Introduction
Cells in senescence are metabolically active and
exhibit senescence-specific phenotypes, including a
senescence-associated secretory phenotype (1,2). These
include the morphological changes in cells being flattened and
enlarged, induction of senescence-associated β-galactosidase
(SΑ-β-Gal) activity and secretion of proinflammatory cytokines,
including interleukin (IL)-6 and IL-8. IL-6 and IL-8 have been
demonstrated to exhibit dual functions in cancer development
(3). Although senescent cells stay
alive, they do not proliferate. The induction of senescence in
cancer cells has previously been established as one of the factors
determining the overall outcome of cancer treatment, and may
represent a potential strategy to suppress cancer growth (4,5). When used
at low concentrations, a variety of chemicals, including
DNA-damaging drugs, including doxorubicin (DOX), etoposide,
cisplatin and camptothecin, are known to induce senescence in
cancer cells (6,7). For example, in in vitro settings,
senescence is induced in cancer cells by treating cells with DOX
for 24 h at submicromolar concentrations followed by subsequent
incubation in DOX-free medium (8–10). DOX
inhibits the proliferation of cancer cells by inducing senescence,
although this does not immediately kill cancer cells. However, the
efficiency of the induction cannot reach 100% and a number of
colonies appear in the incubation (9,10). These
colonies are considered to be generated from cells escaping from
senescence. It would be of clinical value to understand which
conditions can produce such senescence-escaping cells (SECs), as
the occurrence of SECs can significantly influence the outcome of
chemotherapy. In the present study, the relevance of cell cycle
phases of cells treated with DOX and the occurrence of SECs was
examined by monitoring colony formation in DOX-induced
senescence.
Cyclin-dependent kinase 4/6 (Cdk4/6) inhibitors,
including PD0332991, LEE011 and LY2835219, have been used in cancer
treatment (11,12). Cdk4/6 has previously been demonstrated
to be required for the activation of Cdk2, which acts as a key
protein kinase for the transition from the G1 to S phase
(13,14). Therefore, blocking Cdk4/6 is expected
to lead to cell cycle arrest at the G1 phase. Indeed,
G1 arrest has been reported to occur in cells treated
with Cdk4/6 inhibitors (15,16). Since the cell cycle resumes following
the removal of the inhibitors, immediate cell death is not observed
(17–19). On the one hand, cell cycle arrest at
the G1 phase is required for the induction of senescence
(20,21). Therefore, blocking the cell cycle by
the inhibitors may promote DOX-induced senescence and reduce the
occurrence of SECs. In the present study, this assumption was
tested using PD0332991, one of the Cdk4/6 inhibitors.
Materials and methods
Cell lines and cultures
The human colon cancer HCT116 cell line was obtained
from the Riken Cell Bank (Tsukuba, Japan), and was cultured in
McCoy's 5A medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
containing 10% fetal bovine serum (FBS; Sigma-Aldrich; Merck KGaA).
Penicillin and streptomycin (1%) antibiotics (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) were added to the culture
medium. Cells were grown at 37°C with 5% CO2 in a
humidified incubator.
Reagents
Doxorubicin (DOX; 6 mM stock in water;
Sigma-Aldrich; Merck KGaA), nocodazole (5 mg/ml stock in DMSO;
catalog no. 487928; EMD Millipore, Billerica, MA, USA), PD0332991
(5 mM stock in DMSO; catalog no. S1116; Selleckchem, Houston, TX,
USA) and Giemsa solution (catalog no. GS500; Sigma-Aldrich; Merck
KGaA) were used. DOX and PD0332991 were used at various
concentrations (200 and 400 nM for DOX; 50, 100, 200, 400, and 800
nM for PD0332991), which are expressed as D and PD plus numbers,
respectively. For instance, D200 and PD200 represent 200 nM of DOX
and PD0332991, respectively, and D_00 and PD_00 represent the
vehicle of each reagent.
Induction of senescence
A total of 1.5×106 HCT116 cells were
pre-cultured in 6-well plates for 24 h. The cells were subsequently
washed twice with PBS and then incubated in serum-free medium
(McCoy's 5A without FBS) for 24 h. The serum-free medium was
replaced with the standard medium (McCoy's 5A with FBS) containing
DOX and the cells were incubated for an additional 24 h in the
aforementioned culture conditions. Cells were then washed twice
with PBS and were incubated in 2.5 ml DOX-free standard medium at
37°C. Every day 1 ml of the culture medium was replaced with 1 ml
of fresh standard medium. This procedure is designated as the
standard (STD) procedure. For the pre-release (Pre-REL) procedure,
an additional 3–4 h DOX treatment was performed prior to release
from serum starvation. For the post-release (Post-REL) procedure,
the 24 h DOX treatment was performed at 5–6 h post-release from
serum starvation. The procedures of STD, Pre-REL and Post-REL are
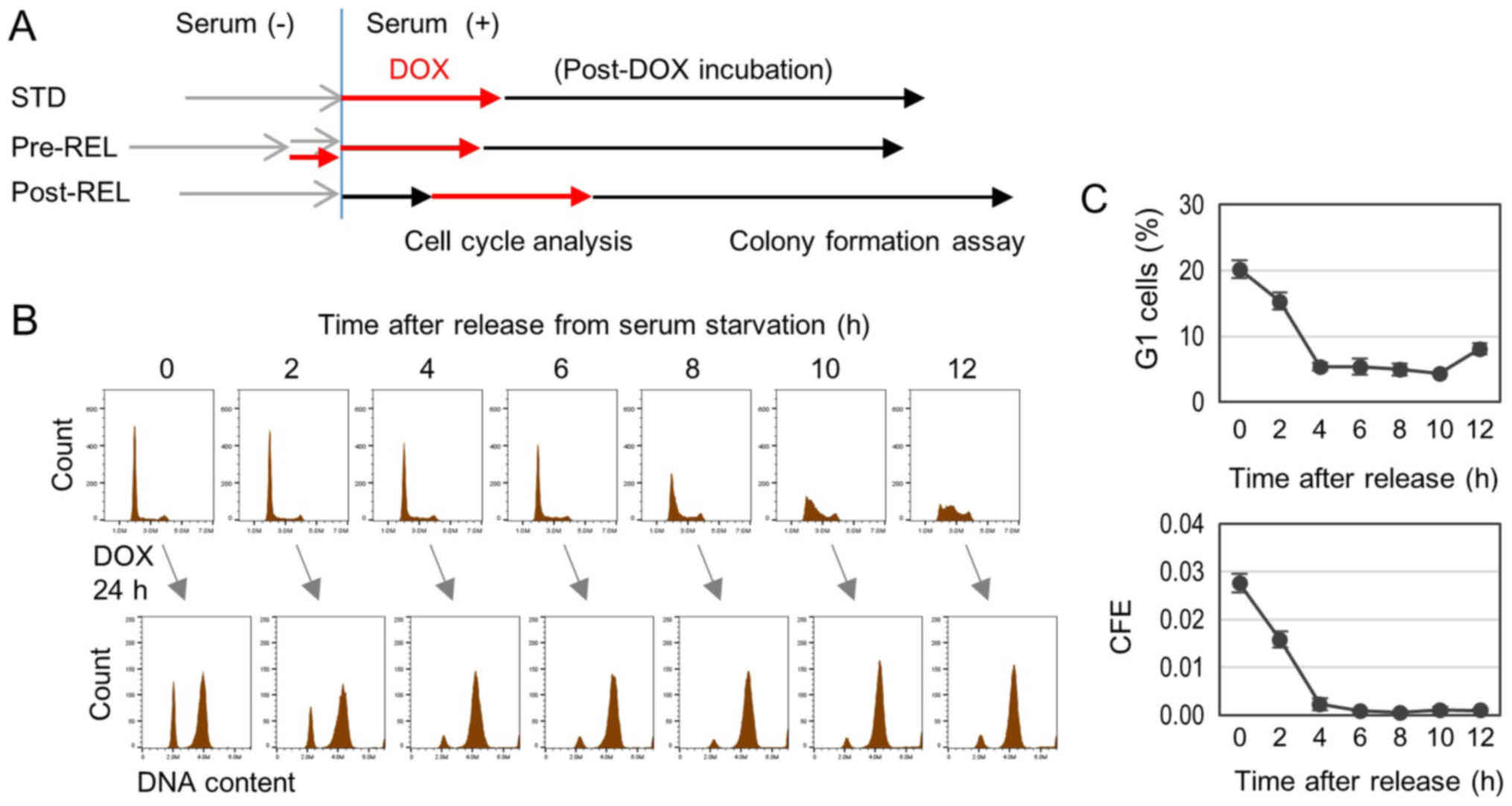
illustrated in Fig. 1A.
Colony formation assay
Treated and control cells were harvested and plated
on 6-well plates (3,000, 6,000, 12,000 and 24,000 cells per well
for treated cells, and 100, 200 and 400 cells per well for control
cells). Following a 14-day incubation, colonies were fixed with
100% methanol for 10 min at room temperature (RT), stained with
Giemsa solution for 1 h at RT and then counted by eye. Colony
formation efficiency (CFE) was expressed as the rate of the number
of colonies formed against the number of cells inoculated.
The effect of PD0332991 on CFE was examined in
DOX-induced senescence in synchronized cells and unsynchronized
growing cells. PD0332991 was co-treated with DOX for 24 h at 37°C.
Induction of senescence in synchronized cells was performed
according to the STD procedure.
SA-β-Gal staining
Treated and untreated cells growing in 6-well plates
were fixed in 3% paraformaldehyde for 5 min at RT and then stained
with 5-bromo-4-chloro-3-indolyl-β-D-galactoside (X-Gal; 1 mg/ml; 50
mg/ml stock in DMSO; Takara Biotechnology, Inc., Otsu, Japan) at pH
6.0 according to a previously described protocol (22). SΑ-β-Gal-positive cells are expressed
as percentage of the total number of cells.
Flow cytometry
Treated and untreated cells were harvested and
washed with cold PBS, and then fixed overnight in cold 70% ethanol.
Fixed cells were twice washed with PBS and stained with propidium
iodide (PI; 0.5 µg/ml; catalog no. P4170; Sigma-Aldrich; Merck
KGaA) for 10 min at RT. The analysis was performed using Accuri C6
flow cytometry (BD Biosciences, San Jose, CA, USA). Cell debris and
doublets were eliminated by gating of forward scatter/side scatter
and fluorescence (FL)-area/FL-width of PI staining. In total,
>8,000 cells were collected for each sample.
For cell cycle analysis of cells treated with
nocodazole, a final concentration of 100 ng/ml nocodazole was added
into the culture medium 24 h prior to the harvest for the
analysis.
Statistical analysis
Cell cycle histograms are representative of
experiments conducted twice in triplicates. Percentages of
G1 cells are presented as the mean ± standard deviation
(SD) (n=3). Colony formation analysis was performed at least twice
in triplicates. CFE data are presented the mean ± SD (n=3). One-way
ANOVA, followed by Tukey post hoc test, and Pearson's correlation
tests were performed in Microsoft Excel 2013 (Microsoft
Corporation, Redmond, WA, USA). The P-value of Pearson's
correlation was calculated using the TDIST function in Microsoft
Excel. P<0.05 was used to indicate a significant difference.
Results
Colony formation in senescence induced
by DOX treatment of cells in different cell cycle phases
It has been reported that cellular senescence can be
induced at high efficiency in HCT116 cells by DOX treatment
(9,20). Using the previously reported procedure
(9), senescence was also induced in
>90% of HCT116 cells in the present study (data not shown). In
addition, HCT116 cells have been used for cell cycle analysis due
to their highly synchronized progression of the cell cycle
(23,24). For these reasons, HCT116 cells were
used in the present study.
The cell cycle progression was synchronized by the
serum-block/release procedure. Cells in different cell cycle phases
were exposed to DOX for 24 h. Following this exposure, the cell
cycle was analyzed by flow cytometry and cells were plated in
culture dishes containing a DOX-free medium. Following a 14-day
incubation, CFE was measured (STD in Fig.
1A). DOX induced cell cycle arrest in the G1 and
G2/M phases (Fig. 1B).
Marked amounts of G1-arrested cells appeared with the
DOX treatment initiated at 0 and 2 h post-release from serum
starvation (Fig. 1B). Since cells
were in the G1 phase at 0–4 h post-release, cells
arrested at the G1 phase were considered to be cells
immediately arrested by the DOX treatment. This was further
confirmed by co-treatment with nocodazole, an M phase-arresting
reagent, which can block the entry of M phase cells into the
G1 phase in the next round of cell cycle (25,26). The
G1-arrested cells were not affected by the addition of
nocodazole (data not shown). The rate of G1 arrested
cells was found to be significantly correlated with CFE (r=0.975,
P=0.000028) (Fig. 1C). The
significant correlation observed strongly suggests that immediate
G1 arrest of cells in the G1 phase by DOX
treatment enhances the colony formation ability. It should be noted
that CFE was markedly low when DOX treatment was initiated at 4 h
and later after the release. This represents the Post-REL procedure
illustrated in Fig. 1A.
Increase in G1-arrested
cells by treatment of G1 cells enhances CFE in
DOX-induced senescence
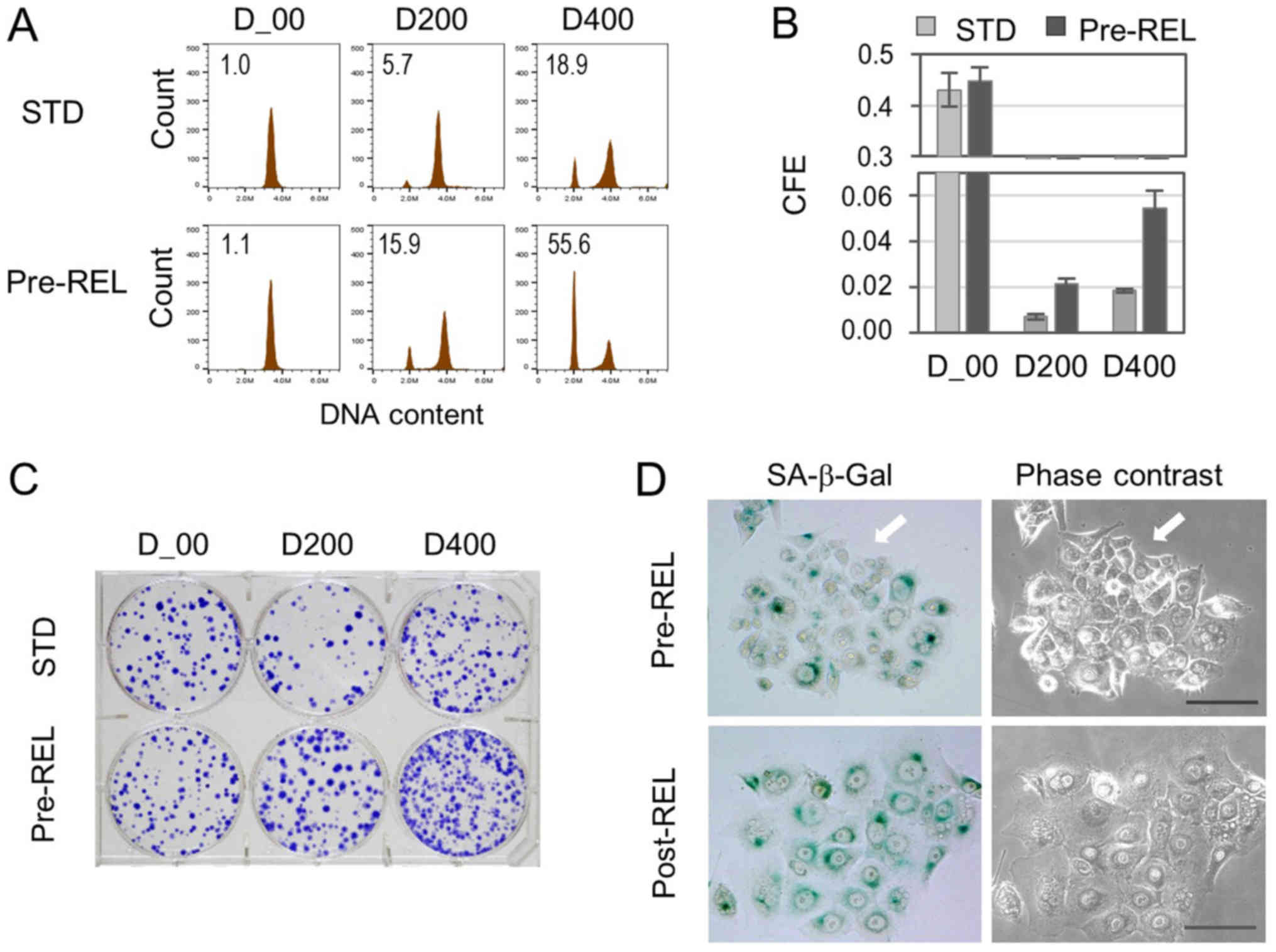
To further examine the association between
G1-treated G1-arrested cells and colony
formation, an attempt was made to increase the number of
G1-treated G1-arrested cells. Serum-starved
cells were pretreated with DOX for 3–4 h prior to release, and the
cell cycle and CFE were examined (Pre-REL in Fig. 1A). Cell cycle analysis demonstrated an
increase of G1-arrested cells, which was not affected by
the addition of nocodazole (Fig. 2A).
In accord with the increase, CFE was significantly enhanced
(Fig. 2B and C). In addition, cell
clusters consisting of 8–15 cells, which exhibited no SA-β-Gal
activity, were detected at 2–3 days of incubation in the Pre-REL
procedure (Fig. 2D). By contrast, in
the Post-REL procedure, such cell clusters were not detected in the
short-term incubation groups (Fig.
2D).
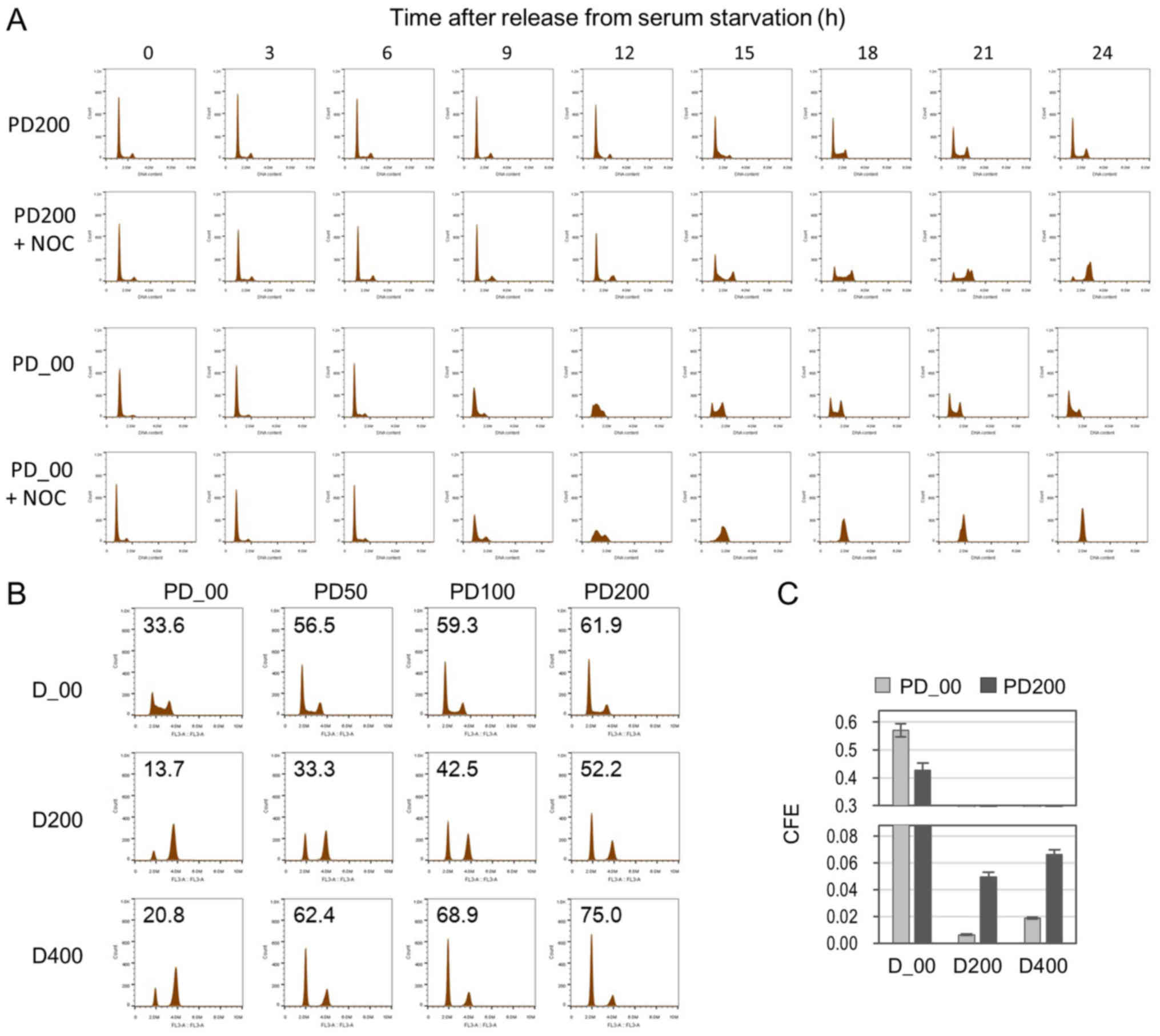
 | Figure 2.G1 arrest by DOX-treatment
of G1 cells enhances CFE in senescence. Cells were
treated with 200 nM (D200) and 400 nM (D400) of DOX and DMSO
vehicle (D_00). (A) Cell cycle distribution at 24 h after treatment
with DOX analyzed by flow cytometry. The numbers in histograms
indicate percentages of G1 cells. For the analysis of
cell cycle distribution, nocodazole (100 ng/m) was added to
completely block the cell cycle progression from M phase to
G1 phase in the next cycle. (B) Cells treated with DOX
were incubated in DOX-free medium, and at 14 days of incubation,
CFE was determined (expressed as the rate of the number of colonies
formed against the number of cells inoculated). (C) Representative
images of colonies stained with Giemsa. (D) Representative images
of senescent cells of day 3+ stained with SA-β-Gal. White arrows
indicate colonies consisting of 12 cells. Scale bar, 100 µm. CFE,
colony formation efficiency; DOX, doxorubicin; SA-β-Gal,
senescence-associated β-galactosidase; Pre-REL, pre-release;
Post-REL, post release; STD, standard. |
Co-treatment of PD0332991 with DOX in
synchronized cells enhances colony formation in the induced
senescence
PD0332991, a CDK4/6 inhibitor, has been demonstrated
to inhibit cell cycle progression and arrest cells in G1
phase (15,16). The present study examined the effects
of PD0332991 on CFE in DOX-induced senescence. First, cells were
arrested in the G1 phase by serum starvation and then
released from the G1 block by medium replacement.
PD0332991 was added at the release from serum starvation and the
cell cycle progression was monitored by flow cytometry (Fig. 3A). Compared with the control,
PD0332991 delayed the progression of cells from the G1
to S phase. This was confirmed in the cell cycle histograms of
cells when co-treated with a mitotic inhibitor nocodazole (+ NOC in
Fig. 3A). The G1 peak
gradually decreased in the incubation and contrastingly, the
G2/M peak increased. Next, the effects of this slow cell
cycle progression in the G1 phase on DOX-induced
senescence were examined. Senescence was induced in cells by DOX
treatment in the presence of PD0332991. As hypothesized, cell cycle
arrest in the G1 phase was enhanced when co-treated with
PD0332991 (Fig. 3B). As a result of
this treatment, CFE was augmented (Fig.
3C). Collectively, slow progression of the cell cycle in the
G1 phase led to an increase in G1-treated
G1-arrested cells and CFE.
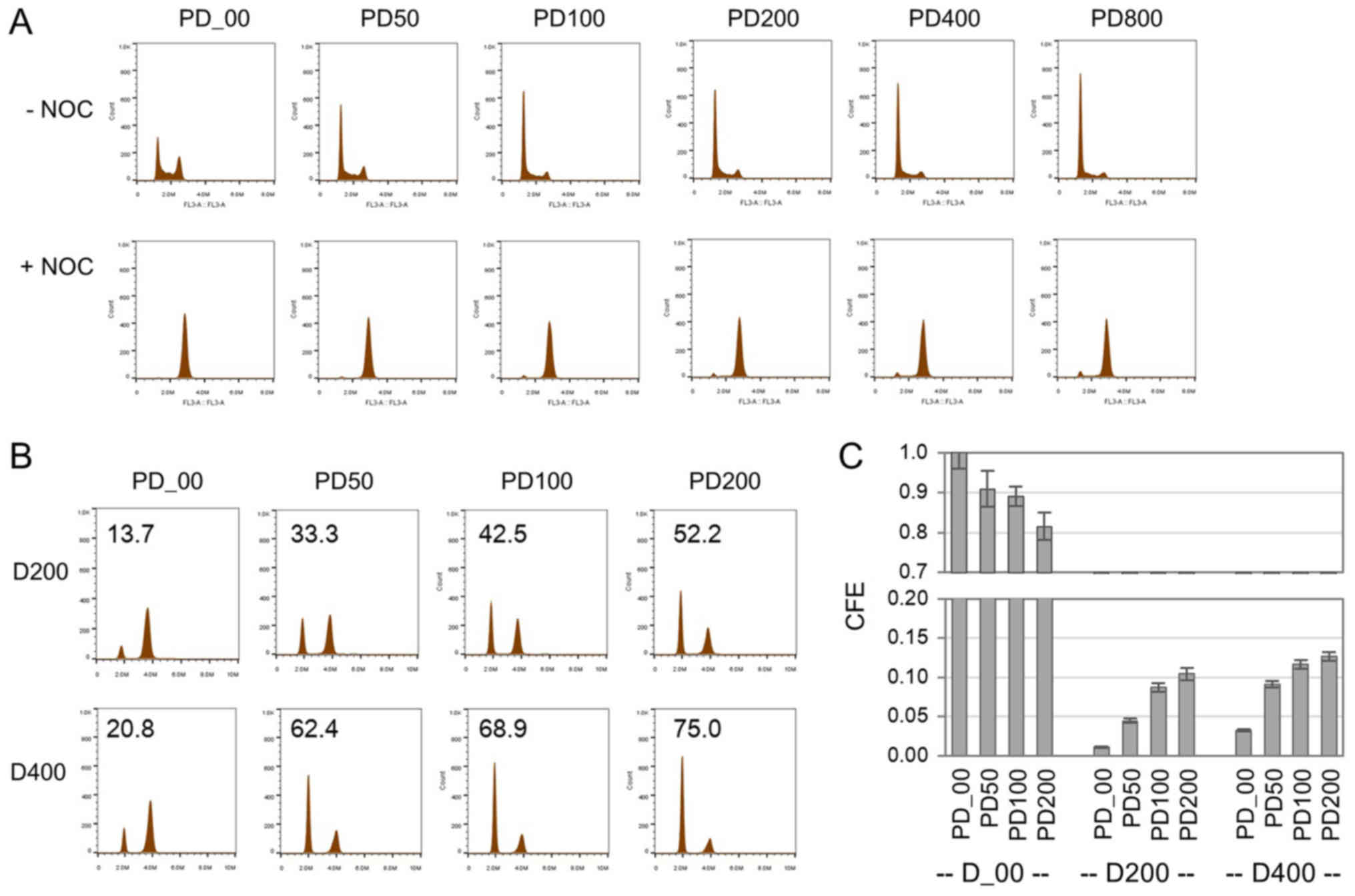
Co-treatment of PD0332991 with DOX in
growing cells enhances colony formation in induced senescence
The effects of PD0332991 on CFE were further
examined using growing cells. Cells were treated with PD0332991 at
concentrations from 50 to 800 nM for 24 h and cell cycle
distributions with or without nocodazole were examined (Fig. 4A). Cells accumulated in the
G1 phase by PD0332991 treatment. Even at a 50 nM
concentration of PD0332991, the accumulation of cells in the
G1 phase was clearly detected. However, these
G1 accumulations were abolished in the presence of
nocodazole, indicating that the G1 accumulations did not
result from complete cell cycle arrest, but rather a slow cell
cycle progression in the G1 phase. Next, senescence was
induced in growing cells by a 24-h DOX treatment in the presence of
PD0332991. The majority of cells (~90%) exhibited SA-β-Gal,
comparable to that of sole treatment with DOX. However, an
increased number of colonies appeared in induced senescence by
co-treatment with DOX and PD0332991, and the increase in CFE was
highly correlated with the increase in G1-arrested cells
by the treatment (r=0.746, n=6, P=0.088457 from the overall
analysis of D200 and D400 data) (Fig. 4B
and C). Even in growing cells, PD0332991 enhanced the colony
formation ability in DOX-induced senescence.
Discussion
The present study describes the relevance of
G1-treated G1-arrested cells to colony
formation in DOX-induced cellular senescence by
increasing/decreasing G1-treated G1-arrested
cells using three different procedures, Pre-REL, Post-REL and STD.
The Pre-REL procedure increased G1-treated
G1-arrested cells and enhanced CFE. Conversely, the
Post-REL procedure decreased G1-treated
G1-arrested cells and reduced CFE. Furthermore, the
ratio of G1-treated G1-arrested cells was
positively associated with the number of colony-forming cells.
Therefore, it is likely that the colony formation ability is
conferred by the G1 arrest of cells treated by DOX in
the G1 phase.
Cell clusters consisting of <15 cells were
detected as early as 2–3 days after DOX treatment in the Pre-REL
procedure. These cells were SA-β-Gal-negative. The cell clusters
were detected as colonies in the subsequent incubation. This
suggested that these colonies were formed from cells that had
escaped from entering senescence during the treatment. It is likely
that these colonies were the result of treatment conditions,
leading to an increase in G1-treated
G1-arrested cells.
DOX induces DNA damage and, in turn, the DNA damage
has been demonstrated to activate the G1 and
G2/M checkpoints, which induces cell cycle arrest
(27,28). During cell cycle arrest, DNA damage is
repaired and the cell cycle resumes following the completion of the
repair. However, cells undergo senescence or apoptosis when the
damage is extensive and not repairable. The present study observed
cell cycle arrest in the G1 and G2/M phases
following treatment with DOX. These arrests are likely induced by
the G1 and G2/M checkpoints, respectively.
Treatment of G1 phase cells, synchronized by serum
starvation, with DOX also induced cell cycle arrest in the
G1 and G2/M phases. The immediate
G1 arrest by DOX treatment of G1 phase cells
can act to protect cells from further damage received in the
subsequent S and G2 phases. G1 arrest has
been reported to protect cells from drugs that selectively kill
dividing cells (29,30) Therefore, the immediate
G1-treated G1 arrest would increase arresting
cells with less and repairable damage. Following the removal of
DOX, such cells can restart the cell cycle and form colonies. The
colonies detected in the present study following treatment with DOX
are hypothesized to be colonies that have resulted from the
transiently arrested cells, which have restarted their cell cycle.
Therefore, an increase of cells in the G1 phase at
treatment with DOX may lead to an increase in SECs.
The treatment of HCT116 cells with PD0332991, a cell
cycle inhibitor, resulted in the accumulation of cells in the
G1 phase. However, the accumulation of G1
cells did not result from complete arrest of the cell cycle in the
G1 phase, but was in fact from the slow cell cycle
progression of cells in the G1 phase. This may explain
why the present study failed to induce senescence in cells treated
with PD0332991, as the induction of senescence is known to require
complete cell cycle arrest (20,21). No
enhancement of DOX-induced senescence was observed by co-treatment
of PD0332991. On the contrary, the co-treatment of PD0332991 with
DOX augmented the number of G1-treated
G1-arrested cells, resulting in an increase in the
number of colonies appearing in DOX-induced senescence. For the
efficient induction of senescence, by reducing the number of SECs,
it is necessary to avoid drug treatment of G1 phase
cells. On this basis, caution would be advised when a drug like
PD0332991, a cell cycle inhibitor, which potentially increases the
cell population in the G1 phase, is considered for
treatment purposes.
Acknowledgements
The authors would like to thank Dr Junji Itou and Dr
Fumiaki Sato (Department of Breast Surgery, Kyoto University,
Kyoto, Japan) and Dr Keiko Iwaisako (Department of Target Oncology,
Kyoto University) for supplying the materials used in the study.
The authors would also like to thank the members of the Breast
Surgery Laboratory for useful discussion and suggestions, and Dr
Ravi Velaga (Department of Breast Surgery, Kyoto University) for
critically revising the manuscript.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
KK designed the study, performed experiments and
wrote the manuscript. FP conducted experiments. MT designed the
study and wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Campisi J: Senescent cells, tumor
suppression, and organismal aging: Good citizens, bad neighbors.
Cell. 120:513–522. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Collado M and Serrano M: The power and the
promise of oncogene-induced senescence markers. Nat Rev Cancer.
6:472–476. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Di Mitri D and Alimonti A:
Non-cell-autonomous regulation of cellular senescence in cancer.
Trends Cell Biol. 26:215–226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X, Tsao SW, Wong YC and Cheung AL:
Induction of senescent-like growth arrest as a new target in
anticancer treatment. Curr Cancer Drug Targets. 3:153–159. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tchkonia T, Zhu Y, van Deursen J, Campisi
J and Kirkland JL: Cellular senescence and the senescent secretory
phenotype: Therapeutic opportunities. J Clin Invest. 123:966–972.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ewald JA, Desotelle JA, Wilding G and
Jarrard DF: Therapy-induced senescence in cancer. J Natl Cancer
Inst. 102:1536–1546. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Petrova NV, Velichko AK, Razin SV and
Kantidze OL: Small molecule compounds that induce cellular
senescence. Aging Cell. 15:999–1017. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elmore LW, Rehder CW, Di X, McChesney PA,
Jackson-Cook CK, Gewirtz DA and Holt SE: Adriamycin-induced
senescence in breast tumor cells involves functional p53 and
telomere dysfunction. J Biol Chem. 277:35509–35515. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sliwinska MA, Mosieniak G, Wolanin K,
Babik A, Piwocka K, Magalska A, Szczepanowska J, Fronk J and Sikora
E: Induction of senescence with doxorubicin leads to increased
genomic instability of HCT116 cells. Mech Ageing Dev. 130:24–32.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Su D, Zhu S, Han X, Feng Y, Huang H, Ren
G, Pan L, Zhang Y, Lu J and Huang B: BMP4-Smad signaling pathway
mediates adriamycin-induced premature senescence in lung cancer
cells. J Biol Chem. 284:12153–12164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hamilton E and Infante JR: Targeting
CDK4/6 in patients with cancer. Cancer Treat Rev. 45:129–138. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu H, Yu S, Liu Q, Yuan X, Mani S, Pestell
RG and Wu K: Recent advances of highly selective CDK4/6 inhibitors
in breast cancer. J Hematol Oncol. 10:972017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Malumbres M, Ortega S and Barbacid M:
Genetic analysis of mammalian cyclin-dependent kinases and their
inhibitors. Biol Chem. 381:827–838. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fry DW, Harvey PJ, Keller PR, Elliott WL,
Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, et
al: Specific inhibition of cyclin-dependent kinase 4/6 by PD
0332991 and associated antitumor activity in human tumor
xenografts. Mol Cancer Ther. 3:1427–1438. 2004.PubMed/NCBI
|
|
16
|
Toogood PL, Harvey PJ, Repine JT, Sheehan
DJ, VanderWel SN, Zhou H, Keller PR, McNamara DJ, Sherry D, Zhu T,
et al: Discovery of a potent and selective inhibitor of
cyclin-dependent kinase 4/6. J Med Chem. 48:2388–2406. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guha M: Cyclin-dependent kinase inhibitors
move into Phase III. Nat Rev Drug Discov. 11:892–894. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kovatcheva M, Liu DD, Dickson MA, Klein
ME, O'Connor R, Wilder FO, Socci ND, Tap WD, Schwartz GK, Singer S,
et al: MDM2 turnover and expression of ATRX determine the choice
between quiescence and senescence in response to CDK4 inhibition.
Oncotarget. 6:8226–8243. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ziemke EK, Dosch JS, Maust JD, Shettigar
A, Sen A, Welling TH, Hardiman KM and Sebolt-Leopold JS:
Sensitivity of KRAS-mutant colorectal cancers to combination
therapy that cotargets MEK and CDK4/6. Clin Cancer Res. 22:405–414.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chang BD, Xuan Y, Broude EV, Zhu H, Schott
B, Fang J and Roninson IB: Role of p53 and p21waf1/cip1 in
senescence-like terminal proliferation arrest induced in human
tumor cells by chemotherapeutic drugs. Oncogene. 18:4808–4818.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ben-Porath I and Weinberg RA: The signals
and pathways activating cellular senescence. Int J Biochem Cell
Biol. 37:961–976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dimri GP, Lee X, Basile G, Acosta M, Scott
G, Roskelley C, Medrano EE, Linskens M, Rubelj I and Pereira-Smith
O: A biomarker that identifies senescent human cells in culture and
in aging skin in vivo. Proc Natl Acad Sci USA. 92:9363–9367. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Trzepacz C, Lowy AM, Kordich JJ and Groden
J: Phosphorylation of the tumor suppressor adenomatous polyposis
coli (APC) by the cyclin-dependent kinase p34. J Biol Chem.
272:21681–21684. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mizuno H, Nakanishi Y, Ishii N, Sarai A
and Kitada K: A signature-based method for indexing cell cycle
phase distribution from microarray profiles. BMC Genomics.
10:1372009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zieve GW, Turnbull D, Mullins JM and
McIntosh JR: Production of large numbers of mitotic mammalian cells
by use of the reversible microtubule inhibitor nocodazole.
Nocodazole accumulated mitotic cells. Exp Cell Res. 126:397–405.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Long BH and Fairchild CR: Paclitaxel
inhibits progression of mitotic cells to G1 phase by interference
with spindle formation without affecting other microtubule
functions during anaphase and telephase. Cancer Res. 54:4355–4361.
1994.PubMed/NCBI
|
|
27
|
Erol A: Deciphering the intricate
regulatory mechanisms for the cellular choice between cell repair,
apoptosis or senescence in response to damaging signals. Cell
Signal. 23:1076–1081. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roos WP, Thomas AD and Kaina B: DNA damage
and the balance between survival and death in cancer biology. Nat
Rev Cancer. 16:20–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen X, Lowe M, Herliczek T, Hall MJ,
Danes C, Lawrence DA and Keyomarsi K: Protection of normal
proliferating cells against chemotherapy by staurosporine-mediated,
selective, and reversible G(1) arrest. J Natl Cancer Inst.
92:1999–2008. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Blagosklonny MV and Pardee AB: Exploiting
cancer cell cycling for selective protection of normal cells.
Cancer Res. 61:4301–4305. 2001.PubMed/NCBI
|