Introduction
Nasopharyngeal carcinoma (NPC) is a distinct type of
head and neck cancer arising from the nasopharynx, which presents
epithelial malignancy (1). In
southern China and Taiwan, this cancer type has a notably high
prevalence, with an annual incidence rate of >20/100,000
(2). The World Health Organization
(WHO) histologically classified NPC into three types: Type I,
squamous cell carcinoma (SCC); type II, non-keratinizing SCC; and
type III, non-keratinizing undifferentiated carcinoma (3). Unlike other head and neck cancer
locations, surgery has notable disadvantages as a treatment of NPC,
due to the high propensity for cervical and lateral retropharyngeal
lymph nodes; thus, radiotherapy and chemotherapy have important
roles in the treatment of NPC (4).
Although numerous randomized trials have confirmed the benefit of
chemotherapy and radiotherapy (5–7), the
majority of patients develop radioresistance and chemoresistance,
despite progress in the research regarding the molecular mechanisms
(8,9).
In our previous study, it was demonstrated that cisplatin treatment
significantly upregulates microRNA-125a and −125b (miR-125a and
−125b), and targeted p53 mRNA, downregulating its protein level
(10).
Accumulating evidence supports the hypothesis that
tumors contain a sub-population of cells that exhibit stemness
properties, termed cancer stem-like cells (CSCs) (11,12). They
are considered to be the sub-population that causes relapse and
metastasis of cancer due to their self-renewal and differentiation
properties (13). CSCs are refractory
to therapy via their quiescent characteristics and expressing
ATP-binding cassette (ABC) transporters (14); thereby, the discovery of potential
mechanisms underlying chemoresistance in CSCs may solve the
clinical curative difficulties, including chemoresistance,
recurrence and metastasis (15–18).
The tumor suppressor protein p53 is known for its
essential roles in multiple signaling pathways, including cell
cycle arrest, apoptosis, senescence, DNA repair and metabolism
(19). Previous research also
demonstrated its regulatory function in the maintenance of stemness
(20). In hematopoietic stem cells
(HSCs), p53 may downregulate Wnt signaling via the miR-34-mediated
inhibition of β-catenin, resulting in the suppression of
self-renewal abilities (20). It has
also been demonstrated that p53 suppresses the self-renewal and
osteogenic differentiation properties, which may be rescued via p53
RNA interference (21). In CSCs, p53
was also determined to regulate the maintenance of stemness
(22). Hegde et al (23) determined that, in leukemia stem cells
(LSC), overexpression of wild-type p53, but not mutant p53 lacking
DNA binding activity, resulted in LSCs gaining properties, such as
proliferating and invasive capacities, and losing their
self-renewal capacity. This raised the question of whether p53
regulates the maintenance of the self-renewal capacity in CSCs, and
whether the induction of chemoresistance is relevant to p53.
In the present study, CSCs were isolated and
incubated by culturing TW01 cells in serum-free medium (SFM) and
the relative expression of miR-215a, miR-125b, p53 and its several
target genes, including B-cell lymphoma 2 (Bcl2), Sco2 and P21,
were measured. It was demonstrated that there is a differential
expression pattern of miR-125a and miR-125b between TW01 and CSCs.
Furthermore, the key regulatory role of p53 in maintaining stemness
of CSCs was determined to be dependent on the DNA binding activity
of p53.
Materials and methods
Cell culture and CSCs
NPC WHO type I, keratinizing SCC cells (TW01;
American Type Culture Collection, Manassas, VA, USA) were
previously frozen in liquid nitrogen and cultured in
10-cm2 dishes using Dulbecco's modified Eagle's medium
(DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
supplemented with 10% fetal bovine serum (FBS; Thermo Fisher
Scientific, Inc.), 1% sodium pyruvate and 1% penicillin and
streptomycin (Biological Industries Israel Beit-Haemek,
Beit-Haemek, Israel). For culturing, dishes were kept in a
humidified atmosphere containing 5% CO2 at 37°C.
To isolate the CSCs from the TW01 cells,
5×105 TW01 cells were suspended and seeded in a 6-well
plate in a SFM-containing DMEM/F12 supplemented with 2% B27 (Thermo
Fisher Scientific, Inc.), 20 ng/ml basic fibroblast growth factor
(bFGF) and 20 ng/ml epidermal growth factor (EGF) (both from
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). After 15 days, the
spheres were collected via filtration through a 70-µm mesh (BD
Biosciences, Franklin Lakes, NJ, USA), and used in subsequent
experiments or passaged.
Side population (SP) assay
The cell concentration was adjusted to
1×106 cells/ml with SFM supplemented with 1% FBS.
Hoechst 33342 (Cat. no. B2261; Sigma-Aldrich; Merck KGaA) was
employed as fluorescent probe and added to produce a final
concentration of 5 µM at room temperature for 5 min in the dark.
The solution was then incubated in a 37°C incubator for 2 h.
Following washing with ice-cold PBS for three times, propidium
iodide (PI; Sigma-Aldrich; Merck KGaA) was added to remove dead
cells at final concentration of 2 µg/ml, incubated for 10 min at
room temperature in darkness. Then stained cells were analyzed by 2
laser Navios flow cytometers (Beckman Coulter, Inc., Brea, CA, USA)
and data was analyzed using FlowJo software 10.5.0. (FlowJo LLC,
Ashland, OR, USA). The control experiments with the ABC transporter
inhibitor were incubated with fumitremorgin C (Sigma-Aldrich; Merck
KGaA) at a final concentration of 10 µM for 30 min at 37°C.
Western blot analysis
Total protein was extracted using
radioimmunoprecipitation assay (25 mM Tris-HCl (pH 7.6), 150 mM
NaCl, 1% NP-40, 1% sodium deoxycholate and 0.1% SDS) at 4°C for 10
min. Subsequently, the cell lysate was centrifugated at 12,000 × g
for 10 min at 4°C, and then the supernatant was collected. Total
protein of cellular lysate was qualified using BCA protein assay
kit (Beyotime Institute of Biotechnology, Beijing, China). Total
protein (20 µg) was separated by electrophoresis in 4–12% SDS-PAGE
gels and then transferred to polyvinylidene fluoride membrane
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and blocked in 5%
bovine serum albumin (Sigma-Aldrich; Merck KGaA) at room
temperature for 30 min. The primary antibodies against p53 (cat.
no. ab1101; dilution, 1:2,000), p21 (cat. no. ab109520; dilution,
1:1,000), Sco2 (cat. no. ab115877; dilution, 1:2,000), Bcl2 (cat.
no. ab32124; dilution, 1:1,000) or β-actin (cat. no. ab8226;
dilution, 1:5,000) were incubated for 1 h at room temperature.
Following three washes, membranes were incubated with Horse radish
peroxidase (HRP) conjugated goat anti-rabbit IgG secondary antibody
(cat. no. ab7090; dilution, 1:5,000) or goat anti-mouse IgG
secondary antibody (cat. no. ab97040; dilution, 1:5,000) for 1 h at
room temperature. All antibodies were purchased from Abcam
(Cambridge, UK). Following three washes, signals were visualized by
chemiluminescence using the ECL Dura Extended Duration Substrate
(Pierce; Thermo Fisher Scientific, Inc.). The membrane was imaged
by Quantity One software (Version: 4.6.9; Bio-Rad Laboratories,
Inc.).
Transfections of miRNA mimics and
antago-miR
miR-125a mimics (cat. no. AM12378;
UCCCUGAGACCCUUUAACCUGUGA), control mimics (scrambled mimics; cat.
no. AM17010; UGACAACCUGGUAGAAAGAGACUUC), antago-miR-125a (cat. no.
MH10389; UCCCUGAGACCCUUAACCUGUG) and control antago-miR
(antago-scrambled; cat. no. 4464076; UCGGCCUUUUGCUCACAGACCA) were
purchased from Thermo Fisher Scientific, Inc. For each
oligonucleotide, 50 nM were transfected using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.)
separately for 15 min at room temperature and then added to target
cell cultures in 12-well plates for 4 h at 37°C; following this,
the medium was replaced. After 48–72 h at 37°C, cells were
harvested, washed with PBS and used for subsequent assays.
Cell Counting Kit-8 (CCK-8) assay
For the CCK-8 assay, cells were seeded in 96-well
plates (4×103 cells/well), and incubated for 24 h for
attach. Cells were then transfected with p53-mutR248Q,
p53 or empty vector, or antago-miR-125a or antago-scrambled as
aforementioned. All plasmids were produced and supplied by
Guangzhou RiboBio Co., Ltd. (Guangzhou, China). At day 1, 2, 3, 4
and 5 after transfection, cell proliferation was measured using a
CCK-8 kit (Beyotime Institute of Biotechnology).
Chromatin immunoprecipitation
(ChIP)
ChIP assay was performed by employing EpiQuik
Chromatin Immunoprecipitation (ChIP)kit (AmyJet Scientific, Wuhan,
China) according to the manufacturer's protocol. Briefly, cells
were cross-linked using 0.25% glutaraldehyde at room temperature
for 10 min, and then sonicated for 5 min. Fractionated chromatin
was immunoprecipitated using anti-FLAG antibody (cat. no. 18230;
dilution, 1:200; Abcam). Precipitated DNA was used for quantitative
PCR using p21 promoter primers: Forward primer,
5′-TCTAGGTGCTCCAGGTGCTT-3′ and reverse primer,
5′-TCTGGCAGGCAAGGATTTAC-3′. 5′ untranslated region (UTR) of
dihydrofolate reductase (DHFR) was considered as negative control
fragments. The primers for 5′UTR of DHFR are as follows: Forward
primer, 5′-CTGATGTCCAGGAGGAGAAAGG-3′ and reverse primer,
5′-AGCCCGACAATGTCAAGGACTG-3′. This was conducted as per the
procedure described in the subsequent paragraph.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay
Total RNA was extracted using TRIzol®
(Thermo Fisher Scientific, Inc.). For RT, the miRNA specific RT
primers were as follows: miR-125a,
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTGGACACT-3′; miR-125b,
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTGAACACT-3′; and let-7,
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCACACCAA-3′. The RT was
performed using a Bulge-Loop™ miRNA qRT-PCR kit
(Guangzhou RiboBio Co., Ltd.), following the manufacturer's
protocols. For qPCR, the reverse primer was universal,
5′-CTCAACTGGTGTCGTGGAGTCG-3′, and the forward primers were:
miR-125a, 5′-TCCCTGAGACCCTTTA-3′; miR-125b, 5′-TCCCTGAGACCCTA-3′;
let-7, 5′-ACACTCCAGCTGGGTGAGGTAGTAGGTACA0-3′; and U6 snRNA,
5′-TGCGGGTGCTCGCTTCGGCAGC-3′. For each reaction, 0.5 µl cDNA was
added to a 20 µl PCR mixture for 40 cycles. Following initiation at
95°C for 3 min, each cycle consisted of 95°C for 30 sec and 60°C
for 30 sec, and then a final extension at 60°C for 5 min. The
amplification signal was detected using SYBR Green I (Guangzhou
RiboBio Co., Ltd., Guangzhou, China) was collected. All genes are
normalized to U6 snoRNA. Analysis of relative gene expression data
using real-time quantitative PCR and the 2−∆∆Cq method
(24).
Dual-luciferase reporter assays
Luciferase reporter assays were performed using the
psiCHECK2 dual-luciferase reporter system (Promega Corporation,
Madison, WI, USA). Transfection is performed using Lipofectamine
2000, following the manufacturer's protocols. PsiCHECK2 and
miR-125A expression vector or empty vector were co-transfected into
293 cells (50 nm; American Type Culture Collection, Manassas, VA,
USA). At 48 h following transfection, Firefly (FF) and Renilla (RE)
luciferase activities were quantified using the Dual-Luciferase
Reporter assay system (Promega Corporation), and RE luciferase
activity was normalized to FF luciferase activity.
Serial replating assay
CSCs cells were harvested and plated in DMEM/F12
containing B27, EGF and bFGF. The colony-forming
units/1×103 plated cells were quantified after 7–10 days
of culture and expressed as the mean and standard error of mean of
at least triplicate experiments. For each passage, the standard
procedure was repeated.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Differences between groups were analyzed using one-way
analysis of variance or Kruskal-Wallis test, with Dunn's multiple
group comparison test. P<0.05 was considered to indicate a
statistically significant difference. Analyses were performed using
Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA,
USA).
Results
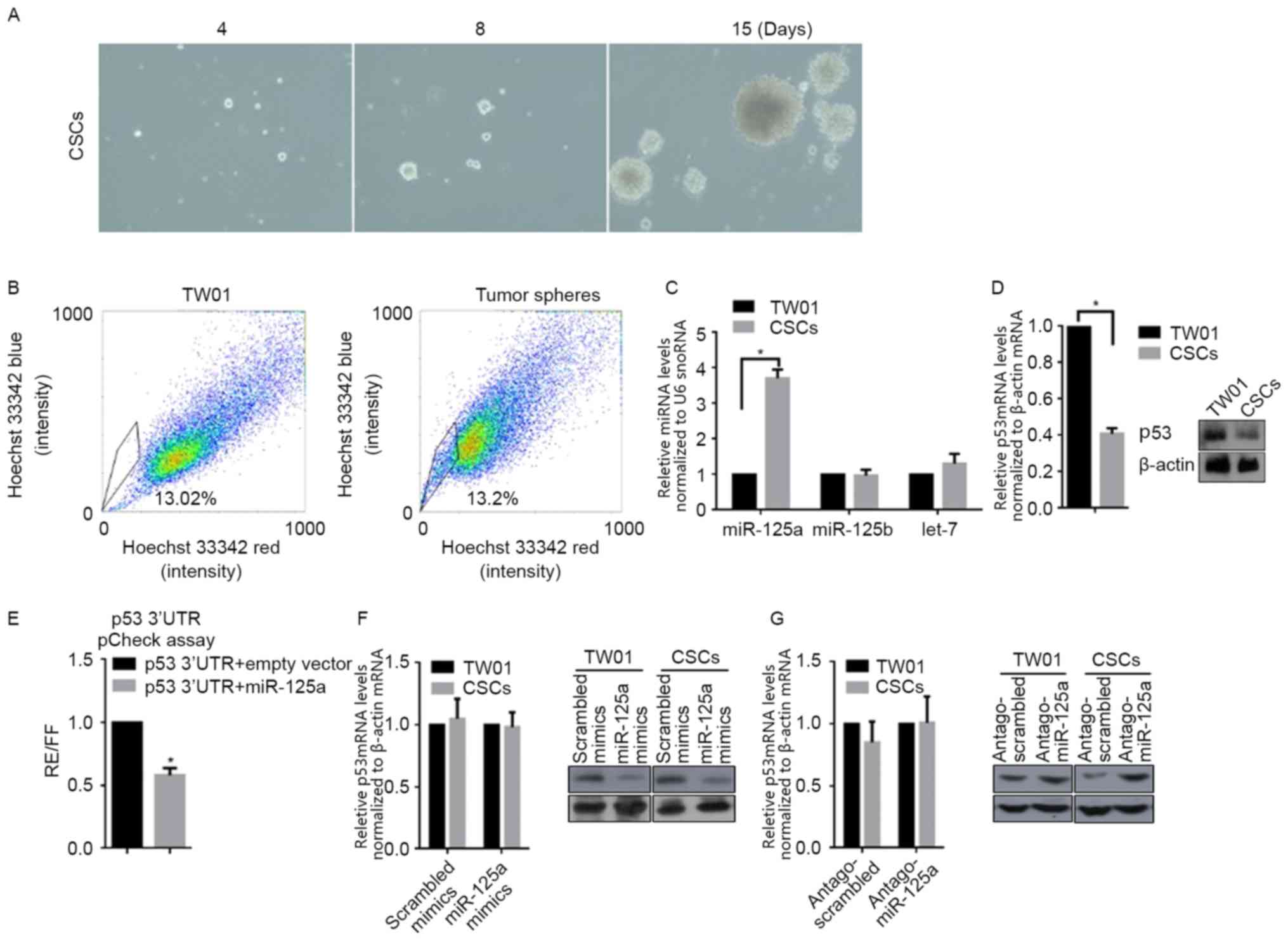
CSCs derived from the self-renewing
NPC cell line TW01, downregulates p53 mRNA and protein levels by
upregulating miR-125a, but not miR-125b
For characterizing specificity of CSCs, cells
derived from TW01, via incubation in SFM for 3 weeks, were used in
self-renewal capacity (serial replating) and SP assays, in order to
identify stemness characteristics. Following tumor sphere selection
for 15 days, notable spheres were detected (Fig. 1A). Subsequently, spheres derived from
TW01 were stained for SP analysis. The percentage of SP was notably
higher in tumor sphere cells compared with parental TW01 cells
(Fig. 1B). These two results
identified the stemness of CSCs isolated with SFM culturing.
According to our previous data, miR-125a and miR-125b are
associated with the malignancy of NPC (10); therefore, the expression level of
miR-125a, miR-125b and a non-relative control let-7 was examined
via RT-qPCR. Notably, miR-125a, but miR-125b and let-7, was
significantly upregulated in CSCs when compared with TW01 cells
(Fig. 1C). By considering the
post-transcriptional regulatory role of miR-125a on p53, RT-qPCR
and western blot analysis were conducted, and as expected, the mRNA
and protein levels of p53 in CSCs were significantly decreased
compared with TW01 cells (Fig. 1D).
To determine if miR-125a binds directly to the 3′UTR of p53, the
miR-125a expression vector and luciferase vectors inserted with a
1,1850-nt long 3′UTR of p53, were co-transfected into 293 cells. As
depicted in Fig. 1E, co-transfection
led to the 42±5.62% reduction in normalized luciferase values
compared with the control. miR-125a mimics, scrambled mimics,
antago-miR-125a or antago-scrambled were transfected into TW01, or
CSCs. RT-qPCR and western blot analysis were then performed to
detect the mRNA and protein levels of p53. As depicted in Fig. 1F, transfection of miR-125a mimics or
scrambled mimics did not significantly affect p53 mRNA levels, but
transfection of miR-125a downregulated p53 protein level markedly
in CSCs compared with the scrambled control. In CSCs, transfection
of antago-miR-125a markedly increased p53 protein levels compared
with the scrambled control, without affecting the p53 mRNA level
(Fig. 1G). Taken together, miR-125a
was demonstrated to be downregulated in CSCs, leading to subsequent
p53 translational downregulation.
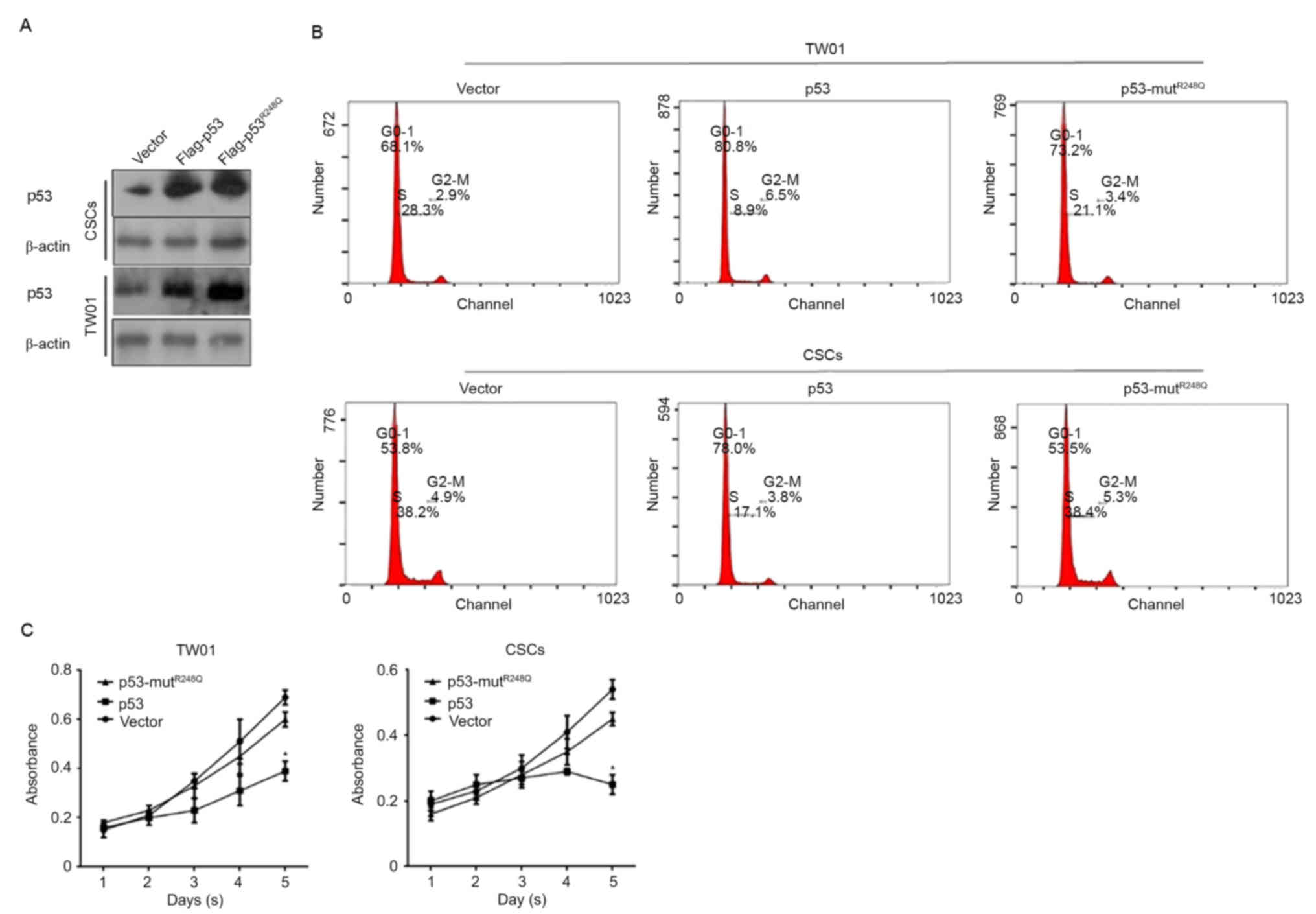
p53 regulates proliferation and
induces cell cycle arrest via its DNA binding activity
p53 functions as an antitumor factor primarily
through its transcriptional regulatory activity, in which DNA
binding activity is necessary. In order to determine how p53
regulates CSCs, two expressing vectors were constructed:
Flag-tagged p53, expressing wild-type p53 fused with Flag peptide;
and Flag-tagged p53-mutR248Q, expressing mutated R to Q
amino acids that cause the complete loss of p53 DNA binding
activity. Successful transfections were confirmed by western blot
analysis 48 h after transfections (Fig.
2A). Following this, cells were stained with PI and analyzed by
flow cytometry. The results demonstrated that overexpressed
Flag-p53 arrested the cell cycle at the G0/G1
phase; however, Flag-p53-mutR248Q revealed no notable
effects on the percentage of cells in the
G0/G1 phase in TW01 and CSCs (Fig. 2B). To further confirm the effect of
overexpressed p53 in TW01 and CSC, a Cell Counting Kit-8 (CCK-8)
assay was used to detect the proliferation changes in the
transfected cells. Consistently, cell cycle arrest, due to the
overexpression of wild type p53, significantly inhibited the
proliferation in TW01 and CSCs compared with the corresponding
vector controls (Fig. 2C).
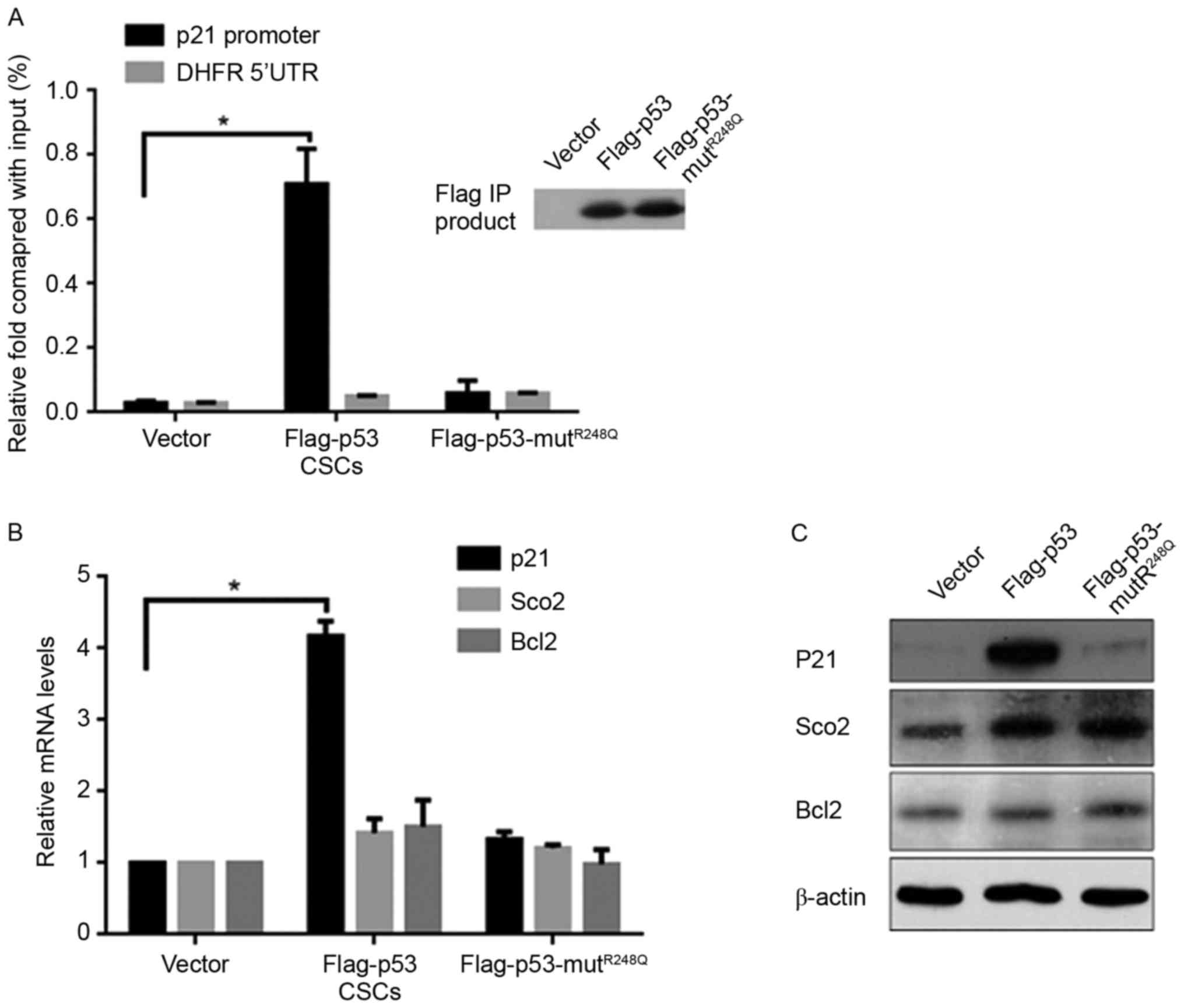
p53 transcriptionally induces the
expression of P21 in CSCs
The regulatory effects of p53 on the cell cycle
primarily occur through the upregulation of its target gene, p21,
indicating the potential regulatory mechanism of p53 on CSCs. The
Chromatin Immunoprecipitation (ChIP) assay demonstrated that
ectopic expression of Flag-p53, but not
Flag-p53-mutR248Q, significantly increased the binding
activity to the p21 promoter compared with the vector (Fig. 3A). It is understood that p53 is
multi-functional, as a result of it transcriptionally regulating
various target genes; therefore, the expression of the
respiration-associated gene Sco2 and mitochondrial
apoptotic-associated gene Bcl2 were measured, which are regulated
by p53. Notably, the mRNA and protein levels of p21 were increased
by p53, but no significant differences in Sco2 or Bcl2 expression
levels were noted compared with the vector control group (Fig. 3B and C).
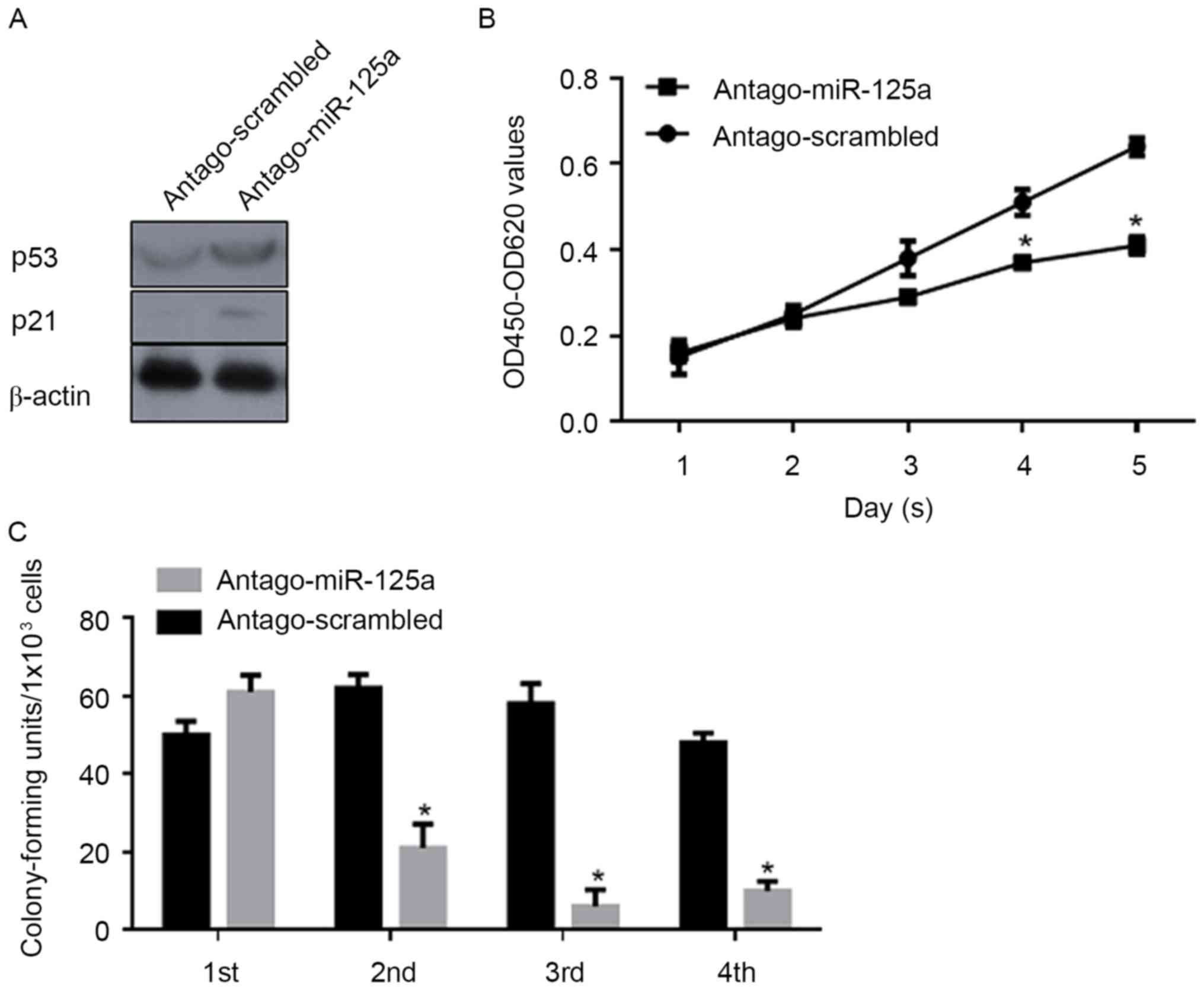
miR-125a post-transcriptionally
regulates p53 and thus promotes cell proliferation and the
self-renewal capacity of CSCs
In order to investigate the regulatory roles of
miR-125a on the proliferation and stemness of CSCs, antago-miR-125a
or antago-scrambled were transfected into CSCs, and CCK-8 and
serial replating assays were performed. Following transfection of
antago-miR-125a, a marked increase in p53 and p21 protein levels
were confirmed by western blot analysis (Fig. 4A). Consistently, proliferation was
significantly decreased compared with the scrambled control
(Fig. 4B). As depicted in Fig. 4C, the self-renewal capacity was also
significantly inhibited by the downregulation of miR-125a. Above
all, the expression of miR-125a is important for the proliferation
and maintenance of stemness in CSCs.
Discussion
According to a previous hypothesis, CSCs are
responsible for tumor initiation, cell survival following
chemotherapy or radiotherapy, metastatic spread and tumor
recurrence (18). As a small
sub-population of tumor cells, CSCs are resistant to numerous
current cancer treatments, particularly chemotherapy and
radiotherapy (25,26), which means instead of eliminating
CSCs, therapies only kill the bulk of tumor. A number of studies
have focused on the effects of therapies on tumor cells and
attempted to reveal the mechanisms of resistance (5–9); however,
further focus should be on revealing the mechanism of
chemoresistance in CSCs. In our previous study, chemotherapy using
cisplatin notably upregulated the expression level of miR-125a and
miR-125b, and thus decreased p53, resulting in resistance (10); therefore, the regulatory role of
miR-125a, miR-125b and p53 in CSCs was investigated.
The present study demonstrated that CSCs derived
from TW01 cells present self-renewal capacity and an increased rate
of SP. The expression levels of miR-125a and miR-125b, which were
previously reported to be upregulated in cisplatin-treated TW01
cells, resulting in chemoresistance, were detected (10). Notably, only the expression level of
miR-125a was upregulated, but not miR-125b in the CSCs in the
present study. Consistently, the upregulation of miR-125a decreased
the mRNA and protein levels of p53 by targeting p53 mRNA. These
results indicated that the change in miR-125a expression levels may
be responsible for the maintenance of stemness of CSCs, via
modifying p53. In order to confirm this, wild-type p53 or mutant
p53, which lacks DNA binding activity in CSCs, was ectopically
introduced. The introduction of wild-type p53 into CSCs
significantly decreased the self-renewal capacity and proliferation
of cells, but proliferation was not decreased as a result of mutant
p53 due to p21 being inactivated,, and caused cell cycle arrest.
Taken together, miR-125a is responsible for maintaining stemness of
CSCs by transcriptionally downregulating p53.
The miR-125 family is composed of three homologs,
miR-125a, miR-125b-1 and miR-125-2, which target different mRNAs
(10). Accumulating evidence
indicates that the expression level of miR-125a, which is located
at 19q13, is frequently downregulated or even deleted in human
malignances, including breast cancer, ovarian cancer, lung cancer,
medulloblastoma and gastric cancer (27–31). Human
miRNA microarrays have indicated that miR-125a is downregulated in
hepatocellular carcinoma tissues (32). miR-125a has the ability to
post-transcriptionally inhibit the mRNA level of p53 in tumors
(33). By targeting p53 mRNA
directly, miR-125a promotes proliferation, migration and invasion
in cancer cells (34). Consistently,
inhibition of miR-125a in multiple myeloma cells presents contrary
effects on proliferation, migration and invasion (35); however, these studies rarely focused
on the expression profile of miR-125a in the CSC subpopulation.
Previous studies demonstrated the association
between p53 and stem cell biology, and indicated the important
effects of the p53 signaling pathway in CSCs (36,37).
Ectopic expression of p53 was demonstrated to decrease the
efficiency of reprogramming somatic cells to induced pluripotent
stem cells (iPSCs) (38,39). The deletion of p53 allows the
suboptimal cells to become iPSCs and simultaneously accelerates
nuclear reprogramming via loss of control of the cell cycle arrest,
by p53 (39). Multiple p53 pathways
are reported to regulate CSCs, including ADP ribosylation
factor/p53 (40), miR34/p53 (41), translationally controlled tumor
protein/p53 (42) and p21/p53
(43). All of these data indicated
the regulation of p53 on CSCs.
In conclusion, the present study revealed the
association between upregulated miR-125a, p53 and stemness of CSCs;
however, the regulation of p53 within stemness and the molecular
mechanism underlying upregulation of miR-125a should be further
investigated for developing novel therapeutic targets for future
anticancer therapies in patients.
Acknowledgements
The authors would like to that Professor Wei Wang
(Sichuan University, Beijing China) for editing the language of
this manuscript.
Funding
Not funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JC designed part of the experiments and performed
gene expressing and cell culture relative experiments. HO and XA
performed gene expression analysis and some cell culture relative
experiments. HO performed analysis and interpretation of data. SL
designed part of experiments and supervised the whole
procedure.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Isayeva T, Li Y, Maswahu D and
Brandwein-Gensler M: Human papillomavirus in non-oropharyngeal head
and neck cancers: A systematic literature review. Head Neck Pathol.
6 Suppl 1:S104–S120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lo KW, To KF and Huang DP: Focus on
nasopharyngeal carcinoma. Cancer Cell. 5:423–428. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheung F, Chan O, Ng WT, Chan L, Lee A and
Pang SW: The prognostic value of histological typing in
nasopharyngeal carcinoma. Oral Oncol. 48:429–433. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spratt DE and Lee N: Current and emerging
treatment options for nasopharyngeal carcinoma. Onco Targets Ther.
5:297–308. 2012.PubMed/NCBI
|
|
5
|
Al-Sarraf M, LeBlanc M, Giri PG, Fu KK,
Cooper J, Vuong T, Forastiere AA, Adams G, Sakr WA, Schuller DE and
Ensley JF: Chemoradiotherapy versus radiotherapy in patients with
advanced nasopharyngeal cancer: Phase III randomized Intergroup
study 0099. J Clin Oncol. 16:1310–1317. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee AW, Lau WH, Tung SY, Chua DT, Chappell
R, Xu L, Siu L, Sze WM, Leung TW, Sham JS, et al: Preliminary
results of a randomized study on therapeutic gain by concurrent
chemotherapy for regionally-advanced nasopharyngeal carcinoma:
NPC-9901 Trial by the Hong Kong Nasopharyngeal Cancer Study Group.
J Clin Oncol. 23:6966–6975. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wee J, Tan EH, Tai BC, Wong HB, Leong SS,
Tan T, Chua ET, Yang E, Lee KM, Fong KW, et al: Randomized trial of
radiotherapy versus concurrent chemoradiotherapy followed by
adjuvant chemotherapy in patients with American Joint Committee on
Cancer/International Union against cancer stage III and IV
nasopharyngeal cancer of the endemic variety. J Clin Oncol.
23:6730–6738. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blanchard P, Lee A, Marguet S, Leclercq J,
Ng WT, Ma J, Chan AT, Huang PY, Benhamou E, Zhu G, et al:
Chemotherapy and radiotherapy in nasopharyngeal carcinoma: An
update of the MAC-NPC meta-analysis. Lancet Oncol. 16:645–655.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Q, Fan H, Liu Y, Yin Z, Cai H, Liu J,
Wang Z, Shao M, Sun X, Diao J, et al: Curcumin enhances the
radiosensitivity in nasopharyngeal carcinoma cells involving the
reversal of differentially expressed long non-coding RNAs. Int J
Oncol. 44:858–864. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen JJ, Liu SX, Chen MZ and Zhao ZY:
Has-miR-125a and 125b are induced by treatment with cisplatin in
nasopharyngeal carcinoma and inhibit apoptosis in a p53-dependent
manner by targeting p53 mRNA. Mol Med Rep. 12:3569–3574. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jordan CT, Guzman ML and Noble M: Cancer
stem cells. N Engl J Med. 355:1253–1261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dalerba P, Cho RW and Clarke MF: Cancer
stem cells: Models and concepts. Annu Rev Med. 58:267–284. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen YA, Wang CY, Hsieh YT, Chen YJ and
Wei YH: Metabolic reprogramming orchestrates cancer stem cell
properties in nasopharyngeal carcinoma. Cell Cycle. 14:86–98. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mueller MT, Hermann PC and Heeschen C:
Cancer stem cells as new therapeutic target to prevent tumour
progression and metastasis. Front Biosci (Elite Ed). 2:602–613.
2010.PubMed/NCBI
|
|
15
|
Tirino V, Desiderio V, Paino F, De Rosa A,
Papaccio F, La Noce M, Laino L, De Francesco F and Papaccio G:
Cancer stem cells in solid tumors: An overview and new approaches
for their isolation and characterization. FASEB J. 27:13–24. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kakarala M and Wicha MS: Cancer stem
cells: Implications for cancer treatment and prevention. Cancer J.
13:271–275. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mannelli G and Gallo O: Cancer stem cells
hypothesis and stem cells in head and neck cancers. Cancer Treat
Rev. 38:515–539. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Levine AJ and Oren M: The first 30 years
of p53: Growing ever more complex. Nat Rev Cancer. 9:749–758. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nii T, Marumoto T and Tani K: Roles of p53
in various biological aspects of hematopoietic stem cells. J Biomed
Biotechnol. 2012:9034352012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoon DS, Choi Y and Lee JW: Cellular
localization of NRF2 determines the self-renewal and osteogenic
differentiation potential of human MSCs via the p53-SIRT1 axis.
Cell Death Dis. 11:e20932016. View Article : Google Scholar
|
|
22
|
Olivos DJ and Mayo LD: Emerging
non-canonical functions and regulation by p53: p53 and stemness.
Int J Mol Sci. 17(pii): E19822016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hegde S, Hankey P and Paulson RF:
Self-renewal of leukemia stem cells in friend virus-induced
erythroleukemia requires proviral insertional activation of Spi1
and hedgehog signaling but not mutation of p53. Stem Cells.
30:121–130. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bi Q, Tang S, Xia L, Du R, Fan R, Gao L,
Jin J, Liang S, Chen Z, Xu G, et al: Ectopic expression of MiR-125a
inhibits the proliferation and metastasis of hepatocellular
carcinoma by targeting MMP11 and VEGF. PLoS One. 7:e401692012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cowden Dahl KD, Dahl R, Kruichak JN and
Hudson LG: The epidermal growth factor receptor responsive miR-125a
represses mesenchymal morphology in ovarian cancer cells.
Neoplasia. 11:1208–1215. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li W, Duan R, Kooy F, Sherman SL, Zhou W
and Jin P: Germline mutation of microRNA-125a is associated with
breast cancer. J Med Genet. 46:358–360. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shang H, Wang T, Shang F, Huang KM and Li
YQ: A germline mutation in the miR-125a coding region reduces
miR-125a expression and is associated with human gastric cancer.
Mol Med Rep. 10:1839–1844. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu Y, Huang Z and Liu Y: Reduced
miR-125a-5p expression is associated with gastric carcinogenesis
through the targeting of E2F3. Mol Med Rep. 10:2601–2608. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Murakami Y, Yasuda T, Saigo K, Urashima T,
Toyoda H, Okanoue T and Shimotohno K: Comprehensive analysis of
microRNA expression patterns in hepatocellular carcinoma and
non-tumorous tissues. Oncogene. 25:2537–2545. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Y, Gao JS, Tang X, Tucker LD,
Quesenberry P, Rigoutsos I and Ramratnam B: MicroRNA 125a and its
regulation of the p53 tumor suppressor gene. FEBS Lett.
583:3725–3730. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gao W, Chan JY and Wong TS: Curcumin
exerts inhibitory effects on undifferentiated nasopharyngeal
carcinoma by inhibiting the expression of miR-125a-5p. Clin Sci
(Lond). 127:571–579. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Alzrigat M and Jernberg-Wiklund H: The
miR-125a and miR-320c are potential tumor suppressor microRNAs
epigenetically silenced by the polycomb repressive complex 2 in
multiple myeloma. RNA Dis. 4(pii): e15292017.PubMed/NCBI
|
|
36
|
Prabhu VV, Allen JE, Hong B, Zhang S,
Cheng H and El-Deiry WS: Therapeutic targeting of the p53 pathway
in cancer stem cells. Expert Opin Ther Targets. 16:1161–1174. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ginestier C, Charafe-Jauffret E and
Birnbaum D: p53 and cancer stem cells: The mevalonate connexion.
Cell Cycle. 11:2583–2584. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hong H, Takahashi K, Ichisaka T, Aoi T,
Kanagawa O, Nakagawa M, Okita K and Yamanaka S: Suppression of
induced pluripotent stem cell generation by the p53-p21 pathway.
Nature. 460:1131–1135. 2009. View Article : Google Scholar
|
|
39
|
Marión RM, Strati K, Li H, Murga M, Blanco
R, Ortega S, Fernandez-Capetillo O, Serrano M and Blasco MA: A
p53-mediated DNA damage response limits reprogramming to ensure iPS
cell genomic integrity. Nature. 460:1149–1153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Grinstein E and Wernet P: Cellular
signaling in normal and cancerous stem cells. Cell Signal.
19:2428–2433. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu C, Kelnar K, Liu B, Chen X,
Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, et
al: The microRNA miR-34a inhibits prostate cancer stem cells and
metastasis by directly repressing CD44. Nat Med. 17:211–215. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Koziol MJ, Garrett N and Gurdon JB: Tpt1
activates transcription of oct4 and nanog in transplanted somatic
nuclei. Curr Biol. 17:801–807. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ligon KL, Huillard E, Mehta S, Kesari S,
Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, et al:
Olig2-regulated lineage-restricted pathway controls replication
competence in neural stem cells and malignant glioma. Neuron.
53:503–517. 2007. View Article : Google Scholar : PubMed/NCBI
|