Introduction
Metformin (Met) has long been used to treat type 2
diabetes; its principle activities have been characterized as the
inhibition of hepatic glucose output and increased glucose uptake
by muscle (1). However, Met has
recently attracted attention as a potential treatment for cancer
due to its strong anticancer effects (2). Evans et al found that the use of
Met by type 2 diabetic patients was associated with a lower cancer
incidence (3). In addition,
cancer-related mortality found to be lower in patients treated with
Met than in those treated with other anti-diabetic drugs (3,4). Following
these impressive epidemiological studies, preclinical studies
revealed Met inhibited cancer cell growth (2,5,6), and induced apoptotic cell death in
diverse solid cancers, including colon (7), ovarian (8), and breast cancers (5,9). It is
widely accepted that inhibition of PI3K/AKT/mTOR pathway via
AMP-activated protein kinase (AMPK) activation underlies the
anticancer activities of Met (10).
More recently, several studies have reported that Met selectively
targets cancer stem cells (CSCs), which are responsible for tumor
growth and failures to respond to chemotherapy and radiotherapy
(11).
Regarding breast cancer, Hirsch et al showed
that Met preferentially reduces the CSC fraction (12), as defined by
CD44high/CD24low expression (12). Since CSCs have known to grow as
tumorspheres (TS) when incubated under non-adherent conditions
(13–15), they also showed that Met suppresses
the number and sizes of TS derived from several breast cancer cell
lines (12). Similarly, other studies
have also reported that Met significantly reduced TS formation, and
the expressions of CSC markers, such as, those of
CD44high/CD24low, ALDH-1, or OCT4 in breast
cancer cells (16–18). Several mechanisms have been proposed
for the targeting of CSCs by Met, including the regulation of
epithelial to mesenchymal transition (EMT) (19), the suppressions of transcription
factors like TGFβ (20), and
inactivation of the PI3K/AKT/mTOR pathway (18,21).
However, the mechanism responsible for the selective targeting of
CSCs by Met remains to be elucidated.
Previously, we showed that TS cultures increased the
quiescent nature of cells, which is a known characteristic of CSCs
(22–24). After optimizing TS cultures to screen
cytotoxic agents in vitro (23), we found that TS derived from breast
cancer cells are resistant to drugs commonly used for chemotherapy
(22–24). Thus, in this study, we utilized TS
cultures to investigate whether Met has the potential to overcome
the drug-resistance of TS generated from 4T1 murine breast cancer
cells and to elucidate the underlying mechanism.
Materials and methods
Monolayer culture
4T1 murine breast cancer cells were obtained from
the American Type Culture Collection (Manassas, VA, USA) and
maintained in DMEM (Welgene, Daegu, Korea) supplemented with 5%
fetal bovine serum (Hyclone Laboratories Inc, South Logan, UT, USA)
and 1% antibiotics antimycotic solution (Welgene).
TS culture
The protocol used for TS culture was as previously
described (22,24). In brief, 4T1 cells were suspended in
serum-free DMEM/F12 (Welgene) supplemented with 1X B27 (Gibco BRL,
Grand Island, NY, USA), 20 ng/ml recombinant human EGF (R&D
Systems, Minneapolis, MN, USA), 10 ng/ml recombinant human FGF
(R&D Systems), 10 µg/ml insulin (Welgene), 10 mM HEPES
(Welgene) and 1% antibiotics antimycotic solution (Welgene) and
cultured in non-adherent plates.
Cell kinetic assay
4T1 cells were seeded into 96-well plates at a
density of 1,000 to 16,000 cells per well and incubated for 4 days
under monolayer or TS culture conditions. To quantify the number of
cells premixed cell proliferation reagent WST-8 (Dojindo
Laboratories, Kumamoto, Japan) was added to each well and the
absorbance of the water-soluble formazan produced by viable cells
was measured at 450 nm according to the manufacturer's
protocol.
In vitro response
To compare the chemosensitivities of monolayer and
TS cultured cells, cells cultured in adherent or non-adherent
96-well plates were treated with various concentrations of
doxorubicin (Dox) (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany),
lapatinib (Lapa) (LC Laboratories, Woburn, MA, USA), or Met
(Sigma-Aldrich; Merck KGaA) for 3 days. Cell viabilities were
determined using reagent WST-8.
Cell cycle analysis
4T1 cells were cultured under monolayer and TS
culture conditions for 4 days, and trypsinized after washing with
PBS. Cells were then centrifuged at 1,000 rpm for 3 min and fixed
with cold 70% ethanol. After centrifugation once again, cells were
washed with PBS containing 2% FBS prior to staining with 20 µg/ml
propidium iodide (PI; Sigma-Aldrich; Merck KGaA) and 200 µg/ml
RNase A (Sigma-Aldrich; Merck KGaA) for 30 min in the dark at room
temperature. Cell cycle was assessed by analyzing the DNA contents
stained with PI using a FACS Calibur II flow cytometer (BD
Biosciences, San Jose, CA, USA).
RNA isolation and reverse
transcription-polymerase chain reaction (PCR)
Total RNA was extracted using the easy-BLUE™ Total
RNA Extraction kit (iNtRON Biotechnology Inc., Sungnam, Korea).
cDNA was synthesized using GoScript™ reverse transcriptase (Promega
Corporation, Madison, WI, USA) and PCR was performed using Taq DNA
polymerase (Thermo Fisher Scientific, Rockford, IL, USA). The used
primer sequences for PCR were as follows: Cyclin D1 (forward)
5′-CTGTGCGCCCTCCGTATCTTA-3′ and cyclin D1 (reverse)
5′-GGCGGCCAGGTTCCACTTGAG-3′; GAPDH (forward)
5′-ACCACAGTCCATGCCATCAC-3′ and GAPDH (reverse)
5′-TCCACCACCCTGTTGCTGTA-3′.
Western blotting
Cells grown as monolayers or TS were harvested and
lysed with RIPA buffer (150 mM NaCl, 1% Triton X-100, 1% sodium
deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 7.5 and 2 mM EDTA)
supplemented with phosphatase and protease inhibitor cocktails
(GenDepot, Barker, TX, USA). Lysates were centrifuged at 13,000 rpm
for 10 min to remove cell debris, and protein concentrations were
determined using bicinchoninic acid reagent (Sigma). Same amounts
of protein were separated by SDS-PAGE and then transferred to
polyvinylidene fluoride (PVDF) membranes, which were then blocked
with 5% non-fat skim milk in 1X TBS-0.1% Tween-20 (TTBS) for 2 h
and incubated with primary antibodies [protein kinase B (AKT),
p-AKT, activator of transcription 3 (STAT3), p-STAT3, GAPDH (Cell
Signaling, Beverly, MA, USA] or β-actin (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA). Blots were then incubated for 1 h at room
temperature with HRP-conjugated secondary anti-rabbit antibody
(Thermo Fisher Scientific) or anti-mouse antibody (Santa Cruz
Biotechnology Inc.) at 1:5,000 in TTBS, and developed using a
Luminescent Image Analyzer LAS-4000 (Fujifilm, Tokyo, Japan).
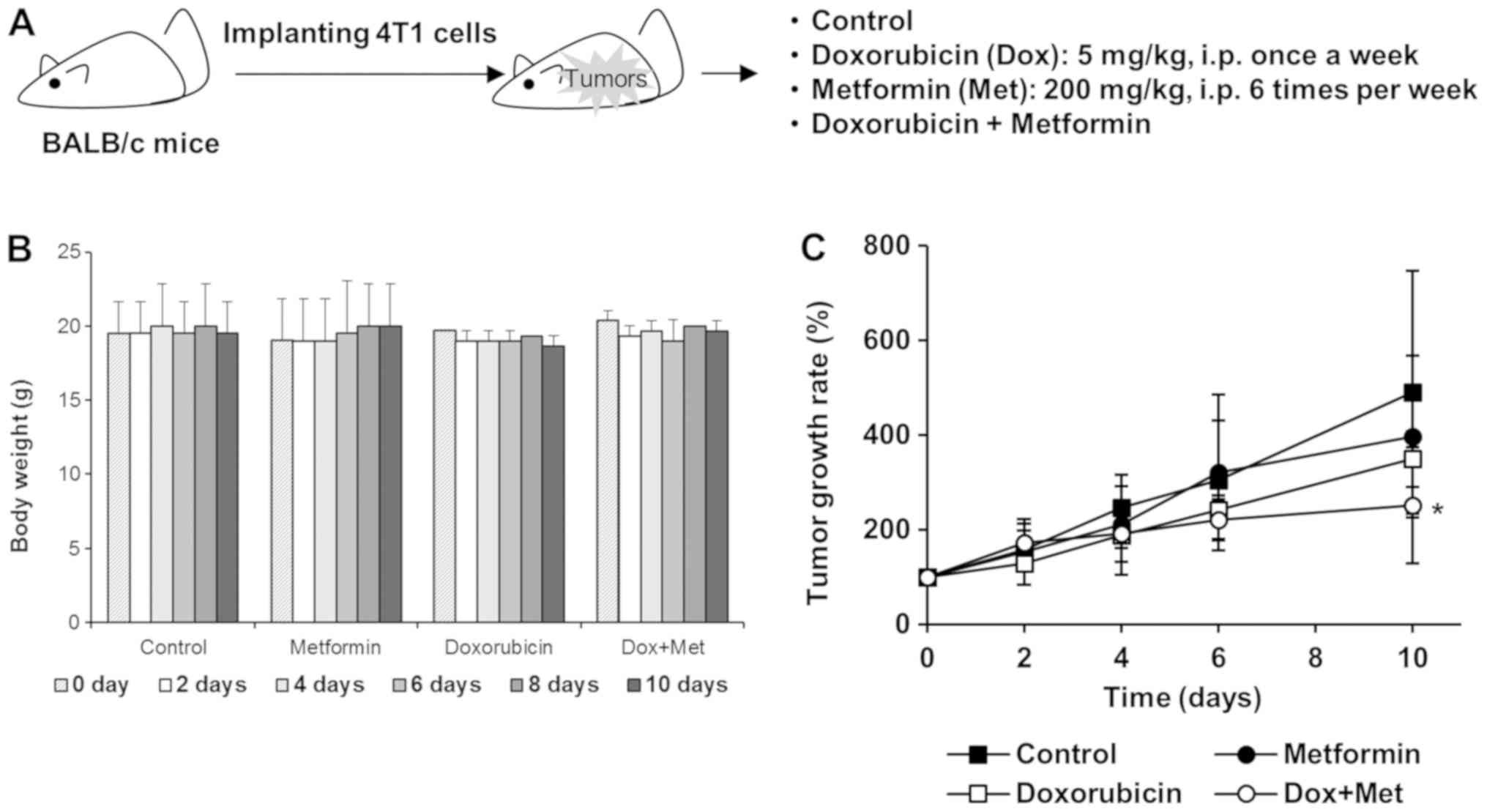
In vivo responses
Four-week-old female BALB/c mice were
purchased from Orient Bio Inc. (Sungnam, Korea) and allowed to
acclimatize under a 12-h light/dark cycle at 25±2°C/RH 50±5% for
two weeks before inoculation. 4T1 cells (1×105) were
implanted into mammary fat pads and when tumor volumes reached ~100
mm3, mice were randomly allocated to one of the
following four groups: a) Control, b) Dox, c) Met, d) Dox plus Met
groups. Mice were intraperitoneally injected with Dox (5 mg/kg,
once a week), Met (200 mg/kg, once a day), or both. Tumor sizes
were measured every on alternate days with a digital caliper and
volumes were calculated using the following formula; tumor volume
(mm3)=(longest length + shortest length2)/2.
To quantify tumor growth rate, we determined relative tumor volume
to the averaged volume of starting tumor (0 day) for each group.
Animal experimentation was performed after obtaining approval from
the Institutional Animal Care and Use Committee at Dongguk
University (IACUC-2016-003).
Statistical analysis
Statistical significance was determined using the
Student's t-test or one-way analysis of variance with Fisher's
least significant difference as the post hoc test. All experiments
were conducted in triplicates, and results are presented as the
mean ± standard deviation.
Results
Quiescence in TS generated from 4T1
cells
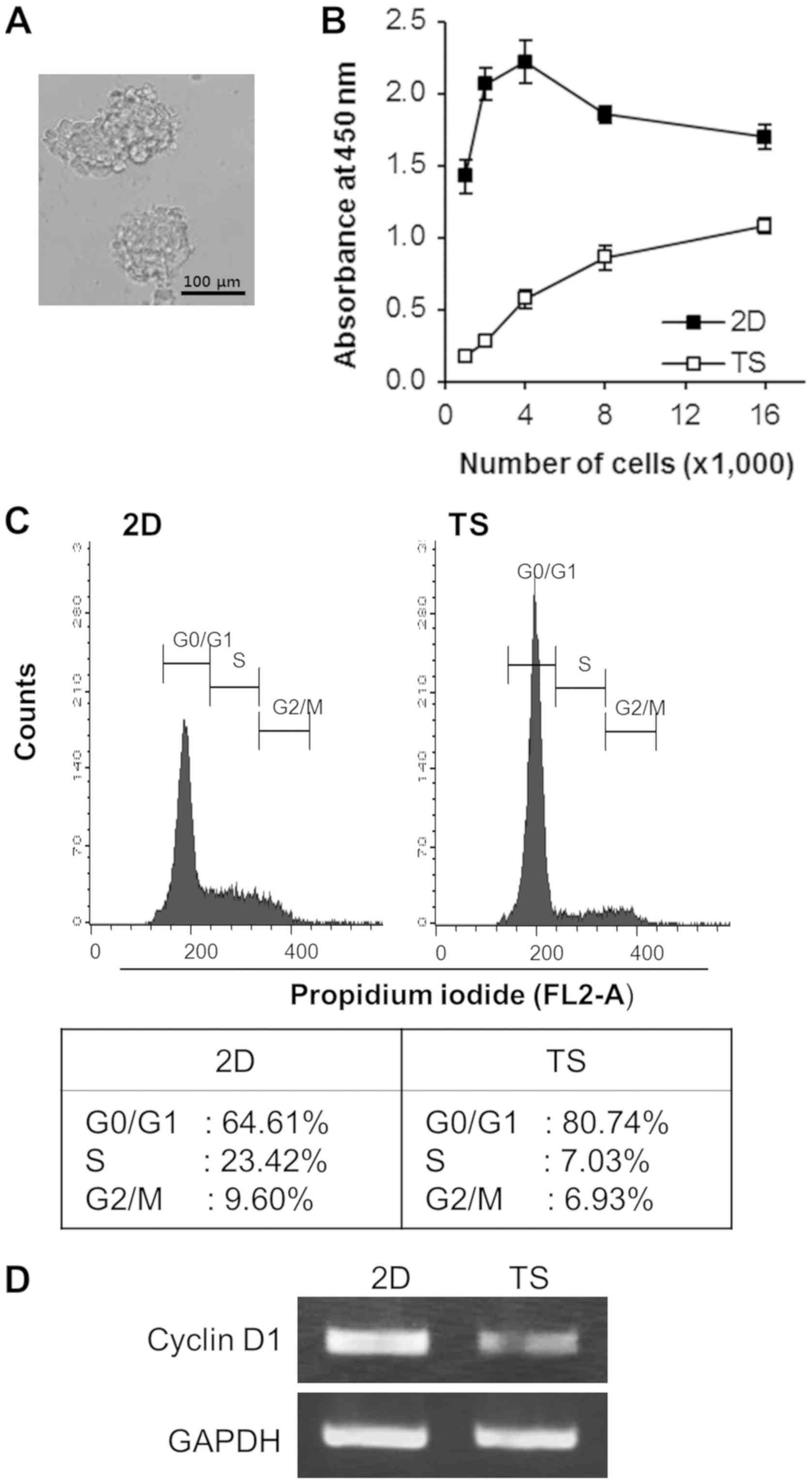
We first tested the ability of 4T1 breast cancer
cells to form TS by culturing them in non-adherent culture
condition for 4 days. The cells successfully formed spheres of
different sizes with a grape-like appearance (Fig. 1A). To compare cell growth rates in TS
and monolayer cultures, various concentrations of cells
(1,000-16,000 cells/well) were plated into 96-well non-adherent or
regular tissue culture plates. After 4 days of culture, the
premixed cell proliferation reagent, WST-8, was added to each plate
and WST-8 absorbance at 450 nm was measured. As shown in Fig. 1B, overall WST-8 absorbance of TS was
almost a forth of that of monolayer cultured cells, indicating the
cell growth rate of TS was slower than that of monolayer cultured
cells. Interestingly, over 80% of the cells in TS accumulated in
the G0/G1 phase, whereas ~65% of monolayer cultured cells did so
(Fig. 1C). Consistent with our cell
cycle analysis results, the mRNA expression of cyclin D1 (an
important regulator of G1 to S phase progression) was also
decreased in TS (Fig. 1D). Taken
together, these results suggest that TS culture conditions increase
the proportion of cells in the quiescent state.
4T1 TS exhibited chemoresistance by
upregulating the STAT3 and AKT signaling pathways
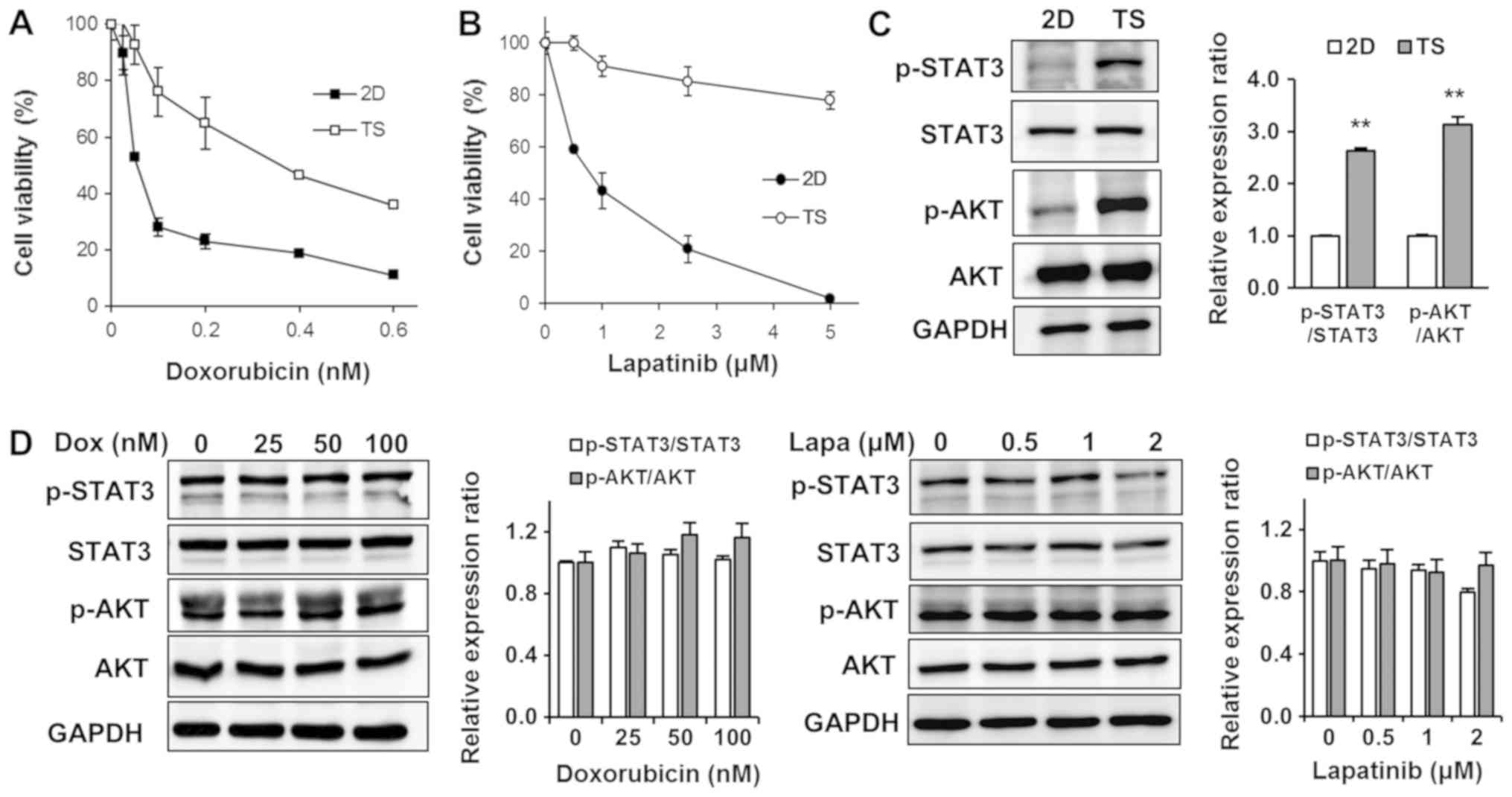
To determine whether TS quiescence was associated
with chemoresistance, we tested cytotoxic effects of Dox on TS. We
treated cells cultured as monolayers or TS with various
concentrations of Dox (0.025–0.6 µM) for 3 days, and determined
cell viabilities using WST-8. As shown in Fig. 2A, 4T1 cells cultured as monolayers
were highly sensitive to Dox (IC50 0.05 µM), whereas
most cells in TS survived at the same concentration (0.05 µM) and
notably the IC50 for Dox was more than ten times higher
(0.6 µM). Since 4T1 breast cancer cells are considered a triple
negative breast cancer model and express epidermal growth factor
receptor (EGFR), we also examined the effects of Lapa (a dual
EGFR/HER2 inhibitor) on TS (Fig. 2B).
Similar to effects of Dox, cells cultured as monolayers were highly
sensitive to Lapa (IC50 1 µM), but over 80% of cells in
TS survived at the highest concentrations tested (5 µM) (Fig. 2B).
To elucidate the molecular mechanism responsible for
the chemoresistance of TS, we first analyzed upregulated signaling
pathways in the TS as compared with monolayer cultured cells. After
checking several main signaling pathways known to regulate cell
survival, proliferation, apoptosis, and chemoresistance, including
the PI3K/AKT, MAPK/extracellular signal-regulated kinase (ERK),
Bcl-2, and STAT3 pathways, we found the signal transducer and STAT3
and AKT signaling pathways were significantly more upregulated in
TS than in monolayer cultured cells (Fig.
2C). However, interestingly, treatment with Dox or Lapa failed
to inhibit enhanced STAT3 and AKT signaling observed in TS
(Fig. 2D), indicating that these
signaling pathways might play important roles in mediating TS
chemoresistance.
Met selectively targets TS rather than
monolayer-cultured cells by inhibiting the STAT3 and AKT signaling
pathways. Several studies have reported that Met dramatically
reduces the sizes and numbers of TS in diverse cancer cell lines,
including breast cancer, thyroid cancer, and hepatocellular
carcinoma cells (16–18). Notably, Liu et al showed Met
induced the death of triple-negative breast cancer cells in
vitro and in vivo (9).
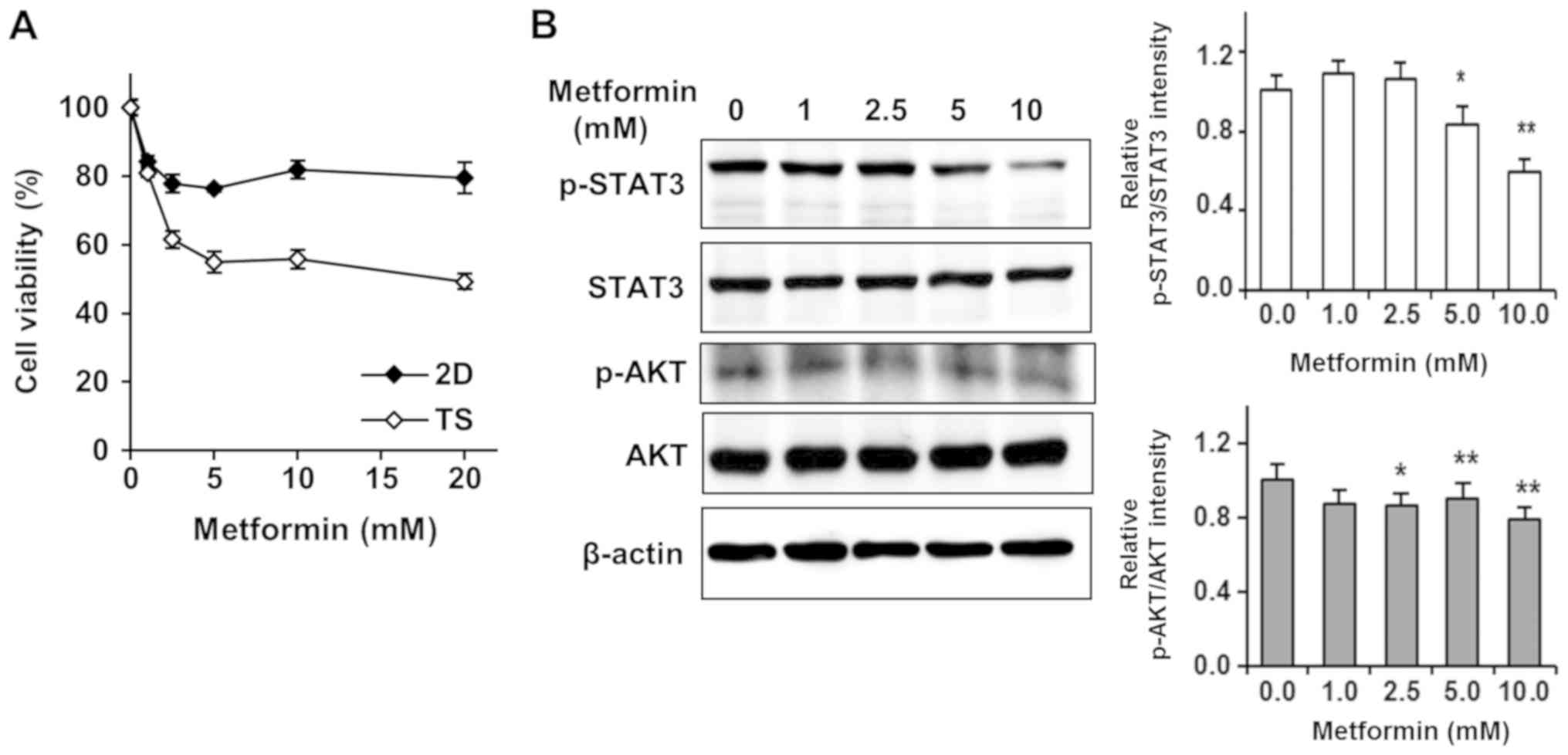
Thus, we tested effects of Met on 4T1 TS to find out a new approach
to overcome chemoresistance of TS. Surprisingly, we found that Met
selectively targeted TS rather than monolayer-cultured cells
(Fig. 3A). In stark contrast to
effects of Dox or Lapa on TS, cell viability in TS treated with 5
mM Met was about 50%, whereas over 80% of cells cultured as
monolayers survived treatment with 20 mM Met (Fig. 3A).
After finding STAT3 and AKT signaling pathway
enhancements were responsible for the chemoresistance of TS, we
tested whether Met affected the phosphorylation of STAT3 and AKT
signaling pathways. As shown in Fig.
3B, Met efficiently and dose-dependently inhibited the
phosphorylations of STAT3 and AKT, suggesting that Met selectively
targeted TS by inhibiting the STAT3 and AKT signaling pathways.
Met sensitizes 4T1 TS to Dox by
inhibiting the STAT3 and AKT signaling pathways
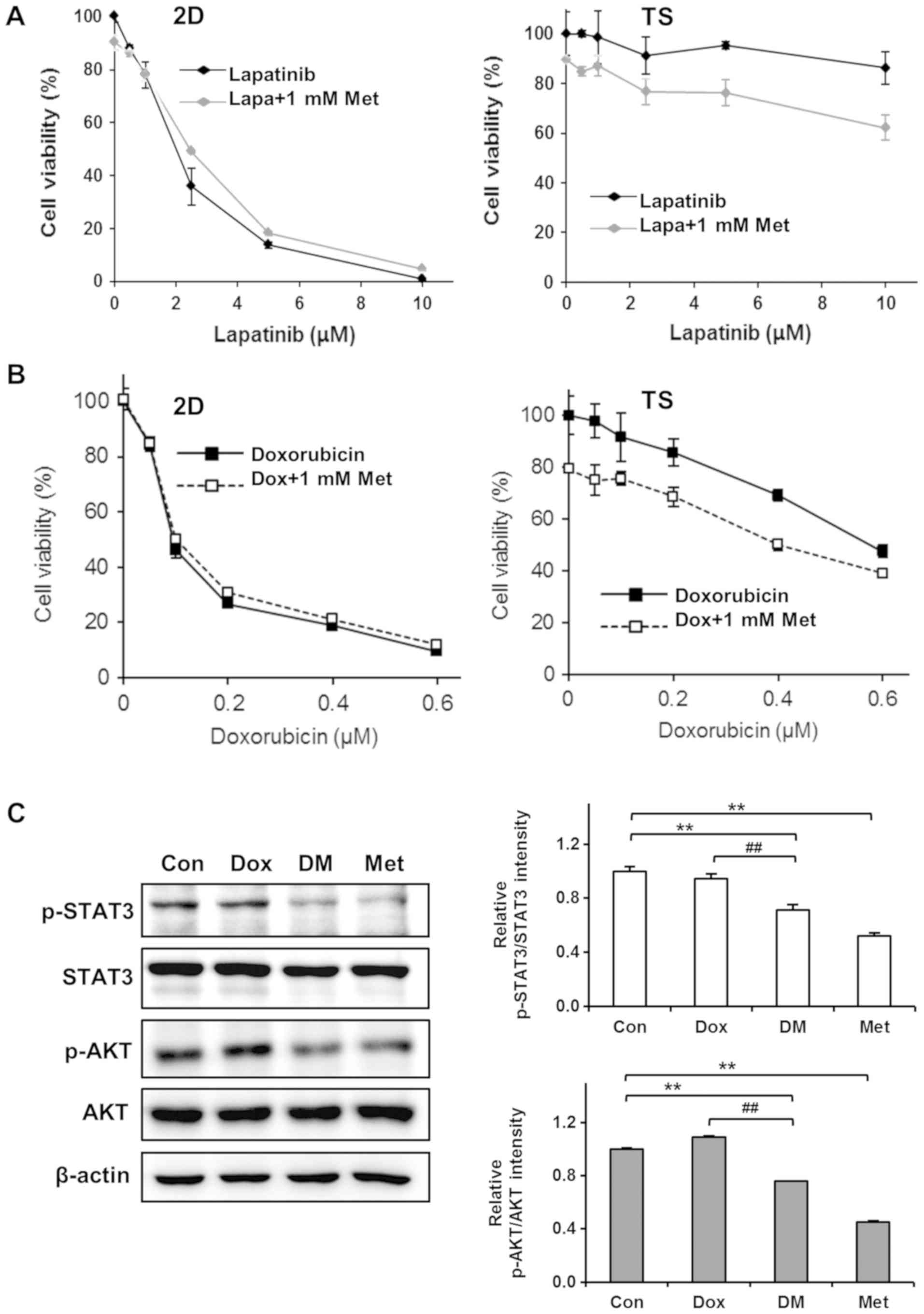
Encouraged by the observation that Met selectively
killed cells in 4T1 TS, we next examined the combined effects of
Met and chemotherapeutic agents on 4T1 TS chemoresistance. Cells
were treated with different concentrations of Lapa or Dox in
presence of 1 mM Met for 3 days and cell viabilities was
determined. Interestingly, Met exhibited synergistic effects with
Lapa or Dox on TS but not on cells cultured as monolayers. As shown
in Fig. 4A, treatment with Lapa and
Met did not enhance the cytotoxicity of Lapa in monolayer cultured
cells but significantly enhanced cytotoxicity in TS; for example,
>90% cell viability observed after treating TS with 10 µM Lapa
for 3 days, but this was reduced to almost 60% when cells were
treated with 10 µM Lapa in the presence of 1 mM Met. Similarly, Met
increased the cytotoxic effects of Dox by almost 20% in TS but no
synergistic effect was observed in monolayer cultured cells
(Fig. 4B).
To understand the mechanisms responsible for
overcoming chemoresistance of TS by Met, we performed western
blotting to investigate the effect of Dox plus Met on the
phosphorylations of STAT3 and AKT in TS. As mentioned above
(Fig. 2D), the phosphorylations of
STAT3 and AKT in TS were not inhibited by Dox. However,
co-treatment with Dox plus Met inhibited the phosphorylations of
STAT3 and of AKT (Fig. 4C). These
observations suggest that Met sensitizes TS to Dox by inhibiting
the STAT3 and AKT signaling pathways.
Met exhibited synergistic antitumor
effects with Dox in 4T1 tumor-bearing mice
We further evaluated the antitumor effects of Met on
4T1 tumor-bearing BALB/c mice. 4T1 cells were implanted into
the mammary gland fat pads of BALB/c female mice and when
mice had developed about 100 mm3 tumors (measured with
calipers), they were injected intraperitoneally with Dox (5 mg/kg,
once a week), Met (200 mg/kg, once a day), or both (Fig. 5A). Mouse body weights were unaffected
by these treatment schedules (Fig.
5B). Tumor growth was mildly suppressed in mice treated with
200 mg/kg Met alone, and mice treated with Dox alone exhibited a
tumor growth reduction of ~30% vs. untreated mice. However, this
antitumor effect of Dox was significantly enhanced when mice were
treated with Dox plus Met (Fig. 5C),
suggesting that synergism between Met and Dox accelerated tumor
regression.
Discussion
Initially, TS culture was proposed to detect and
propagate CSCs in the stem cell biology (13–15).
Although there is ongoing debate regarding the enrichment of CSCs
in TS (25,26), the generation of TS confers
interesting and unique features on cells, such as, quiescence
(27–29). We previously observed that sphere
cultures exhibited higher proportions of quiescent cells (22–24), and
in the present, we also observed that the majority of cells in 4T1
TS are quiescent, showing the accumulations of cells in the G0/G1
phase in TS as compared to cells in monolayer culture. This
increased quiescent nature of cells in 4T1 TS confers
chemoresistance to Dox and Lapa.
Recently, it was demonstrated that CSCs are
resistant to chemotherapeutics and served as the root cause of
disease recurrence and metastasis (30–32). Thus,
the elimination of CSCs is considered an effective strategy to
improve clinical response in breast cancer. Interestingly, Met has
been shown to selectively target CSCs, showing the decrease in the
CD44high/CD24low or ALDH-1 CSC fractions and
the suppression of the number and size of TS (12,17,18).
However, the mechanisms underlying the selective targeting of CSCs
by Met have not been fully elucidated. In the present study, we
also observed that Met preferentially targeted 4T1 TS rather than
cells in monolayer culture, and that it enhanced the sensitivity of
TS to Dox and Lapa. In addition, it was found this enhancement of
the antitumor effects of chemotherapeutics by Met appeared to be
mediated by downregulations of the STAT3 and AKT signaling
pathways.
PI3K/AKT/mTOR signaling pathway is a well-known
intracellular pathway, and its activation leads to the survival and
proliferation of cancer cells. Recently published evidences suggest
that activation of the PI3K/AKT/mTOR pathway is required for the
viability and maintenance of breast CSCs (21,33,34). Zhou
et al reported that CSC fractions were significantly reduced
after silencing the PI3K/AKT/mTOR pathway using a lentivirus-based
short-hairpin RNA (shRNA) (21). In
another study, suppression of the PI3K/AKT/mTOR pathway was found
to be preferentially toxic to CSCs (33). Met has been shown to inactivate the
PI3K/AKT/mTOR pathway through AMPK activation and that this
inactivation results in cell cycle arrest, apoptosis, and inhibits
tumorigenesis. Met can also directly target mTOR independently of
AMPK (2,35). In the present study, we were unable to
detect AMPK activation by Met because endogenous AMPK expression
was undetectable in 4T1 TS (data not shown). However, Met
efficiently and dose-dependently inhibited the phosphorylation of
AKT, which implies that Met directly targets the PI3K/AKT/mTOR
pathway in 4T1 TS without activating AMPK.
Like the role played by the PI3K/AKT/mTOR pathway,
the STAT3 signaling pathway has been associated with the generation
and maintenance CSCs in breast cancer (21,36,37). STAT3
is an important transcription factor that maintains embryonic stem
cells (ESCs) in an undifferentiated state (38,39). When
STAT proteins are fully activated, they dimerize and translocate
into nucleus to regulate gene expression. Regarding the role of
STAT3 in CSCs, it has been shown STAT3 is preferentially active in
CD44high/CD24low human breast cancer cells
(36), and that STAT3 agonist expands
the proportion of CD44high/CD24low CSCs
fraction (40). Wei et al used
a lentiviral fluorescent STAT3 signaling reporter system to
identify STAT3-mediated transcriptional activity, and found that
cells displaying highly activated STAT3 signaling are enriched for
in vitro TS formation potential and tumorigenic potential in
an in vivo transplantation assay (37). Although STAT3 is not a well-known
target of Met, in the present study, Met efficiently suppressed
STAT3 phosphorylation in 4T1 TS. Similarly, Deng et al
reported that Met targets STAT3 to inhibit cell growth and induce
apoptosis in triple negative breast cancer (41). Beside the STAT3, we also checked other
transcription factors including NF-κB and c-fos. However, c-fos was
not detected in Met-treated 4T1 TS, and the expression of NF-κB was
not affected by Met (data not shown), implying that STAT3 is a
major transcription factor targeted by Met in 4T1 TS.
Considering the vital roles of AKT and STAT3 in
CSCs, it might be that the selective cytotoxic effect of Met on 4T1
TS was derived from its inhibitory effects on the AKT and STAT3
signaling pathways. The link between the PI3K/AKT/mTOR pathway and
the STAT3 signaling pathway within 4T1 TS was not elucidated in the
present study, but we consider that inhibition of AKT
phosphorylation by Met probably leads to the inactivation of STAT3
signaling in 4T1 TS, because it has been proposed that the
PI3K/AKT/mTOR pathway is a positive regulator of STAT3 signaling
(21).
In the present study, we found that 4T1 TS were
resistant to Dox or Lapa because of upregulations of the STAT3 and
AKT signaling pathways, and that Met overcame chemoresistance by
inhibiting the phosphorylations of AKT and STAT3 in Dox-treated TS.
Furthermore, Met exhibited synergistic antitumor effects with Dox
in 4T1 tumor-bearing BALB/c mice. Accordingly, our findings
suggest that Met and cytotoxic anticancer drugs when used in
combination offer benefits for the treatment of drug-resistant
breast cancer. But these findings have limitations to elucidate
full mechanisms on synergistic antitumor effect of Met and
anticancer drugs, so further study is needed.
In summary, we show that the inhibitions of AKT and
STAT3 activation underlies the selective cytotoxic effects of Met
on TS. We hope that these results provide clues regarding the
mechanism responsible for the targeting of CSCs by Met and a basis
for the use of combinations of Met and chemotherapeutics to improve
clinical response in breast cancer patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National R&D Program for Cancer Control, the Korean Ministry of
Health & Welfare (grant no. 1320060) and by a grant from the
Basic Science Research Program, Korean National Research Foundation
(NRF) funded by the Ministry of Education, Science and Technology,
Republic of Korea (grant no. NRF-2015R1D1A1A01059738).
Availability of data and materials
All data generated during this study are included in
this published article.
Authors' contributions
YSK contributed to the conception and design of the
study, and the acquisition of data, and wrote the manuscript. SYC
performed experiments to acquire the data and analyzed data. HYN
analyzed and interpreted the data. KSN contributed to the
conception and design of the study. CL contributed to the
conception and design of the manuscript, and was involved in
drafting and revising the manuscript. SK contributed to the
conception and design of the manuscript, drafted and wrote the
manuscript, and gave final approval of the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Animal experimentation was performed after obtaining
approval from the Institutional Animal Care and Use Committee at
Dongguk University (approval no. IACUC-2016-003; Gyeongju-si,
Korea).
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AKT
|
protein kinase B
|
|
AMPK
|
AMP-activated protein kinase
|
|
CSC
|
cancer stem cells
|
|
Dox
|
doxorubicin
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
Lapa
|
lapatinib
|
|
Met
|
metformin
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
TS
|
tumorspheres
|
References
|
1
|
Hundal HS, Ramlal T, Reyes R, Leiter LA
and Klip A: Cellular mechanism of metformin action involves glucose
transporter translocation from an intracellular pool to the plasma
membrane in L6 muscle cells. Endocrinology. 131:1165–1173. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dowling RJ, Goodwin PJ and Stambolic V:
Understanding the benefit of metformin use in cancer treatment. BMC
Med. 9:332011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bowker SL, Majumdar SR, Veugelers P and
Johnson JA: Increased cancer-related mortality for patients with
type 2 diabetes who use sulfonylureas or insulin. Diabetes Care.
29:254–258. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dowling RJ, Zakikhani M, Fantus IG, Pollak
M and Sonenberg N: Metformin inhibits mammalian target of
rapamycin-dependent translation initiation in breast cancer cells.
Cancer Res. 67:10804–10812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iliopoulos D, Hirsch HA and Struhl K:
Metformin decreases the dose of chemotherapy for prolonging tumor
remission in mouse xenografts involving multiple cancer cell types.
Cancer Res. 71:3196–3201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Buzzai M, Jones RG, Amaravadi RK, Lum JJ,
DeBerardinis RJ, Zhao F, Viollet B and Thompson CB: Systemic
treatment with the antidiabetic drug metformin selectively impairs
p53-deficient tumor cell growth. Cancer Res. 67:6745–6752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shank JJ, Yang K, Ghannam J, Cabrera L,
Johnston CJ, Reynolds RK and Buckanovich RJ: Metformin targets
ovarian cancer stem cells in vitro and in vivo. Gynecol Oncol.
127:390–397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu B, Fan Z, Edgerton SM, Deng XS,
Alimova IN, Lind SE and Thor AD: Metformin induces unique
biological and molecular responses in triple negative breast cancer
cells. Cell Cycle. 8:2031–2040. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rattan R, Ali Fehmi R and Munkarah A:
Metformin: An emerging new therapeutic option for targeting cancer
stem cells and metastasis. J Oncol. 2012:9281272012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hirsch HA, Iliopoulos D, Tsichlis PN and
Struhl K: Metformin selectively targets cancer stem cells, and acts
together with chemotherapy to block tumor growth and prolong
remission. Cancer Res. 69:7507–7511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dontu G, Abdallah WM, Foley JM, Jackson
KW, Clarke MF, Kawamura MJ and Wicha MS: In vitro propagation and
transcriptional profiling of human mammary stem/progenitor cells.
Genes Dev. 17:1253–1270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ponti D, Costa A, Zaffaroni N, Pratesi G,
Petrangolini G, Coradini D, Pilotti S, Pierotti MA and Daidone MG:
Isolation and in vitro propagation of tumorigenic breast cancer
cells with stem/progenitor cell properties. Cancer Res.
65:5506–5511. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang L, Jiao M, Li L, Wu D, Wu K, Li X,
Zhu G, Dang Q, Wang X, Hsieh JT and He D: Tumorspheres derived from
prostate cancer cells possess chemoresistant and cancer stem cell
properties. J Cancer Res Clin Oncol. 138:675–686. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen G, Xu S, Renko K and Derwahl M:
Metformin inhibits growth of thyroid carcinoma cells, suppresses
self-renewal of derived cancer stem cells, and potentiates the
effect of chemotherapeutic agents. J Clin Endocrinol Metab.
97:E510–E520. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jung JW, Park SB, Lee SJ, Seo MS, Trosko
JE and Kang KS: Metformin represses self-renewal of the human
breast carcinoma stem cells via inhibition of estrogen
receptor-mediated OCT4 expression. PLoS One. 6:e280682011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song CW, Lee H, Dings RP, Williams B,
Powers J, Santos TD, Choi BH and Park HJ: Metformin kills and
radiosensitizes cancer cells and preferentially kills cancer stem
cells. Sci Rep. 2:3622012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vazquez-Martin A, Oliveras-Ferraros C,
Cufi S, Del Barco S, Martin-Castillo B and Menendez JA: Metformin
regulates breast cancer stem cell ontogeny by transcriptional
regulation of the epithelial-mesenchymal transition (EMT) status.
Cell Cycle. 9:3807–3814. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cufí S, Vazquez-Martin A,
Oliveras-Ferraros C, Martin-Castillo B, Joven J and Menendez JA:
Metformin against TGFβ-induced epithelial-to-mesenchymal transition
(EMT): From cancer stem cells to aging-associated fibrosis. Cell
Cycle. 9:4461–4468. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang
Y, Deng J, Margolick JB, Liotta LA, Petricoin E III and Zhang Y:
Activation of the PTEN/mTOR/STAT3 pathway in breast cancer
stem-like cells is required for viability and maintenance. Proc
Natl Acad Sci USA. 104:16158–16163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chun SY, Kwon YS, Nam KS and Kim S:
Lapatinib enhances the cytotoxic effects of doxorubicin in MCF-7
tumorspheres by inhibiting the drug efflux function of ABC
transporters. Biomed Pharmacother. 72:37–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim S and Alexander CM: Tumorsphere assay
provides more accurate prediction of in vivo responses to
chemotherapeutics. Biotechnol Lett. 36:481–488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kwon YS, Chun SY, Nam KS and Kim S:
Lapatinib sensitizes quiescent MDA-MB-231 breast cancer cells to
doxorubicin by inhibiting the expression of multidrug
resistance-associated protein-1. Oncol Rep. 34:884–890. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Calvet CY, André FM and Mir LM: The
culture of cancer cell lines as tumorspheres does not
systematically result in cancer stem cell enrichment. PLoS One.
9:e896442014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pastrana E, Silva-Vargas V and Doetsch F:
Eyes wide open: A critical review of sphere-formation as an assay
for stem cells. Cell Stem Cell. 8:486–498. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guttilla IK, Phoenix KN, Hong X, Tirnauer
JS, Claffey KP and White BA: Prolonged mammosphere culture of MCF-7
cells induces an EMT and repression of the estrogen receptor by
microRNAs. Breast Cancer Res Treat. 132:75–85. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Manuel Iglesias J, Beloqui I,
Garcia-Garcia F, Leis O, Vazquez-Martin A, Eguiara A, Cufi S, Pavon
A, Menendez JA, Dopazo J and Martin AG: Mammosphere formation in
breast carcinoma cell lines depends upon expression of E-cadherin.
PLoS One. 8:e772812013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Uchida Y, Tanaka S, Aihara A, Adikrisna R,
Yoshitake K, Matsumura S, Mitsunori Y, Murakata A, Noguchi N, Irie
T, et al: Analogy between sphere forming ability and stemness of
human hepatoma cells. Oncol Rep. 24:1147–1151. 2010.PubMed/NCBI
|
|
30
|
Li L and Bhatia R: Stem cell quiescence.
Clin Cancer Res. 17:4936–4941. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moore N and Lyle S: Quiescent,
slow-cycling stem cell populations in cancer: A review of the
evidence and discussion of significance. J Oncol. 2011:3960762011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tirino V, Desiderio V, Paino F, De Rosa A,
Papaccio F, La Noce M, Laino L, De Francesco F and Papaccio G:
Cancer stem cells in solid tumors: An overview and new approaches
for their isolation and characterization. FASEB J. 27:13–24. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eyler CE, Foo WC, LaFiura KM, McLendon RE,
Hjelmeland AB and Rich JN: Brain cancer stem cells display
preferential sensitivity to Akt inhibition. Stem Cells.
26:3027–3036. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Korkaya H, Paulson A, Charafe-Jauffret E,
Ginestier C, Brown M, Dutcher J, Clouthier SG and Wicha MS:
Regulation of mammary stem/progenitor cells by
PTEN/Akt/beta-catenin signaling. PLoS Biol. 7:e10001212009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kalender A, Selvaraj A, Kim SY, Gulati P,
Brûlé S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et
al: Metformin, independent of AMPK, inhibits mTORC1 in a rag
GTPase-dependent manner. Cell Metab. 11:390–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Marotta LL, Almendro V, Marusyk A,
Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ,
Choudhury SA, Maruyama R, et al: The JAK2/STAT3 signaling pathway
is required for growth of CD44+CD24+ stem
cell-like breast cancer cells in human tumors. J Clin Invest.
121:2723–2735. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wei W, Tweardy DJ, Zhang M, Zhang X,
Landua J, Petrovic I, Bu W, Roarty K, Hilsenbeck SG, Rosen JM and
Lewis MT: STAT3 signaling is activated preferentially in
tumor-initiating cells in claudin-low models of human breast
cancer. Stem Cells. 32:2571–2582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Matsuda T, Nakamura T, Nakao K, Arai T,
Katsuki M, Heike T and Yokota T: STAT3 activation is sufficient to
maintain an undifferentiated state of mouse embryonic stem cells.
EMBO J. 18:4261–4269. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Raz R, Lee CK, Cannizzaro LA, d'Eustachio
P and Levy DE: Essential role of STAT3 for embryonic stem cell
pluripotency. Proc Natl Acad Sci USA. 96:2846–2851. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Iliopoulos D, Hirsch HA, Wang G and Struhl
K: Inducible formation of breast cancer stem cells and their
dynamic equilibrium with non-stem cancer cells via IL6 secretion.
Proc Natl Acad Sci USA. 108:1397–1402. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deng XS, Wang S, Deng A, Liu B, Edgerton
SM, Lind SE, Wahdan-Alaswad R and Thor AD: Metformin targets Stat3
to inhibit cell growth and induce apoptosis in triple-negative
breast cancers. Cell Cycle. 11:367–376. 2012. View Article : Google Scholar : PubMed/NCBI
|