Introduction
Germline transmission of variant forms of tumor
suppressor genes or oncogenes greatly increases the risk of
developing certain types of cancer. These genetic variants
associated with cancer predisposition have been identified through
studies of familial cancers. However, the majority of cancers
develop sporadically, and the epidemiological data suggest that
cancer susceptibility in the general population is affected by
multiple low-penetrant tumor susceptibility genes (1,2). Mouse
cancer models are powerful tools for identification of these tumor
susceptibility genes. As they have various advantages such as
well-controlled genetic background and environment, mouse models
have been utilized to analyze cancer susceptibility, and many
genetic loci associated with cancer susceptibility have been mapped
(3,4).
Susceptibility to the two-stage skin carcinogenesis
protocol varies among mouse strains (5,6), and
genetic approaches have been used to identify the specific loci
related to the differences in tumor susceptibility among strains
(3,4,7–11). For example, 15 skin tumor
susceptibility loci, Skts1-15 have been mapped by analysis
of NIH and Mus spretus cross, in which Mus spretus
showed the dominant resistance against NIH (12,13).
Psl1-4 were mapped by crossing of the resistant C57BL/6J
(B6) strain and the susceptible DBA strain (14,15).
Skts-fp1-3 were found by crossing the resistant PWK strain
and the susceptible FVB strain (8),
and Stmm1-3 were identified by crossing the resistant MSM/Ms
strain and the susceptible FVB strain (11). Over the past 10 years, a number of
responsible genes at each locus have been identified. Skts13
and Skts14 were identified as Aurka and Tgfb1
genes, respectively (16,17). Recently, Okumura et al reported
that the parathyroid hormone (Pth) gene is one of the genes
responsible for Stmm1 (18).
However, many of the responsible genes within the
remaining loci have not yet been identified. As the contribution of
each tumor susceptibility locus to the phenotype was relatively low
and most of the loci were mapped to broad intervals, it is
difficult to identify the precise locations of these tumor
susceptibility loci. Derivation of congenic mice by multiple rounds
of breeding to transfer the regions containing the variant from a
resistant strain to a susceptible strain is a reliable approach for
fine mapping of the responsible genes. However, the standard
genotype-driven congenic approach, in which mice that have
inherited the resistance allele on susceptible loci are selected
and bred with a susceptible strain to generate the next generation,
may not be appropriate for precise mapping of low-penetrant genes.
For example, we have shown that the contribution of the D4Mit26
locus to the Skts-fp1 locus is approximately 15% of the PWK
resistant phenotype, and therefore congenic mice carrying the PWK
allele only at this locus in the FVB mouse background cannot be
expected to show the resistant phenotype (8).
We used a phenotype-driven approach in the present
study, in which mice were subjected to the two-stage skin
carcinogenesis protocol in each generation. Mice showing the
resistant phenotype were bred to generate progeny.
Materials and methods
Animals
The mice used in this study were bred in the
specific pathogen-free (SPF) facility of the Department of
Laboratory Animals at Roswell Park Cancer Institute (Buffalo, NY,
USA) and were treated in accordance with Institutional Animal Care
and Use Committee (IACUC) regulations. FVB/N mice were purchased
from Taconic (Germantown, NY, USA). PWK/Rbrc inbred mice were
obtained from RIKEN BioResource Center, (Tsukuba, Japan).
(FVB/NxPWK/Rbrc) F1 males were backcrossed with FVB/N females to
generate 198 F1 backcross progeny, abbreviated as FxFP. All were
subjected to the two-stage skin carcinogenesis protocol, and then
resistant FxFP mice were selected for further phenotype-driven
backcrossing.
Skin carcinogenesis
7,12-Dimethylbenz(a)anthracene (DMBA) and
12-O-tetradecanoylphorbol-13-acetate (TPA) were purchased from
Sigma-Aldrich; Merck KGaA, (Darmstadt, Germany) for use as a
carcinogen and a promoter, respectively. The back skin of each
animal was carefully shaved with an electric shaver at 8–11 weeks
old. Two days after shaving, 200 µl of DMBA (0.125 mg/ml solution
in acetone) was applied to the back of each mouse. A total of 97.4
nmol of DMBA was applied to each mouse. One week after the
treatment, mice were treated with 400 µl of TPA (5×10−5
M solution in acetone), i.e., 32.4 nmol of TPA, twice a week for 20
weeks. Animals were assessed twice a week for the appearance of
papilloma. The number of papillomas was counted every other week
until 20 weeks after the initial DMBA treatment. The incidence of
papilloma during the promotion phase and papilloma multiplicity 12
weeks after initiation were used for assessment of skin tumor
susceptibility. Male mice of FxFP and congenic strains were
subjected to the two-stages skin carcinogenesis protocol. After the
final treatment with TPA, most of the animals were maintained for
more than 10 months to obtain skin squamous cell carcinoma (SCC)
tissues.
Genotyping
Genomic DNA was isolated from the tails of the mice
using NaOH solution. Briefly, 2–3 mm sections of the tails were
boiled in 300 µl of 50 mM NaOH for 30 min, and then 25 µl of 1 M
Tris-HCl (ph 8.0) was added. Genomic DNA prepared from 199 FxFP
backcross mice was genotyped based on 196 polymorphic
microsatellite markers (MIT) spaced at approximately 8-cM intervals
through chromosomes 1 to 19 by polymerase chain reaction (PCR).
Information regarding the MIT markers used in this study is
available upon request. PCR was carried out using a Biometra
thermocyler (Analytik Jena AG, Jena, Germany), and PCR products
were separated by electrophoresis through 4% Nusieve-GTG
low-melting temperature agarose gels (FMC) in 0.5 × TBE buffer.
Analysis of allelic imbalances in skin
tumor tissues
Genome DNA was prepared from skin tumor tissues
developed in FxFP mice or from N5-N7 congenic
mice sharing the PWK allele on chromosome 7. Genomic DNA extracted
from the tails of F1 mice was utilized as a standard. PCR was
performed to amplify the microsatellite markers on chromosome 7 and
the PCR products were separated by electrophoresis as described
above. The densities of the DNA bands for FVB and PWK alleles were
measured using ImageJ software (National Institutes of Health,
Bethesda, MD, USA) (19), and allelic
imbalances were analyzed as described previously (20), with a modification. In the present
study, differences of ≥25% in the band intensity ratios of the two
alleles in tumor DNAs compared to F1 mouse tail DNA were considered
to be allelic imbalances. Although previous report used ≥50%
threshold, we realized that genome deletions or amplifications
seemed to be too noncontiguous when we analyzed our data using ≥50%
threshold. Therefore we used ≥25% threshold in the present
study.
Statistical analysis
Interval mapping was performed using R/qtl software,
an add-on package for the R statistical system, to map QTLs related
to susceptibility for chemically induced skin tumors. One thousand
permutations were performed to estimate the empirical threshold
value for mapping. Statistical analyses were performed using
one-way ANOVA and the Tukey's post hoc test using JMP software
v11.2 (SAS Institute, Inc., Cary, NC, USA). Data are presented as
means ± SD from at least three independent experiments. In all
analyses, P<0.05 was considered to indicate a statistically
significant difference.
Results
Linkages for papilloma multiplicity
mapped to chromosome 3–7, 14, and 17 by analyzing FxFP male
mice
Using the standard two-stage skin carcinogenesis
protocol, 126 (63.6%) of 198 FxFP backcross males (N2
progeny) developed at least one papilloma 12 weeks after the
initial DMBA treatment. Average multiplicity was 5.08±0.55.
Genome-wide linkage mapping using 196 polymorphic markers revealed
a highly significant linkage (P<0.01) located on chromosome 4,
significant linkages (P<0.05) on chromosomes 5 and 17, and
suggestive linkages (P<0.63) on chromosomes 3, 6, 7, and 14 for
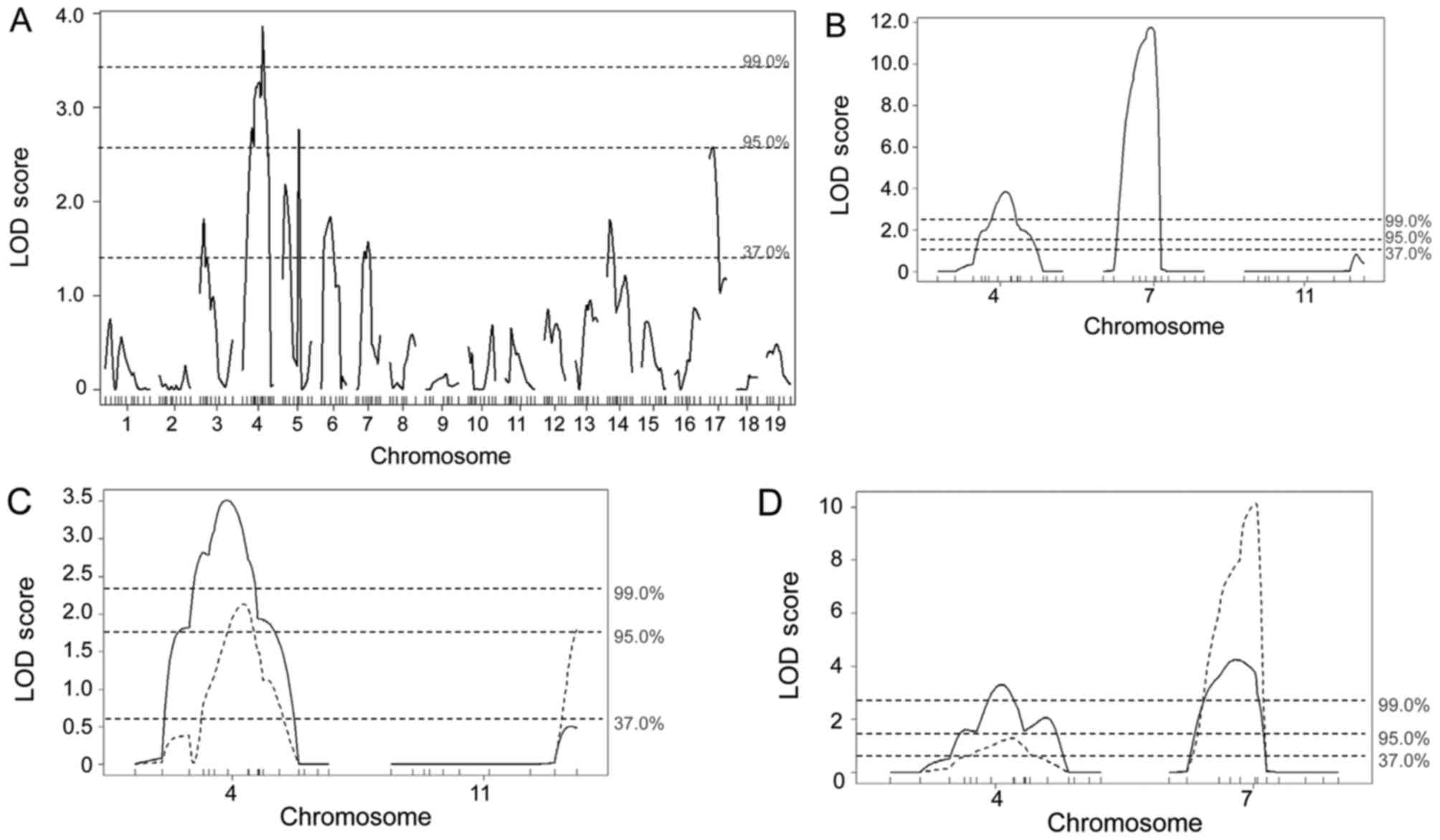
papilloma multiplicity (Fig. 1A).
N6 progeny of congenic
strains obtained by phenotype-driven backcrossing
Forty-seven (23.7%) of 198 FxFP mice did not develop
any skin tumors until at least until 1 year old. To generate a
congenic strain with a skin tumor resistance phenotype, one of the
completely resistant mice was picked and subjected to further
phenotype-driven backcrossing. This mouse was crossed with FVB
females. The resultant male offspring were processed for the
two-stage carcinogenesis protocol, and individuals showing the
resistant phenotype (12 weeks after the initial treatment) were
again picked to produce the next generation. This procedure was
repeated to finally obtain N6 progeny. The genotypes and
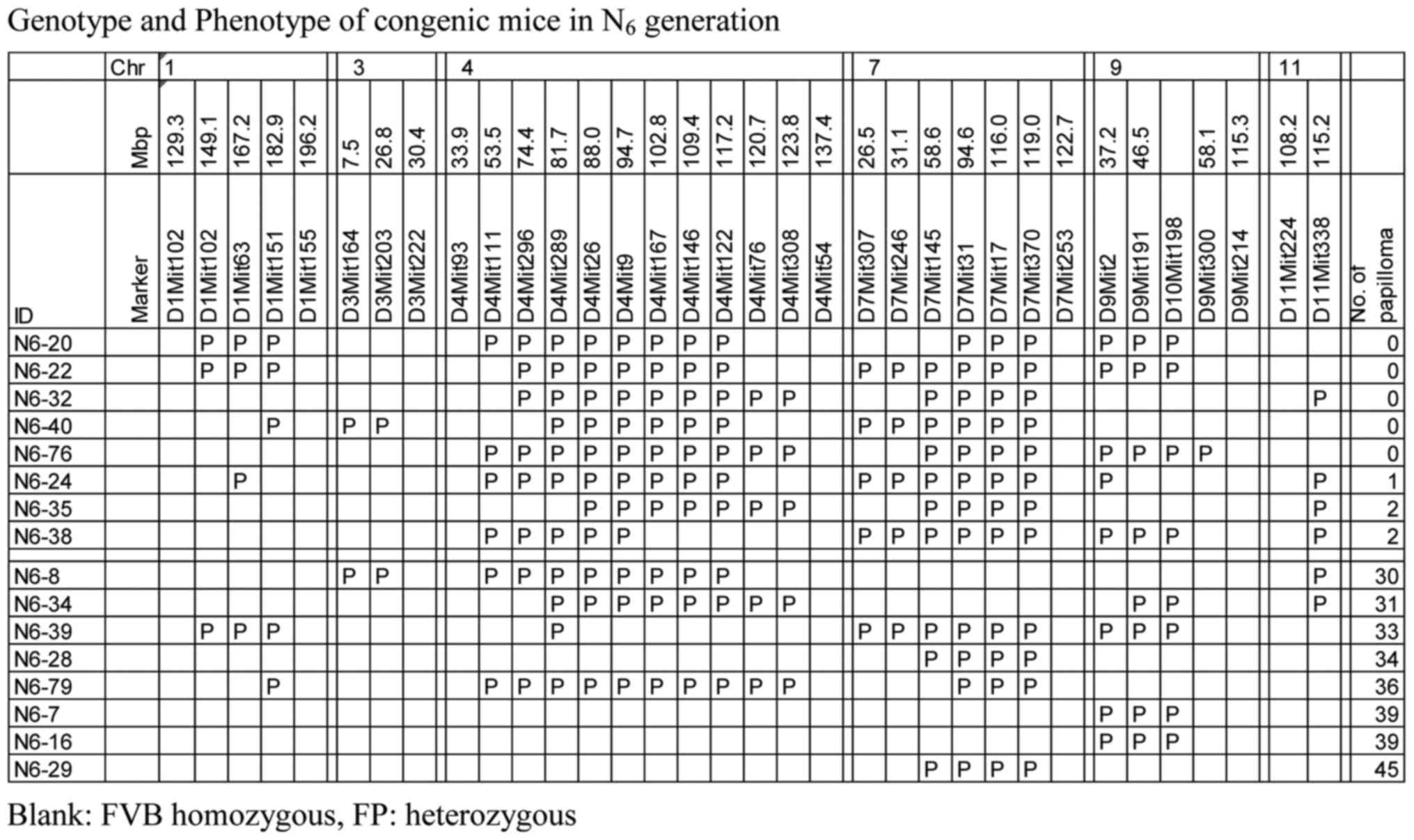
phenotypes of each of eight N6 progeny mice exhibiting
the highest resistance or susceptibility are shown in Table I. Five of 69 males did not develop
papillomas and all carried the PWK allele on chromosomes 4 and 7.
In addition, they also harbored at least one PWK allele on
chromosome 1, 3, 9, or 11 (Table
I).
 | Table I.Genotype and phenotype of congenic
mice in N6 generation. |
Table I.
Genotype and phenotype of congenic
mice in N6 generation.
Linkage analysis of N7
progeny exhibits significant linkage for papilloma multiplicity on
chromosome 11 only in mice with the PWK allele on chromosome 7
To examine the possible effect of each PWK allele,
N7 congenic progeny were produced by crossing
N6−32 male, which retained the PWK allele on chromosomes
4, 7, and 11, with FVB females. Among 95 N7 males, 88
(92.6%) developed papillomas 12 weeks after the initial DMBA
treatment and the average multiplicity was 18.78±1.36. Linkage
analysis of N7 males indicated a highly significant
linkage (P<0.01) for papilloma multiplicity mapped to
chromosomes 4 and 7, whereas neither significant nor suggestive
linkages were mapped to chromosome 11 (Fig. 1B). When the animals were grouped
depending on the genotype at the D7Mit31 locus, a significant
linkage was mapped to chromosome 11 only in N7 progeny
with the PWK allele at the D7Mit31 locus (Fig. 1C). Consistent with this, linkage on
chromosome 7 was obviously higher in N7 progeny with the
PWK allele at the D11Mit338 locus than those homozygous for FVB at
this locus (Fig. 1D).
Association studies confirmed the
contribution of the remaining PWK allele to the resistant phenotype
of the N7 progeny
In association study, one-way ANOVA showed that at
least one of the remaining PWK alleles significantly reduced
papilloma number in N7 progeny (P<0.0001). The post
hoc Tukey test to examine the effect of each remaining allele
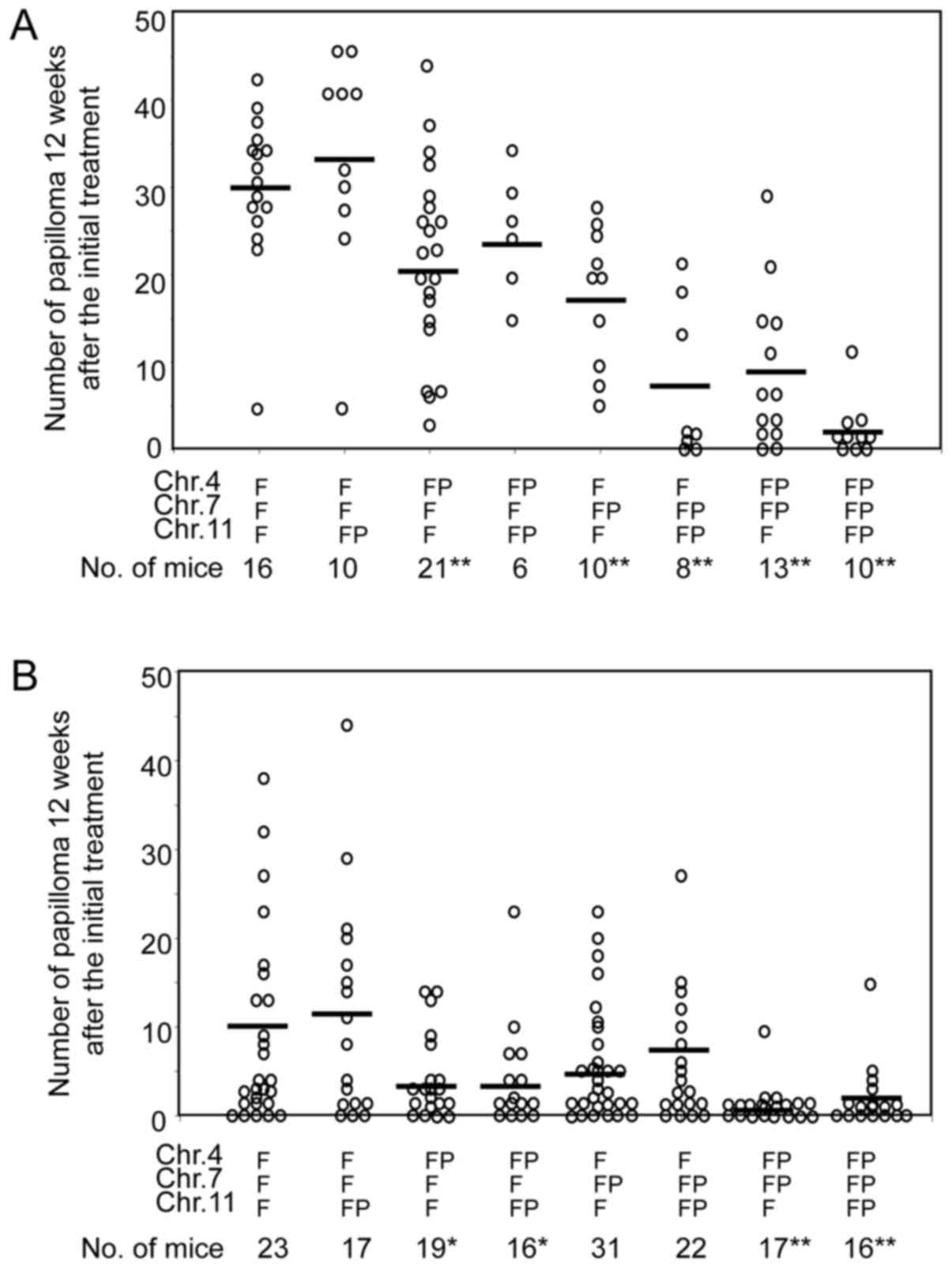
demonstrated that the mice carrying the PWK allele at D7Mit31 loci
developed a significantly reduced number of papillomas compared to
the mice without the PWK allele (Fig.
2A). Although it was not significant, the papilloma
multiplicity of mice carrying the PWK allele at both D7Mit31 and
D11Mit338 loci tended to be decreased compared to mice harboring
the PWK allele only at the D7Mit31 locus. In addition, the number
of papillomas in mice containing the PWK allele at the
above-mentioned three loci also seemed to be lower than that of
mice carrying the PWK allele at both D4Mit26 and D7Mit31 loci, but
not at D11Mit338. In contrast, mice carrying the PWK allele only at
D11Mit338 developed almost the same number of papilloma as mice
without the PWK allele (Fig. 2A).
For FxFP mice, one-way ANOVA showed that at least
one of the 3 PWK alleles significantly reduced papilloma number
(P<0.0005), and Tukey test revealed that the presence of both
PWK alleles at the D4Mit26 and D7Mit31 loci is needed to
significantly reduce the number of papillomas as compared to mice
with the FVB allele at the indicated three loci (P<0.05)
(Fig. 2B). The presence of the PWK
allele at the D11Mit338 locus had no detectable effect on papilloma
multiplicity regardless of the genotype at the D7Mit31 locus
(Fig. 2B). Consistent with these
results, the degree of penetrance of the PWK allele in seven
N7 progeny that did not develop papilloma until at least
12 weeks after the initial DMBA treatment were 57.1, 100 and 71.4%
for D4Mit26, D7Mit31 and D11Mit338, respectively. On the other
hand, they were 60.3, 54.8 and 39.7% for D4Mit26, D7Mit31 and
D11Mit338, respectively, in 73 of N2 progeny that did
not develop papillomas until at least 12 weeks after the initial
treatment.
Allele imbalances in favor of FVB
allele re more frequent compared with those in favor of PWK on
chromosome 7 in tumor genome DNA obtained from congenic males
As the resistance allele-specific loss or
susceptibility allele-specific amplification of chromosome 7 in the
tumor genome DNA was frequently observed in NIH-spretus crosses
(9,20), we sought to narrow down the putative
susceptible loci on chromosome 7 utilizing skin tumors that
developed in the congenic mice. Genomic DNA was purified from skin
tumors that developed in N5-N7 congenic mice,
which shared 40–122 Mb of the PWK allele, and allelic imbalances
were analyzed using nine informative microsatellite markers
(Fig. 3A). From the analysis of 27
skin tumor tissues, including nine papillomas and 18 skin cancers,
all markers showed allelic imbalance, and the allele imbalances in
favor of the FVB allele, i.e., loss of the PWK allele or
amplification of the FVB allele, were observed more frequently than
those in favor of the PWK allele.
Allele imbalance pattern at D7Mit31 is
affected by the genotype at the D11Mit338 locus in tumor genome DNA
obtained from congenic males
To examine whether the genotype of the D11Mit338
locus could affect the allelic imbalance, tumor tissues obtained
from FVB mice homozygous and heterozygous at D11Mit338 were
analyzed separately. From the analysis of 13 tumors, consisting of
five papillomas and eight skin cancers, obtained from mice with FVB
homozygous genotype in the D11Mit338 region, we found that the
extent of allelic imbalance at D7Mit31 in favor of the FVB allele
was similar to that of the PWK allele (Fig. 3B). On the other hand, analysis of 12
heterozygous tumors, consisting of eight skin cancers and four
papillomas, indicated that 45 and 9.1% of tumors showed allelic
imbalance at the D7Mit31 locus in favor of FVB and PWK alleles,
respectively (Fig. 3C).
Differences in allele imbalance
patterns at D7Mit31 are not observed in tumor genome DNA obtained
from FxFP males with or without the PWK allele at D11Mit338
Finally, we examined 43 skin cancer tissues obtained
from FxFP progeny carrying the PWK allele on chromosome 7. As shown
in Fig. 3D, the allele imbalance in
favor of the FVB allele was approximately four times higher than
that in favor of the FVB allele through the entire chromosome 7.
Cancer samples from D11Mit338 homozygous and heterozygous mice were
analyzed separately, and no significant differences in patterns of
allelic imbalance at the D7Mit31 locus were detected between the
two groups (Fig. 3E and F).
Discussion
PWK is a dominant-resistant strain against two-stage
skin carcinogenesis as shown by the crossing with the highly
susceptible strain, FVB (8). As PWK
is a wild-derived strain (5,6), we sought to identify the large genetic
variation associated with skin tumor susceptibility between FVB and
PWK, as seen between NIH and Mus spretus (12). Our previous study using FPxF
backcrosses demonstrated the presence of only one significant
linkage, Skts-fp1, and the suggestive linkage mapped to
chromosome 4 and chromosomes 1/3/11/12/14, respectively (8). In the present study, we generated a
congenic strain carrying the PWK resistance allele in the FVB
strain background by utilizing a congenic-driven approach to narrow
down the candidate loci and find corresponding genes. To obtain
sufficient numbers of progeny in each generation, we performed FxFP
crosses, in which FVB females were crossed with FP males, rather
than FPxF crosses. Our association study using N7
progeny derived from N6−32 mouse with PWK alleles on
chromosomes 4, 7, and 11 indicated that both chromosomes 4 and 7
can significantly reduce the number of papillomas, whereas
chromosome 11 required the PWK allele on chromosome 7 for the
acquisition of this phenotype. These results indicated that there
is at least one tumor resistance locus on chromosome 7, which may
be regulated by certain genes(s) on chromosome 11.
In analysis of the FxFP backcross (N2
generation), we did not observe a significant linkage on chromosome
11, and also a close association between papilloma multiplicity and
chromosome 11 regardless of the genotype at D7Mit31. According to
our previous results obtained from FPxF backcross, a suggestive
linkage, Skts-fp2, was mapped to chromosome 11, and this
linkage became evident in mice homozygous for the FVB allele at the
Skts-fp3 locus on chromosome 16 (8). Although Skts-fp2 was present in
the region between D11Mit155 (58.8 Mbp) and D11Mit178 (85.2 Mbp),
the remaining PWK allele in the present congenic strain was present
in the region between D11Mit338 (115.4 Mbp) and the telomere. These
differences may have been due to putative effects of the other
remaining resistance allele in the N2 generation. As the
N2 generation theoretically retains around 50% of the
PWK allele, the average papilloma number was estimated to be
significantly lower than that in the N7 generation.
Thus, it is possible that the remaining resistance alleles of PWK
made it difficult to detect the effect of each resistance allele in
the N2 generation.
It has been reported that loss of the allele
inherited from a susceptible strain or amplification of the allele
from a susceptible strain is frequently observed in tumors obtained
from the progeny of F1 backcrosses between resistant and
susceptible strains (9,20,21). Based
on these observations, we sought to narrow down the tumor
susceptibility loci by analyzing the allelic imbalances in skin
tumors. These analyses indicated that the allele imbalances in
favor of the FVB chromosomes are much more frequent than those in
favor of PWK chromosomes in both of the congenic strains and FxFP
mice. Using D7Mit31 as a marker, we detected one of the highly
frequent allele-specific imbalances in both the congenic and
N2 progeny, indicating that at least one cancer
susceptibility locus exists around this region. The allelic
imbalance in favor of the FVB allele detected using this marker was
observed more frequently than that in favor of the PWK allele in
tumors carrying the heterozygous allele at D11Mit338 but not in
tumors with the homozygous allele at D11Mit338 in congenic mice. On
the other hand, we did not detect a clear difference in the allelic
imbalance pattern at the D7Mit31 locus between D11Mit338 homozygous
and heterozygous N2 progeny. These results suggest that,
around the D7Mit31 locus, there may be at least one tumor
resistance gene, the function of which could be affected by the
genotypes of certain genes located around D11Mit338. To adequately
address this issue and identify the corresponding genes, further
analyses, such as global gene expression analysis and allelic
imbalance analysis with increased number of skin cancer tissues to
narrow down the candidate loci are needed.
Although we did not analyze other N6
congenic strains with the PWK allele on chromosome 1, 3, or 9 in
the present study, it is likely that these mice will contribute to
identification of novel gene(s) as well as alternative genetic
interaction(s) affecting cancer susceptibility.
Acknowledgements
The authors thank Ms. T. Triplet, Dr. S. Mahmood
(both from the Department of Cancer Genetics, Roswell Park Cancer
Institute), Ms. D. Tabaczynski (Department of Laboratory Animal
Resource, Roswell Park Cancer Institute), and Ms. Asako Oguni
(Department of Medicine, Division of General Medicine, Nihon
University School of Medicine) for technical support and Ms. Paula
Jones (Department of Cancer Genetics, Roswell Park Cancer
Institute) and Ms. Kaoru Tagata (Department of Laboratory Animal
Resource, Roswell Park Cancer Institute) for editorial and
secretarial assistance.
Funding
The present study was supported in part by a grant
from the National Institute of Environmental Health Services (grant
no. ES012249; received by HN), the Roswell Park Alliance Foundation
(grant no. 55-5263-26), the NCI Cancer Center Support Grant to
Roswell Park Cancer Institute (grant no. CA016156), and the
Academic Frontier Project for 2006 Project for Private Universities
matching fund subsidy from MEXT (grant no. 2006-9; received by
HN).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
KF and HN planned the experiments. KF performed the
experiments. YI and MS performed statistical analysis of the data.
KF wrote the paper. TO made substantial contribution to analysis
and interpretation of data, revised the paper critically, and gave
final approval of the paper to be published.
Ethics approval and consent to
participate
All of the animal experiments in this study had
ethics approval from the Institutional Animal Care and Use
Committee (IACUC).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Li FP: Identification and management of
inherited cancer susceptibility. Environ Health Perspect. 103 Suppl
8:S297–S300. 1995. View
Article : Google Scholar
|
|
2
|
Houlston RS and Peto J: The search for
low-penetrance cancer susceptibility alleles. Oncogene.
23:6471–6476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Demant P: Cancer susceptibility in the
mouse: Genetics, biology and implications for human cancer. Nat Rev
Genet. 4:721–734. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mao JH and Balmain A: Genomic approaches
to identification of tumour-susceptibility genes using mouse
models. Curr Opin Genet Dev. 13:14–19. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bangrazi C, Mouton D, Neveu T, Saran A,
Covelli V, Doria G and Biozzi G: Genetics of chemical
carcinogenesis. 1. Bidirectional selective breeding of susceptible
and resistant lines of mice to two-stage skin carcinogenesis.
Carcinogenesis. 11:1711–1719. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ashman LK, Murray AW, Cook MG and
Kotlarski I: Two-stage skin carcinogenesis in sensitive and
resistant mouse strains. Carcinogenesis. 3:99–102. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Balmain A: Cancer as a complex genetic
trait: Tumor susceptibility in humans and mouse models. Cell.
108:145–152. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fujiwara K, Igarashi J, Irahara N, Kimura
M and Nagase H: New chemically induced skin tumour susceptibility
loci identified in a mouse backcross between FVB and dominant
resistant PWK. BMC Genet. 8:392007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Koning JP, Wakabayashi Y, Nagase H, Mao
JH and Balmain A: Convergence of congenic mapping and
allele-specific alterations in tumors for the resolution of the
Skts1 skin tumor susceptibility locus. Oncogene. 26:4171–4178.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fujiwara K, Wie B, Elliott R and Nagase H:
New outbred colony derived from Mus musculus castaneus to identify
skin tumor susceptibility loci. Mol Carcinog. 49:653–661.
2010.PubMed/NCBI
|
|
11
|
Okumura K, Sato M, Saito M, Miura I,
Wakana S, Mao JH, Miyasaka Y, Kominami R and Wakabayashi Y:
Independent genetic control of early and late stages of chemically
induced skin tumors in a cross of a Japanese wild-derived inbred
mouse strain, MSM/Ms. Carcinogenesis. 33:2260–2268. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nagase H, Bryson S, Cordell H, Kemp CJ,
Fee F and Balmain A: Distinct genetic loci control development of
benign and malignant skin tumours in mice. Nat Genet. 10:424–429.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nagase H, Mao JH and Balmain A: A subset
of skin tumor modifier loci determines survival time of
tumor-bearing mice. Proc Natl Acad Sci USA. 96:15032–15037. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Angel JM, Beltrán L, Minda K, Rupp T and
DiGiovanni J: Association of a murine chromosome 9 locus (Psl1)
with susceptibility to mouse skin tumor promotion by
12-O-tetradecanoylphorbol-13-acetate. Mol Carcinog. 20:162–167.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Angel JM, Caballero M and DiGiovanni J:
Identification of novel genetic loci contributing to
12-O-tetradecanoylphorbol-13-acetate skin tumor promotion
susceptibility in DBA/2 and C57BL/6 mice. Cancer Res. 63:2747–2751.
2003.PubMed/NCBI
|
|
16
|
Ewart-Toland A, Briassouli P, de Koning
JP, Mao JH, Yuan J, Chan F, MacCarthy-Morrogh L, Ponder BA, Nagase
H, Burn J, et al: Identification of Stk6/STK15 as a candidate
low-penetrance tumor-susceptibility gene in mouse and human. Nat
Genet. 34:403–412. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mao JH, Saunier EF, de Koning JP, McKinnon
MM, Higgins MN, Nicklas K, Yang HT, Balmain A and Akhurst RJ:
Genetic variants of Tgfb1 act as context-dependent modifiers of
mouse skin tumor susceptibility. Proc Natl Acad Sci USA.
103:8125–8130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Okumura K, Saito M, Yoshizawa Y, Munakata
H, Isogai E, Miura I, Wakana S, Yamaguchi M, Shitara H, Taya C, et
al: The parathyroid hormone regulates skin tumour susceptibility in
mice. Sci Rep. 7:112082017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rasband WS and Image J: U. S. National
institutes of health; bethesda, maryland, USA: https://imagej.nih.gov/ij/1997–2016
|
|
20
|
Nagase H, Mao JH and Balmain A:
Allele-specific Hras mutations and genetic alterations at tumor
susceptibility loci in skin carcinomas from interspecific hybrid
mice. Cancer Res. 63:4849–4853. 2003.PubMed/NCBI
|
|
21
|
Saito M, Okumura K, Miura I, Wakana S,
Kominami R and Wakabayashi Y: Identification of Stmm3 locus
conferring resistance to late-stage chemically induced skin
papillomas on mouse chromosome 4 by congenic mapping and
allele-specific alteration analysis. Exp Anim. 63:339–348. 2014.
View Article : Google Scholar : PubMed/NCBI
|