Introduction
Glioblastoma (GBM) is the most fatal primary
malignant tumor of the central nervous system in adults, and
accounts for 46.1% of all cases of malignant brain tumors (1). Patients who are newly diagnosed with GBM
receive surgery as standard, followed by concurrent
radiochemotherapy and maintenance temozolomide chemotherapy
(2). Despite this aggressive
treatment strategy, relapse is common and the median overall
survival (OS) of patients with GBM is ~15 months (3). No other effective agents against GBM
have been developed over the past decade since the approval of
temozolomide for GBM treatment in 2004. Furthermore, the use of
currently available agents has been hindered due to limited
information on the molecular mechanisms involved in GBM development
or treatment response. Therefore, it may be beneficial to elucidate
the mechanisms underlying GBM and consequently develop novel
therapeutic strategies.
Numerous studies have investigated the genes
involved in GBM. A previous study by Yeom et al (4) indicated that the expression of the
guanosine-5′-triphosphate-binding protein Ras related glycolysis
inhibitor and calcium channel regulator is correlated with
temozolomide resistance, and contributes to the poor survival of
patients with GBM. The inhibitor of nuclear factor κ-B kinase
subunit ε (IKBKE) is overexpressed in human GBM, and the inhibition
of IKBKE markedly suppresses the proliferative and invasive
activity of GBM cells (5). High
expression levels of hypoxia-inducible factor-1α promote the
activation of glioma cell motility by affecting molecules
associated with invasion (6).
Recombinant expression of HMG-CoA reductase (HMGCR) promotes the
growth and migration of U251 and U373 cells, whereas the knockdown
of HMGCR expression inhibits the growth, migration and metastasis
of GBM cells (7). Lymphoid enhancer
factor-1 maintains the state of proliferation and migration in GBM
cells, and the GBM stem-cell-like self-renewal ability of U251
cells (8). However, the current
understanding of the mechanisms underlying GBM remains limited.
In 2006, Sun et al (9) published a study in which 157 primary
human glioma and 23 nontumor human brain samples underwent mRNA
expression profiling, in order to verify whether overexpression of
stem cell factors was associated with the poor prognosis of
patients with glioma. In the current study, microarray analysis was
conducted to screen differentially expressed genes (DEGs) in GBM
samples. Hub genes, in addition to significant modules and
pathways, were identified using comprehensive bioinformatics
methods. The present study aimed to identify the candidate genes
and associated pathways of GBM, in order to elucidate the molecular
mechanisms underlying this malignancy.
Materials and methods
Microarray data
The gene expression profiles of GSE4290 were
downloaded from the public functional genomics data repository Gene
Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/), which is based on
the Affymetrix (Thermo Fisher Scientific, Inc., Waltham, MA, US)
Human Genome U133 Plus 2.0 Array. These gene expression files were
deposited by Sun et al (9).
The gene expression profiles of 77 GBM tissue samples and 23
nontumor brain samples from patients with epilepsy were retrieved
from the GSE4290 dataset.
DEG screening
GEO2R is an interactive online tool based on the R
programming language, which allows for comparisons between two
groups of samples in a GEO series to be made (10). Adjusted P-values were utilized to
decrease the false-positive rate through the default Benjamini and
Hochberg false discovery rate method. An adjusted P<0.05 and
|logFC|≥2 were considered to indicate a statistically significant
difference.
Functional enrichment analysis
Gene Ontology (GO) analysis may be applied in
large-scale functional studies on genomic or transcriptomic data
(11). The Kyoto Encyclopedia of
Genes and Genomes (KEGG) is the major recognized pathway-associated
database, which contains information on gene networks in various
organisms (12). Previous studies
have claimed that the analysis of upregulated and downregulated
genes separately may allow for the identification of additional
pathways, compared with combined analysis (13–15). In
the present study, specific pathways involved in tumor occurrence
and development were used; hence, separate analysis was performed.
GO functional and KEGG pathway enrichment analyses were conducted
separately for upregulated and downregulated genes using the
Database for Annotation, Visualization, and Integrated Discovery
software (DAVID version 6.8; http://david.ncifcrf.gov/) (16). P<0.05 was considered to indicate a
statistically significant difference.
Integration of protein-protein
interaction (PPI) network and module analysis
The STRING (https://string-db.org/) database is an online tool for
the assessment and integration of PPIs, including direct (physical)
and indirect (functional) associations. STRING version 10.5
encompasses 9,643,763 proteins from 2,031 organisms (17). PPI associations amongst DEGs were
searched for using the STRING database with a default required
confidence of >0.4. The PPI networks of the DEGs were
constructed using Cytoscape software version 3.6.0 (http://www.cytoscape.org/). The plug-in Molecular
Complex Detection (MCODE) was used to screen important modules with
established scores of >3 and nodes of >4. GO and KEGG
analyses were also conducted using the genes in these modules. In
the PPI network, the number of edges involved determined the
degrees of the nodes, and nodes with high degrees were determined
to be hub genes. Hub genes were also mapped to STRING in order to
evaluate their PPI information.
Expression and survival analyses of
hub genes
Gene Expression Profiling Interactive Analysis
(GEPIA) is an online tool used to analyze the RNA sequencing
expression data of 9,736 tumors and 8,587 healthy samples from The
Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression
(GTEx) databases (18). GEPIA was
used to perform the tumor/healthy differential expression and
survival analyses of hub genes. The method of Kaplan-Meier for
survival analysis was conducted in GEPIA between the high and low
expression groups, with a cut-off value of 50%. The hazard ratio
with 95% confidence intervals and the log-rank P-value were
calculated, and the results are displayed as a plot. P<0.05 was
considered to indicate a statistically significant difference.
Results
Identification of DEGs
The comparative GEO2R analysis of the DEGs in the
GBM samples and healthy controls revealed 1,801 DEGs, including 620
upregulated and 1,181 downregulated genes.
GO function and KEGG pathway
enrichment analysis
The upregulated and downregulated DEGs were imported
into DAVID for GO analysis. The GO analysis results revealed that
the upregulated DEGs were significantly enriched in the terms
‘mitotic cell cycle process’, ‘mitotic cell cycle’ and ‘cell cycle
process’ (Table I). Downregulated
genes were enriched in ‘trans-synaptic signaling’, ‘anterograde
trans-synaptic signaling’ and ‘synaptic signaling’ (Table I).
 | Table I.GO analysis of differentially
expressed genes associated with glioblastoma. |
Table I.
GO analysis of differentially
expressed genes associated with glioblastoma.
| A, upregulated
genes |
|---|
|
|---|
| Term | Count | % | P-value | FDR |
|---|
| GO:1903047: Mitotic
cell cycle process | 80 | 17.699 |
9.95×10−25 |
1.90×10−21 |
| GO:0000278: Mitotic
cell cycle | 83 | 18.362 |
2.43×10−24 |
4.64×10−21 |
| GO:0022402: Cell
cycle process | 98 | 21.681 |
2.27×10−23 |
4.35×10−20 |
| GO:0007049: Cell
cycle | 107 | 23.672 |
1.40×10−21 |
2.68×10−18 |
| GO:0051301: Cell
division | 59 | 13.053 |
7.57×10−21 |
1.45×10−17 |
| GO:0007067: Mitotic
nuclear division | 48 | 10.619 |
2.22×10−18 |
4.25×10−15 |
| GO:0044770: Cell
cycle phase transition | 54 | 11.946 |
2.47×10−18 |
4.72×10−15 |
| GO:0044772: Mitotic
cell cycle phase transition | 52 | 11.504 |
4.17×10−18 |
7.96×10−15 |
| GO:0000819: Sister
chromatid segregation | 35 | 7.743 |
5.93×10−18 |
1.13×10−14 |
| GO:0000280: Nuclear
division | 52 | 11.504 |
9.96×10−16 |
1.91×10−12 |
|
| B, downregulated
genes |
|
| Term | Count | % | P-value | FDR |
|
| GO:0099537:
Trans-synaptic signaling | 137 | 16.707 |
1.13×10−64 |
2.15×10−61 |
| GO:0098916:
Anterograde trans-synaptic signaling | 137 | 16.707 |
1.13×10−64 |
2.15×10−61 |
| GO:0099536:
Synaptic signaling | 137 | 16.707 |
1.13×10−64 |
2.15×10−61 |
| GO:0007268:
Chemical synaptic transmission | 137 | 16.707 |
1.13×10−64 |
2.15×10−61 |
| GO:0007399: Nervous
system development | 227 | 27.682 |
4.23×10−45 |
8.05×10−42 |
| GO:0007267:
Cell-cell signaling | 178 | 21.707 |
9.70×10−41 |
1.85×10−37 |
| GO:0050804:
Modulation of synaptic transmission | 66 | 8.048 |
1.69×10−31 |
3.21×10−28 |
| GO:0048666: Neuron
development | 124 | 15.121 |
2.61×10−31 |
4.98×10−28 |
| GO:0031175: Neuron
projection development | 111 | 13.536 |
5.22×10−30 |
9.94×10−27 |
| GO:0007610:
Behavior | 90 | 10.975 |
9.43×10−30 |
1.80×10−26 |
The most significantly enriched KEGG pathways of the
upregulated and downregulated DEGs are displayed in Table II. The upregulated DEGs were enriched
in ‘cell cycle’, ‘ECM-receptor interaction’, ‘PI3K-Akt signaling
pathway, ‘p53 signaling pathway’ and ‘focal adhesion’.
Downregulated DEGs were enriched in ‘morphine addiction’,
‘GABAergic synapse’, ‘retrograde endocannabinoid signaling’,
‘calcium signaling pathway’ and ‘glutamatergic synapse’.
| Table II.KEGG pathway analysis of
differentially expressed genes associated with glioblastoma. |
Table II.
KEGG pathway analysis of
differentially expressed genes associated with glioblastoma.
| A, upregulated
genes |
|---|
|
|---|
| Term | Count | % | P-value |
|---|
| hsa04110: Cell
cycle | 19 | 4.203 |
7.83×10−10 |
| hsa04512:
ECM-receptor interaction | 16 | 3.539 |
1.89×10−09 |
| hsa04151: PI3K-Akt
signaling pathway | 28 | 6.194 |
5.08×10−08 |
| hsa04115: p53
signaling pathway | 13 | 2.876 |
5.41×10−08 |
| hsa04510: Focal
adhesion | 19 | 4.203 |
2.20×10−06 |
| hsa05205:
Proteoglycans in cancer | 17 | 3.761 |
2.54×10−05 |
| hsa04610:
Complement and coagulation cascades | 10 | 2.212 |
3.88×10−05 |
| hsa05150:
Staphylococcus aureus infection | 9 | 1.991 |
4.15×10−05 |
| hsa05166: HTLV-I
infection | 18 | 3.982 |
1.49×10−04 |
|
| B, downregulated
genes |
|
| Term | Count | % | P-value |
|
| hsa05032: Morphine
addiction | 32 | 3.902 |
5.34×10−21 |
| hsa04727: GABAergic
synapse | 30 | 3.658 |
9.98×10−20 |
| hsa04723:
Retrograde endocannabinoid signaling | 31 | 3.780 |
1.97×10−18 |
| hsa04020: Calcium
signaling pathway | 39 | 4.756 |
1.40×10−17 |
| hsa04724:
Glutamatergic synapse | 30 | 3.658 |
8.56×10−16 |
| hsa05033: Nicotine
addiction | 19 | 2.317 |
3.78×10−15 |
| hsa04080:
Neuroactive ligand-receptor interaction | 44 | 5.365 |
1.82×10−14 |
| hsa04024: cAMP
signaling pathway | 35 | 4.268 |
6.72×10−13 |
| hsa04713: Circadian
entrainment | 24 | 2.926 |
2.85×10−12 |
| hsa05031:
Amphetamine addiction | 19 | 2.317 |
8.49×10−11 |
| hsa05032: Morphine
addiction | 32 | 3.902 |
5.34×10−21 |
PPI network and module analyses
The PPI network constructed for the DEGs had 993
nodes and 7,810 interactions, in which two of the most significant
modules were identified by MCODE (Fig.
1). Module 1 had 59 nodes and 1,576 interactions, whereas
Module 2 had 32 nodes and 496 interactions. DEGs in these modules
were also enriched in ‘cell cycle’ and ‘neuroactive ligand-receptor
interaction’ of the KEGG pathways (Fig.
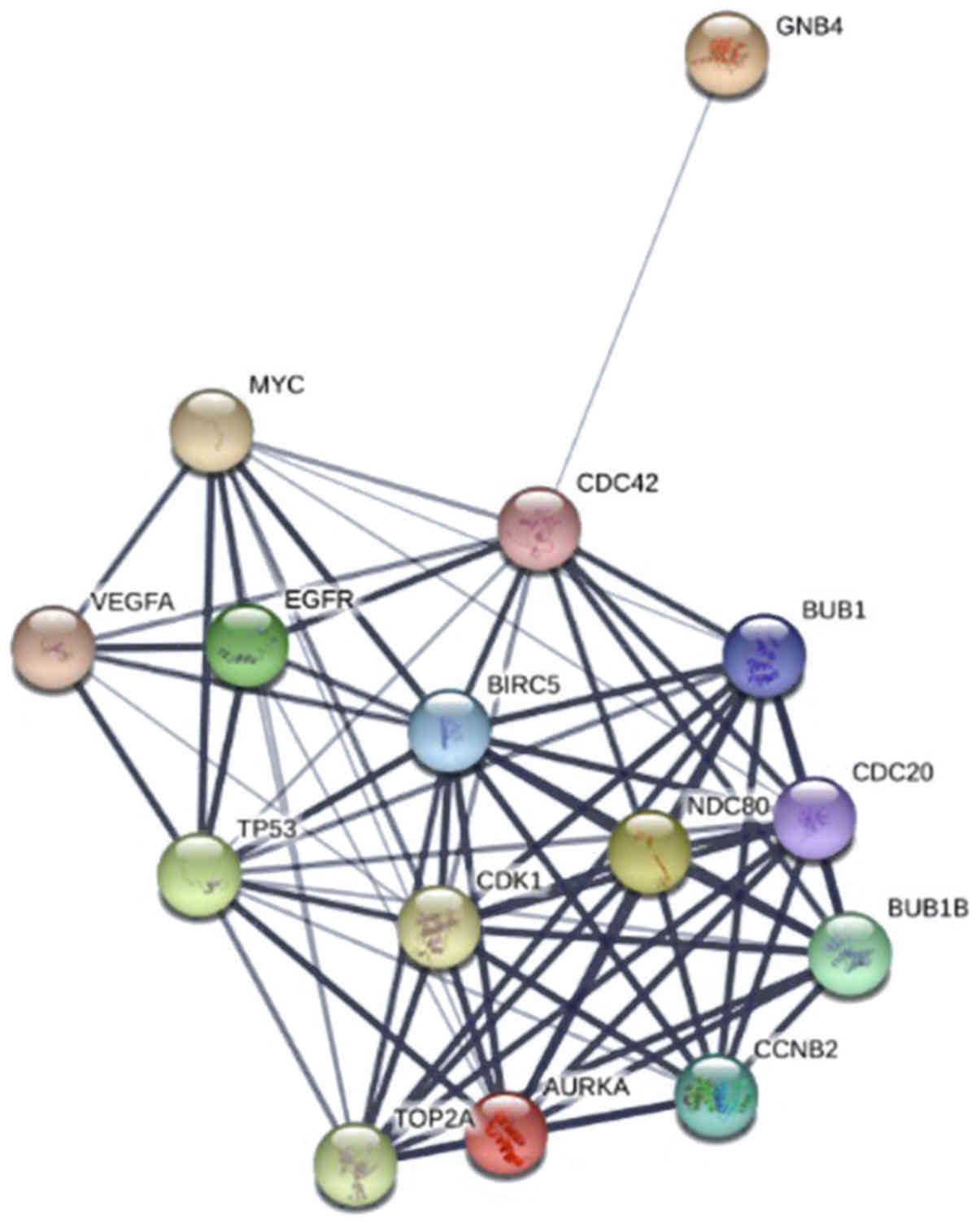
1). The top 15 hub genes were selected by the PPI network, with
a degree of >81 (Fig. 2).
Expression level and Kaplan-Meier plot
of hub genes
The expression levels of all 15 hub genes in
patients with GBM were upregulated relative to those in the healthy
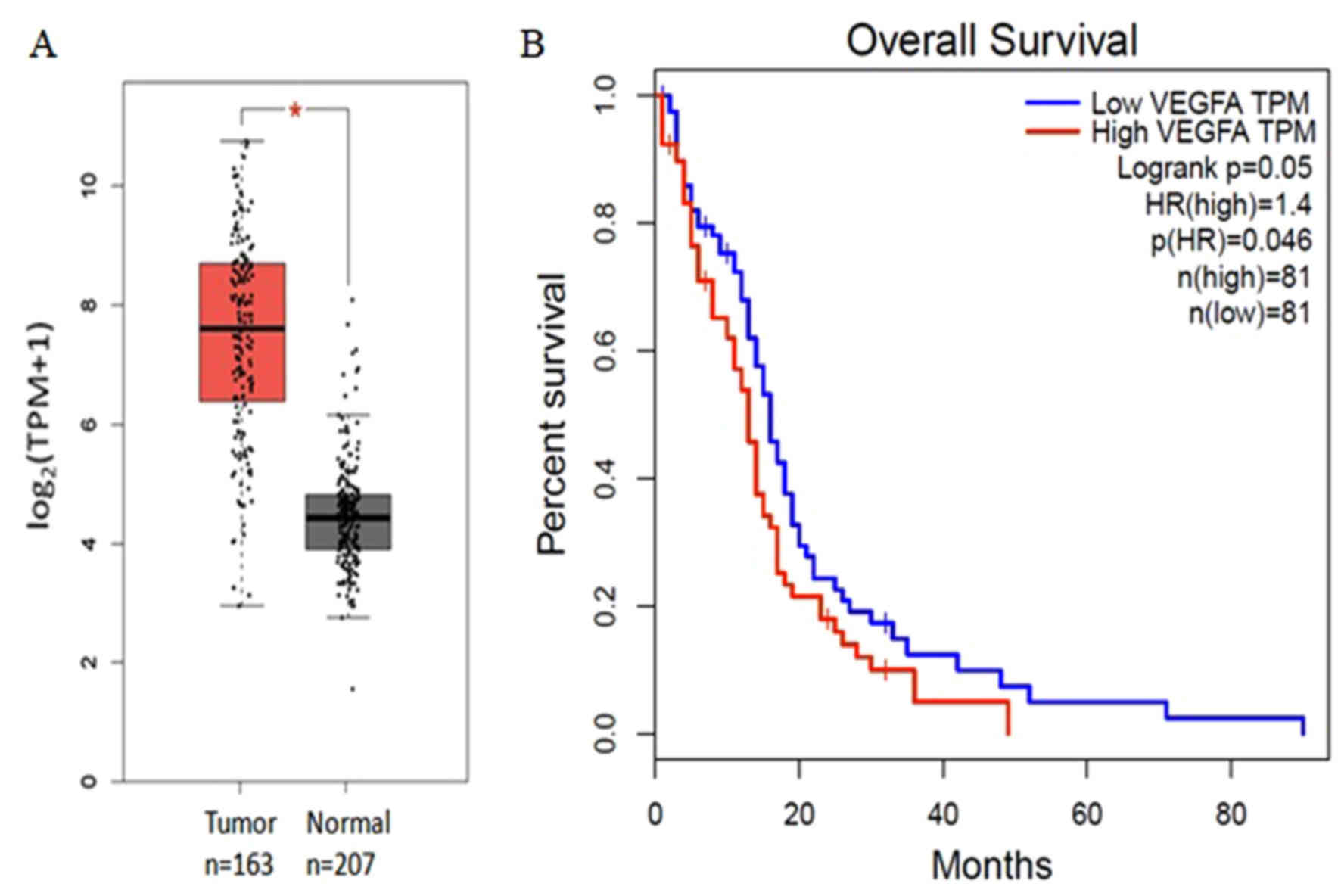
controls (P<0.05). High expression levels of vascular
endothelial growth factor A (VEGFA) was associated with poor
prognosis of patients with GBMs, whereas no significant difference
was observed for the remaining 14 genes (Fig. 3). The hub genes cyclin-dependent
kinase 1 (CDK1), cell-division cycle protein 20 (CDC20), aurora
kinase A (AURKA), and budding uninhibited by benzimidazoles 1
(BUB1) were also enriched in the top three modules.
Discussion
GBM is the most common primary malignant tumor of
the brain. However, the molecular mechanisms underlying the
progression of GBM remains unclear. In the present study, DEGs
between GBM and healthy samples were identified, and a series of
bioinformatics analytical methods applied in order to determine the
key genes and pathways associated with GBM. A total of 1,801 DEGs
were identified. These DEGs included 620 upregulated and 1,181
downregulated genes. Subjecting the DEGs to bioinformatics
analysis, including GO enrichment, KEGG pathway, PPI network and
survival analyses, revealed that GBM-associated genes and pathways
may serve an important role in the initiation and progression of
cancer.
GO term enrichment analysis indicated that the
upregulated DEGs were significantly enriched in the terms ‘mitotic
cell cycle process’, ‘mitotic cell cycle’ and ‘cell cycle process’.
Deregulation of the cell cycle serves a critical role in the
proliferation of malignant glioma cells. Genetic analyses of
primary human brain tumors detected common mutations in genes
encoding proteins critical for cell cycle regulation. These genes
include retinoblastoma protein, INK4A and CDK4 (19–22). The
downregulated DEGs were enriched in pathways involved in
trans-synaptic signaling and synaptic signaling. Yu et al
(23) reported that metabotropic
glutamate receptors, which are involved in synaptic signaling, are
also involved in the transformation and maintenance of various
cancer types, including glioma, melanoma skin cancer, breast cancer
and prostate cancer. The WW and C2 domain-containing protein (WWC)
family serves important roles in regulating cell proliferation,
cell migration and synaptic signaling. The overexpression of WWC3
inhibits glioma cell proliferation, migration and invasion
(24).
KEGG pathway analysis indicated that the functions
of the upregulated genes were enriched in ‘cell cycle’,
‘ECM-receptor interaction’, ‘PI3K-Akt signaling pathway’, ‘p53
signaling pathway’ and ‘focal adhesion’. Extracellular matrix (ECM)
rigidity may mediate the invasion of GBM multiforme cells through
actomyosin contractility (25,26). The
PI3K-Akt signaling pathway serves an important role in glioma
formation, through the suppression of cell death (27,28). p53
is a tumor suppressor factor which initiates DNA repair, cell cycle
arrest and apoptosis, and responds to numerous types of cancer
therapy (29,30). Downregulated DEGs were enriched in
‘morphine addiction’, ‘GABAergic synapse’, ‘retrograde
endocannabinoid signaling’, ‘calcium signaling pathway’ and
‘glutamatergic synapse’. Calcium signaling has notable functions in
numerous signaling processes involved in the proliferation and
motility of GBM cells (31).
Analysis of the top two modules from the PPI network
indicated that GBM was associated with the cell cycle and
neuroactive ligand-receptor interaction. Pal et al (32) recently demonstrated that patients who
have GBM in combination with a defective neuroactive
ligand-receptor interaction pathway have a poor prognosis
(P<0.0001). Therefore, monitoring these signaling pathways may
help predict tumor occurrence and progression. The top 15 hub genes
were identified from the network. Although these hub genes were all
upregulated in GBM, VEGFA is the only gene which was significantly
associated with the poor prognosis of patients with GBMs. GBMs are
highly vascularized tumors, and VEGFA is highly expressed in the
endothelial cells of blood vessels (33). Bevacizumab, a monoclonal antibody
against VEGFA, improves the progression-free survival of patients
with GBM, however it does not prolong the OS of patients compared
with the historical control (34).
Antiangiogenic treatment does not improve the OS of patients with
GBM compared with standard cytotoxic treatment, thus, an in-depth
understanding of the molecular mechanism underlying all of the hub
genes of GBM, including CDK1, CDC20, AURKA, and BUB1, is required.
CDK1, which is enriched in the module of Cluster 1, serves vital
roles in regulating oncogenesis and cell cycle progression
(35,36). The overexpression of CDC20 is
associated with temozolomide resistance in glioma cells (37). AURKA regulates the self-renewal and
tumorigenicity of glioma-initiating cells through the stabilization
of β-catenin (38).
Similar bioinformatics studies also used the
expression profile of GSE4290 for their analysis. However, previous
studies applied alternative bioinformatics methods to those used in
the present study, and thus obtained different results. For
example, two separate studies conducted bioinformatics analysis in
accordance with pathological grading (e.g. astrocytoma, GBM and
oligodendroglioma). One study reported that long-term potentiation
and ECM-receptor interaction may have important roles in the
occurrence and development of glioma, whereas the other study
focused on the involvement of the Wnt and p53 signaling pathways in
glioma (39,40). Li et al (41) compared 81 GBM samples with 23 controls
from GSE4290 and identified significant MAPK and cell cycle
signaling pathways. Furthermore, they reported that a number of
genes, including neuroblastoma RAS viral oncogene homolog, CDK2,
fibroblast growth factor receptor 2, and cyclin D1, were associated
with GBM (41). Wei et al
(42) used a method similar to that
used in the present study, in order to analyze 23 nontumor and 77
GBM (Grade 4) tumor samples of GSE4290. Through GO analysis, it was
discovered that DEGs were enriched in ‘synaptic transmission’,
‘regulation of vesicle-mediated transport’ and ‘ion-gated channel
activity’. KEGG analysis results indicated that DEGs were enriched
in ‘neuroactive ligand-receptor interaction’, ‘calcium signaling
pathway’, ‘p53 signaling pathway’ and ‘cell cycle’. The study also
identified vital transcription factors, including tumor protein
p53, specificity protein 1, JUN proto-oncogene AP-1 transcription
factor subunit, signal transducer and activator of transcription 3,
and transcription factor PU.1 (42).
In the present study, only GBM samples were included, and not
astrocytoma or oligodendroglioma. Expression and survival analyses
were also conducted using the novel GEPIA tool and the TCGA and
GTEx databases. Therefore, the results of the present study expand
on the current knowledge and understanding of the molecular
mechanisms of GBM.
In conclusion, bioinformatics analysis identified
hub genes and pathways that may have central roles in the
occurrence, development and prognosis of GBM. VEGFA, CDK1, CDC20,
AURKA and BUB1, the hub genes of GBM, may serve important roles in
the diagnosis and treatment of GBM.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets analyzed during the current study are
available in the GEO repository (http://www.ncbi.nlm.nih.gov/geo/).
Author's contributions
WL was in charge of study design. SY and KG were in
charge of data analysis and article publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dolecek TA, Propp JM, Stroup NE and
Kruchko C: CBTRUS statistical report: Primary brain and central
nervous system tumors diagnosed in the United States in 2005-2009.
Neuro Oncol. 14 Suppl 5:v1–v49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yeom SY, Nam DH and Park C: RRAD promotes
EGFR-mediated STAT3 activation and induces temozolomide resistance
of malignant glioblastoma. Mol Cancer Ther. 13:3049–3061. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Z, Lu J, Guo G, Yang Y, Dong S, Liu
Y, Nan Y, Zhong Y, Yu K and Huang Q: IKBKE promotes glioblastoma
progression by establishing the regulatory feedback loop of
IKBKE/YAP1/miR-Let-7b/i. Tumour Biol. 39:10104283177055752017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaynar MY, Sanus GZ, Hnimoglu H, Kacira T,
Kemerdere R, Atukeren P, Gumustas K, Canbaz B and Tanriverdi T:
Expression of hypoxia inducible factor-1alpha in tumors of patients
with glioblastoma multiforme and transitional meningioma. J Clin
Neurosci. 15:1036–1042. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qiu Z, Yuan W, Chen T, Zhou C, Liu C,
Huang Y, Han D and Huang Q: HMGCR positively regulated the growth
and migration of glioblastoma cells. Gene. 576:22–27. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao X, Mi Y, Ma Y and Jin W: LEF1
regulates glioblastoma cell proliferation, migration, invasion, and
cancer stem-like cell self-renewal. Tumour Biol. 35:11505–11511.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun L, Hui AM, Su Q, Vortmeyer A,
Kotliarov Y, Pastorino S, Passaniti A, Menon J, Walling J, Bailey
R, et al: Neuronal and glioma-derived stem cell factor induces
angiogenesis within the brain. Cancer Cell. 9:287–300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davis S and Meltzer PS: GEOquery: A bridge
between the gene expression omnibus (GEO) and BioConductor.
Bioinformatics. 23:1846–1847. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Deng L, Xiong P, Luo Y, Bu X, Qian S and
Zhong W: Bioinformatics analysis of the molecular mechanism of
diffuse intrinsic pontine glioma. Oncol Lett. 12:2524–2530. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma K, Cheng Z, Sun L and Li H:
Identification of potential therapeutic targets for gliomas by
bioinformatics analysis. Oncol Lett. 14:5203–5210. 2017.PubMed/NCBI
|
|
15
|
Sun C, Yuan Q, Wu D, Meng X and Wang B:
Identification of core genes and outcome in gastric cancer using
bioinformatics analysis. Oncotarget. 8:70271–70280. 2017.PubMed/NCBI
|
|
16
|
Jiao X, Sherman BT, Huang da W, Stephens
R, Baseler MW, Lane HC and Lempicki RA: DAVID-WS: A stateful web
service to facilitate gene/protein list analysis. Bioinformatics.
28:1805–1806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:(Database Issue). D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Behin A, Hoang-Xuan K, Carpentier AF and
Delattre JY: Primary brain tumours in adults. Lancet. 361:323–331.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fueyo J, Gomez-Manzano C, Alemany R, Lee
PS, McDonnell TJ, Mitlianga P, Shi YX, Levin VA, Yung WK and
Kyritsis AP: A mutant oncolytic adenovirus targeting the Rb pathway
produces anti-glioma effect in vivo. Oncogene. 19:2–12. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fueyo J, Gomez-Manzano C, Liu TJ and Yung
WK: Delivery of cell cycle genes to block astrocytoma growth. J
Neurooncol. 51:277–287. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alexiou GA, Vartholomatos G, Goussia A,
Batistatou A, Tsamis K, Voulgaris S and Kyritsis AP: Fast cell
cycle analysis for intraoperative characterization of brain tumor
margins and malignancy. J Clin Neurosci. 22:129–132. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu LJ, Wall BA, Wangari-Talbot J and Chen
S: Metabotropic glutamate receptors in cancer. Neuropharmacology.
115:193–202. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, Jiang M, Yao Y and Cai Z: WWC3
inhibits glioma cell proliferation through suppressing the
Wnt/β-catenin signaling pathway. DNA Cell Biol. 37:31–37. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ulrich TA, de Juan Pardo EM and Kumar S:
The mechanical rigidity of the extracellular matrix regulates the
structure, motility, and proliferation of glioma cells. Cancer Res.
69:4167–4174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bellail AC, Hunter SB, Brat DJ, Tan C and
Van Meir EG: Microregional extracellular matrix heterogeneity in
brain modulates glioma cell invasion. Int J Biochem Cell Biol.
36:1046–1069. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cantrell DA: Phosphoinositide 3-kinase
signalling pathways. J Cell Sci. 114:1439–1445. 2001.PubMed/NCBI
|
|
28
|
Ashcroft M, Ludwig RL, Woods DB, Copeland
TD, Weber HO, Macrae EJ and Vousden KH: Phosphorylation of HDM2 by
Akt. Oncogene. 21:1955–1962. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vassilev LT, Vu BT, Graves B, Carvajal D,
Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et
al: In vivo activation of the p53 pathway by small-molecule
antagonists of MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bond GL, Hu W, Bond EE, Robins H, Lutzker
SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, et al: A
single nucleotide polymorphism in the MDM2 promoter attenuates the
p53 tumor suppressor pathway and accelerates tumor formation in
humans. Cell. 119:591–602. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kang SS, Han KS, Ku BM, Lee YK, Hong J,
Shin HY, Almonte AG, Woo DH, Brat DJ, Hwang EM, et al:
Caffeine-mediated inhibition of calcium release channel inositol
1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion
and extends survival. Cancer Res. 70:1173–1183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pal J, Patil V, Kumar A, Kaur K, Sarkar C
and Somasundaram K: Loss-of-function mutations in Calcitonin
receptor (CALCR) identify highly aggressive glioblastoma with poor
outcome. Clin Cancer Res. 24:1448–1458. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wong ML, Prawira A, Kaye AH and Hovens CM:
Tumour angiogenesis: Its mechanism and therapeutic implications in
malignant gliomas. J Clin Neurosci. 16:1119–1130. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang SB, Gao KD, Jiang T, Cheng SJ and Li
WB: Bevacizumab combined with chemotherapy for glioblastoma: A
meta-analysis of randomized controlled trials. Oncotarget.
8:57337–57344. 2017.PubMed/NCBI
|
|
35
|
Paruthiyil S, Cvoro A, Tagliaferri M,
Cohen I, Shtivelman E and Leitman DC: Estrogen receptor β causes a
G2 cell cycle arrest by inhibiting CDK1 activity through the
regulation of cyclin B1, GADD45A, and BTG2. Breast Cancer Res
Treat. 129:777–784. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fan X and Chen JJ: Role of Cdk1 in DNA
damage-induced G1 checkpoint abrogation by the human papillomavirus
E7 oncogene. Cell Cycle. 13:3249–3259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang J, Zhou F, Li Y, Li Q, Wu Z, Yu L,
Yuan F, Liu J, Tian Y, Cao Y, et al: Cdc20 overexpression is
involved in temozolomide-resistant glioma cells with
epithelial-mesenchymal transition. Cell Cycle. 16:2355–2365. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xia Z, Wei P, Zhang H, Ding Z, Yang L,
Huang Z and Zhang N: AURKA governs self-renewal capacity in
glioma-initiating cells via stabilization/activation of
β-catenin/Wnt signaling. Mol Cancer Res. 11:1101–1111. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang R, Wei J, Li Z, Tian Y and Du C:
Bioinformatical analysis of gene expression signatures of different
glioma subtypes. Oncol Lett. 15:2807–2814. 2018.PubMed/NCBI
|
|
40
|
Hu G, Wei B, Wang L, Wang L, Kong D, Jin Y
and Sun Z: Analysis of gene expression profiles associated with
glioma progression. Mol Med Rep. 12:1884–1890. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li W, Li K, Zhao L and Zou H:
Bioinformatics analysis reveals disturbance mechanism of MAPK
signaling pathway and cell cycle in Glioblastoma multiforme. Gene.
547:346–350. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wei B, Wang L, Du C, Hu G, Wang L, Jin Y
and Kong D: Identification of differentially expressed genes
regulated by transcription factors in glioblastomas by
bioinformatics analysis. Mol Med Report. 11:2548–2554. 2015.
View Article : Google Scholar
|