caspases in apoptosis and
beyond. Oncogene. 27:6194–6206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tian H, Li X, Jiang W, Lv C, Sun W, Huang
C and Chen R: High expression of AKR1C1 is associated with
proliferation and migration of small-cell lung cancer cells. Lung
Cancer (Auckl). 7:53–61. 2016.PubMed/NCBI
|
|
25
|
Penning TM: The aldo-keto reductases
(AKRs): Overview. Chem Biol Interact. 234:236–246. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kobayashi A, Kang MI, Okawa H, Ohtsuji M,
Zenke Y, Chiba T, Igarashi K and Yamamoto M: Oxidative stress
sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to
regulate proteasomal degradation of Nrf2. Mol Cell Biol.
24:7130–7139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jaiswal AK: Nrf2 signaling in coordinated
activation of antioxidant gene expression. Free Radic Biol Med.
36:1199–1207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Homma S, Ishii Y, Morishima Y, Yamadori T,
Matsuno Y, Haraguchi N, Kikuchi N, Satoh H, Sakamoto T, Hizawa N,
et al: Nrf2 enhances cell proliferation and resistance to
anticancer drugs in human lung cancer. Clin Cancer Res.
15:3423–3432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kaspar JW, Niture SK and Jaiswal AK:
Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol
Med. 47:1304–1309. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nguyen T, Yang CS and Pickett CB: The
pathways and molecular mechanisms regulating Nrf2 activation in
response to chemical stress. Free Radic Biol Med. 37:433–441. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
de Vries HE, Witte M, Hondius D,
Rozemuller AJ, Drukarch B, Hoozemans J and van Horssen J:
Nrf2-induced antioxidant protection: A promising target to
counteract ROS-mediated damage in neurodegenerative disease? Free
Radic Biol Med. 45:1375–1383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zenkov NK, Menshchikova EB and Tkachev VO:
Keap1/Nrf2/ARE redox-sensitive signaling system as a
pharmacological target. Biochemistry (Mosc). 78:19–36. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu JB, Shi J, Zhang Y, Gong LR, Dong SA,
Cao XS, Wu LL and Wu LN: Electroacupuncture ameliorates acute renal
injury in lipopolysaccharide-stimulated rabbits via induction of
HO-1 through the PI3K/Akt/Nrf2 pathways. PLoS One. 10:e01416222015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tao Y, Pinzi V, Bourhis J and Deutsch E:
Mechanisms of disease: Signaling of the insulin-like growth factor
1 receptor pathway-therapeutic perspectives in cancer. Nat Clin
Pract Oncol. 4:591–602. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nurwidya F, Andarini S, Takahashi F,
Syahruddin E and Takahashi K: Implications of insulin-like growth
factor 1 receptor activation in lung cancer. Malays J Med Sci.
23:9–21. 2016.PubMed/NCBI
|
|
36
|
Tao Y1, Pinzi V, Bourhis J and Deutsch E:
Expression and distribution of insulin-like growth factor-1
receptor pathway-therapeutic perspectives in cancer. Nat Clin Pract
Oncol. 4:591–602. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yi CH and Yuan AJ: The Jekyll and Hyde
functions of caspases. Dev Cell. 16:21–34. 2009. View Article : Google Scholar : PubMed/NCBI
|