Introduction
Breast cancer is the most common malignancy among
women and estrogen is an essential stimulant of estrogen receptor
(ER)-positive breast cancer (1,2).
Tamoxifen (TAM), a non-steroidal anti-estrogen, is the most
commonly used therapeutic or preventative agent for ER-positive
breast cancer patients (3). However,
acquired resistance to TAM is a critical problem with long-term TAM
use, and the mechanism underlying this resistance remains unclear
(4). We previously established an
MCF-7-derived TAM-resistant cell line (TAMR-MCF-7 cells) by
long-term incubation with 4-hydroxytamoxifen (5), and determined that TAMR-MCF-7 cells
lost polarity and acquired migratory and invasive properties with
higher expression of epithelial mesenchymal transition (EMT) marker
proteins (6–8). This process is considered a
prerequisite for tumor infiltration and metastasis (9). TAMR-MCF-7 cells have also been shown to
exhibit enhanced vascular endothelial growth factor (VEGF)
expression levels, leading to the promotion of angiogenesis
(10).
Several receptors and transcription factors are
actively involved in the regulation of cell proliferation and
acquisition of migratory and invasive properties in diverse cancer
cell types. Among these, the Janus kinase 2 (JAK2)-signal
transducer and activator of transcription (STAT) 3 pathway
transfers extracellular signals from cytokines and growth factor
receptors, including the interleukin-6 receptor (IL-6R) and
platelet-derived growth factor receptor (PDGFR), to its downstream
target genes (11–13). Activation of the JAK-STAT pathway in
cancer cells activates the transcription of several oncogenic
genes, including c-myc, ccnd1, and VEGF (14–16).
Several studies have indicated that the JAK2-STAT3 signaling
pathway is a crucial activator of cell migration and cancer
metastasis (17–19).
Ruxolitinib (Jakafi), a potent JAK1/2 inhibitor, was
approved by the United States Food and Drug Administration (FDA) in
2011 for the treatment of patients with myeloproliferative
neoplasia (MPN). Ruxolitinib directly inhibits JAK1 and JAK2
kinases, with IC50 values of 3.3 and 2.8 nM, respectively (20). Ruxolitinib reduced protein levels of
phosphorylated JAK2 and subsequently inhibited protein expression
of its downstream target genes, c-Myc, cyclin D1 and Bcl-2
(21). In preclinical studies,
ruxolitinib was shown to inhibit the proliferation of
JAK2V617F-positive Ba/F3 cells showing a constitutively active
mutant form of JAK2, and to alleviate MPN symptoms in
JAK2V617F-transgenic mice (20).
Moreover, involvement of JAK2 in cell proliferation was
experimentally verified in various cancer cell lines, lung cancer
cells (H661, H1975, H1563, ADOR, NSCLC1), breast cancer cells
(MCF-7, MCF-7-HER18, SUM149, BT474, BT549, SKBR3) and glioblastoma
cells (GBM6, GBM12) (21–23). However, the therapeutic effects of
ruxolitinib on chemo-resistant breast cancer cells, and especially
TAM-resistant cells, remain obscure. In the present study, we
demonstrated for the first time that TAM-resistant MCF-7 cells
showed higher sensitivity to ruxolitinib compared to parental MCF-7
cells, and the JAK inhibitor blocked the EMT process and VEGF
production, consequently suppressing TAMR-MCF-7 cell migration and
angiogenesis.
Materials and methods
Cell culture and reagents
MCF-7 (ER-positive human breast cancer cell line)
cells were obtained from Korea Cell Line Bank (#30022, 2005), and
cultured at 37°C in Dulbecco's modified Eagle's medium (DMEM)
containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, and
100 µg/ml streptomycin. TAMR-MCF-7 cells were established as
previously described (5,24).
The antibodies used in the present study were as
follow: phospho-STAT3 (p-STAT3) (Y705, cat. no. 9131S), STAT3 (cat.
no. 4904S), Bcl-2 (cat. no. 2876S) and hypoxia inducible factor
(HIF)-1α (cat. no. 3716S) were purchased from Cell Signaling
Technology (Beverly, MA, USA), c-Myc (cat. no. sc-40), c-Jun (cat.
no. sc-1694), Cyclin B1 (cat. no. sc-245), Cyclin D1 (cat. no.
sc-753), vimentin (cat. no. sc-32322), twist (cat. no. sc-81417)
were obtained from Santa Cruz Biotechnology (Dallas, TX, USA), and
glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Calbiochem,
Gibbstown, NJ, USA; cat. no. CB-1001), β-actin (Sigma-Aldrich, St.
Louis, MO, USA; cat. no. a2228), E-cadherin (BD Biosciences, San
Jose, CA, USA; cat. no. 610181), N-cadherin (BD Biosciences; cat.
no. 610920), snail (Abcam, Cambridge, UK; cat. no. ab53519) were
used. VEGF-luc plasmids were kindly donated from Dr. Lee (Chonnam
National University, Gwangju, Korea).
Western blot analysis
Cells were lysed with lysis buffer containing 20 mM
Tris-Cl (pH 7.5), 1% Triton X-100, 137 mM sodium chloride, 10%
glycerol, 2 mM EDTA, 1 mM sodium orthovanadate, 25 mM
β-glycerophosphate, 2 mM sodium pyrophosphate, 1 mM
phenylmethylsulfonylfluoride and 1 µg/ml leupeptin. The cell
lysates were centrifuged at 13,000 × g for 15 min to remove
insoluble material, the supernatants were quantified using the
Bradford method (25) and
fractionated using polyacrylamide gel, and electrophoretically
transferred to nitrocellulose membranes. Horseradish peroxidase
(HRP)-conjugated anti-IgG antibodies were used as the secondary
antibodies (cat. no. 7076S, 7074S, 1:5,000 dilution; Cell Signaling
Technology). The nitrocellulose papers were developed using an ECL
chemiluminescence system (Milipore, Billerica, MA, USA). For ECL
chemiluminescence detection, LAS-3000 mini system (Fujifilm, Tokyo,
Japan) was used.
Immunocytochemistry
MCF-7 and TAMR-MCF-7 cells were cultured overnight
on coverslips. After treatment with 10 µM ruxolitinib or vehicle,
the cells were fixed with 4% paraformaldehyde and blocked with 1%
bovine serum albumin at room temperature for 1 h. The cells were
incubated with Alexa Fluor 488-conjugated Phalloidin antibody
(1:200 dilution, Thermo Fisher Scientific, Waltham, MA, USA) in
0.1% Tween-20-containing phosphate-buffered saline at 4°C
overnight. Mouse monoclonal E-cadherin or N-cadherin antibody
(1:200 dilution, BD Biosciences) were also incubated in the same
buffer at 4°C overnight. After twice washing with PBS, the
coverslips were incubated with goat anti-mouse Alexa Fluor 568 IgG
(1:2,000 dilution; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) at room temperature for 1 h. Finally, the coverslips were
washed thoroughly with PBS and then mounted with ProLong Gold
Antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI;
Invitrogen, Carlsbad, CA, USA). Images were obtained using CELENAS
Digital Imaging System (Logos Biosystems, Anyang, South Korea).
Cell proliferation assay
MCF-7 and TAMR-MCF-7 cells were seeded in 96-well
plate (3×103 cells/well) and cultured for 96 h in the presence or
absence of ruxolitinib (0.1–10 µM). The kinetics of viable cell
numbers were counted using IncuCyte ZOOM live-cell analysis system
(Essen BioScience, Ann Arbor, MI, USA) and monitored using the
IncuCyte label-free cell monolayer confluence algorithm. IncuCyte
provides the ability to acquire phase-contrast images and an
integrated confluence metric as a surrogate for cell number.
Transwell migration assay
TAMR-MCF-7 cells were seeded in the upper chamber of
the transwell plate (Essen BioScience) and the lower side of the
upper chamber was covered with type I collagen (Sigma-Aldrich). The
lower chamber was filled with 200 µl FBS-containing culture media.
The cells were incubated at 5% CO2 for 18 h. The migrated cell
numbers were counted using IncuCyte ZOOM live-cell analysis
system.
Semi-quantitative polymerase chain
reaction (sqPCR)
Total RNA was isolated from MCF-7 and TAMR-MCF-7
cells using Trizol® reagent (Invitrogen), and cDNA was
synthesized by reverse transcriptase using an oligo (dT) primer.
PCR was performed using the selective primers for human VEGF
(sense: 5′-GCTACTGCCATCCAATCGAG-3′, antisense:
5′-TGCATTCACATTTGTTGTGC-3′), human GAPDH (sense:
5′-AAGGCTGAGAACGGGAAG-3′, antisense: 5′-GCCCCACTTGATTTTGGA-3′) as
the housekeeping gene. sqPCR was performed for 40 cycles
(denaturation at 98°C for 30 sec, annealing at 60°C for 30 sec, and
then a final extension at 60°C for 5 min). After PCR, 6 µl samples
of the products were subjected to 2.0% agarose gel electrophoresis
and stained with ethidium bromide. Densitometric analyses were
performed using Quantity One 1-D Analysis Software version 4.6.2
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Reporter gene assay
MCF-7 and TAMR-MCF-7 cells (1×105 cells/well) were
seeded in 48-well plates, then transfected with luciferase reporter
plasmids containing VEGF-luc reporter plasmid containing the
luciferase structural gene and the human VEGF promoter and pRL-SV
plasmids (Renilla luciferase expression for normalization). The
transfected cells were exposed to compound for 24 h, and the
promoter activity was measured using a dual-luciferase reporter
assay system (Promega, Madison, WI, USA). The firefly and Renilla
luciferase activities were measured using a luminometer (LB941,
Berthold Technologies, Bad Wildbad, Germany).
Chick chorioallantoic membrane (CAM)
assay
Fertilized eggs were purchased from Siprigol Poultry
Farm (Gyeongsan, South Korea) and the CAM was prepared as described
previously (26–28). The surfaces of 10-day-old
post-fertilization chick eggs were sterilized and the CAM was
exposed by cutting a window (1 cm2) on one side of the egg using
the false air sac technique. TAMR-MCF-7 cells were placed on the
exposed CAM and the windows were sealed with transparent tape. The
eggs were then incubated in a humidified incubator at 37°C. The
CAMs were examined at 3 days after inoculation using a SV6
stereomicroscope (Carl Zeiss, Hamburg, Germany) at 50 ×
magnification. The CAM experiments were terminated at 13th
embryonic day. After extraction of chorioallantoic membrane, chick
embryos should be dead due to the excess hemorrhage. Then the dead
cadaver was disposed in accordance with the biological waste
treatment procedures. Digital images of CAM sections were collected
using a 3-charge coupled device color video camera system (Toshiba,
Tokyo, Japan). The number of vessel branch points contained in a
circular region was counted. The experimental studies were approved
by the Institutional Animal Care and Use Committee of Yeungnam
University (Gyeongsangbuk, Korea).
Statistical analysis
Statistical analysis was performed using Sigma plot
12.0 (Systat Software, Inc., San Jose, CA, USA) with one-way
analysis of variance and Tukey's post hoc multiple comparisons test
to determine the differences. P<0.05 was considered to indicate
a statistically significant difference.
Results
Ruxolitinib inhibits STAT3 activation
in TAMR-MCF-7 cells
We compared JAK2 activity between MCF-7 and
TAMR-MCF-7 cells by determining the extent of Tyr705 STAT3
phosphorylation. Immunoblot results showed that STAT3
phosphorylation was much higher in TAMR-MCF-7 cells than in control
MCF-7 cells (Fig. 1A). We then
assessed whether ruxolitinib disturbed the JAK2-STAT3 signaling
pathway in both breast cancer cell types, and found that p-STAT3
levels decreased with ruxolitinib treatment (0.1–10 µM) in a
concentration-dependent manner in both MCF-7 and TAMR-MCF-7 cells
(Fig. 1B). Many studies have
reported that STAT3 activation is required for the transactivation
of several oncoproteins and survival factors, including c-Myc,
c-Jun, Cyclin B1, Cyclin D1, Bcl-2, and HIF-1α (29,30). In
comparison to protein levels of MCF-7 cells, those of c-Myc, c-Jun,
Cyclin B1, Cyclin D1, Bcl-2, and HIF-1α were upregulated in
TAMR-MCF-7 cells, and this enhanced expression was markedly
inhibited by ruxolitinib (0.1–10 µM, Fig. 1C). These results demonstrate that
ruxolitinib attenuates activation of the JAK2-STAT3 pathway in
TAMR-MCF-7 cells.
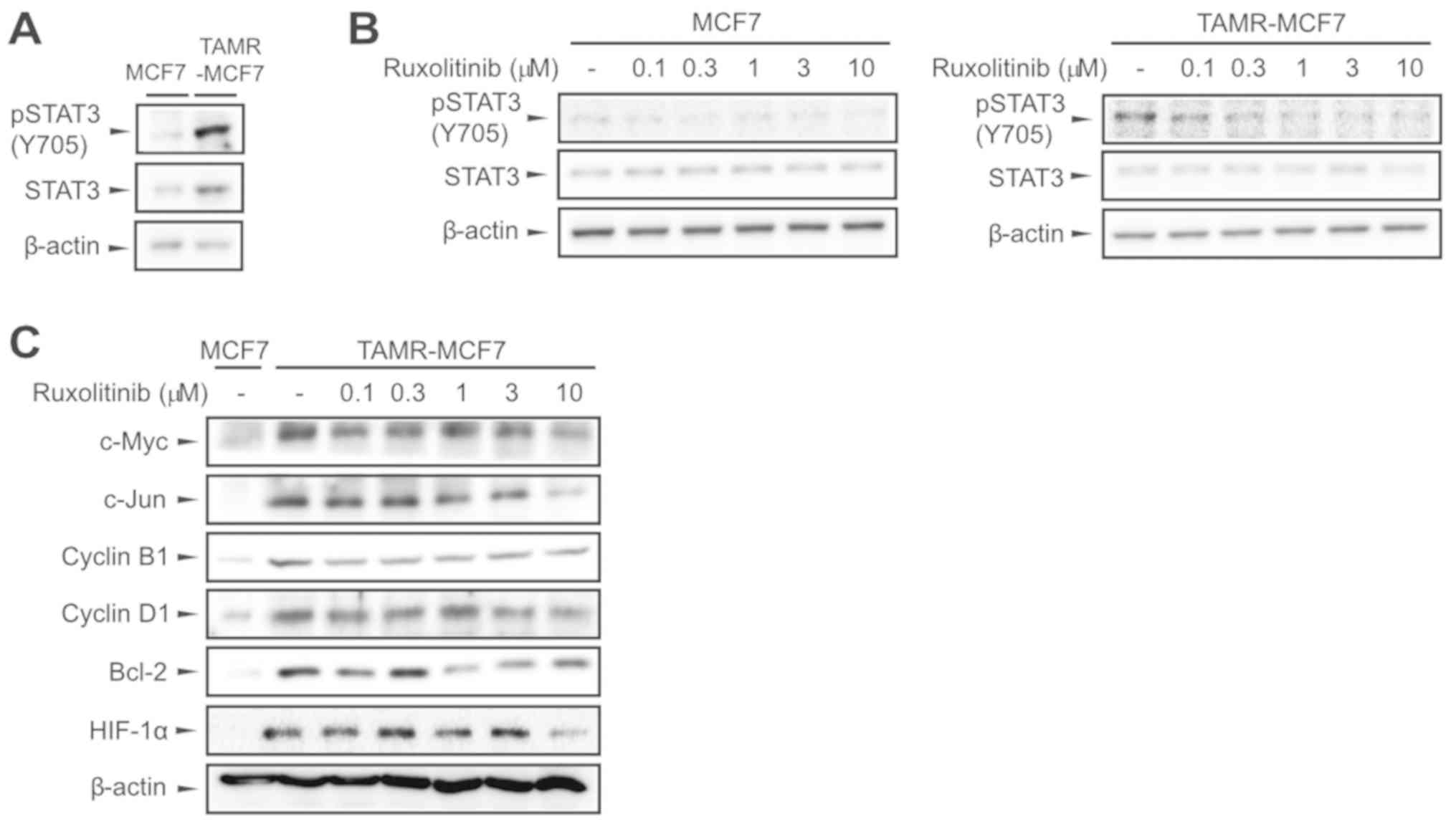 | Figure 1.Inhibition of the STAT3 signaling
pathway by ruxolitinib in TAMR-MCF-7 cells. (A) Expression of
pSTAT3 and STAT3 in MCF-7 and TAMR-MCF-7 cells. β-actin levels were
utilized as the loading controls. (B) Inhibition of STAT3
phosphorylation by ruxolitinib. MCF-7 and TAMR-MCF-7 cells were
treated with vehicle or ruxolitinib (0.1–10 µM) for 24 h. (C)
Effects of ruxolitinib on the expression of downstream target genes
of the JAK-STAT pathway. The protein levels of c-Myc, c-Jun, Cyclin
B1, Cyclin D1, Bcl-2 and HIF-1α were determined in MCF-7 and
TAMR-MCF-7 cells 24 h following ruxolitinib treatment (0.1–10 µM).
All of the experiments were repeated at least three times. JAK,
Janus kinase; STAT, signal transducer and activator of
transcription; pSTAT, phosphorylated STAT; HIF-1α, hypoxia
inducible factor-1α; Bcl-2, B-cell lymphoma 2; TAMR, tamoxifen
resistant. |
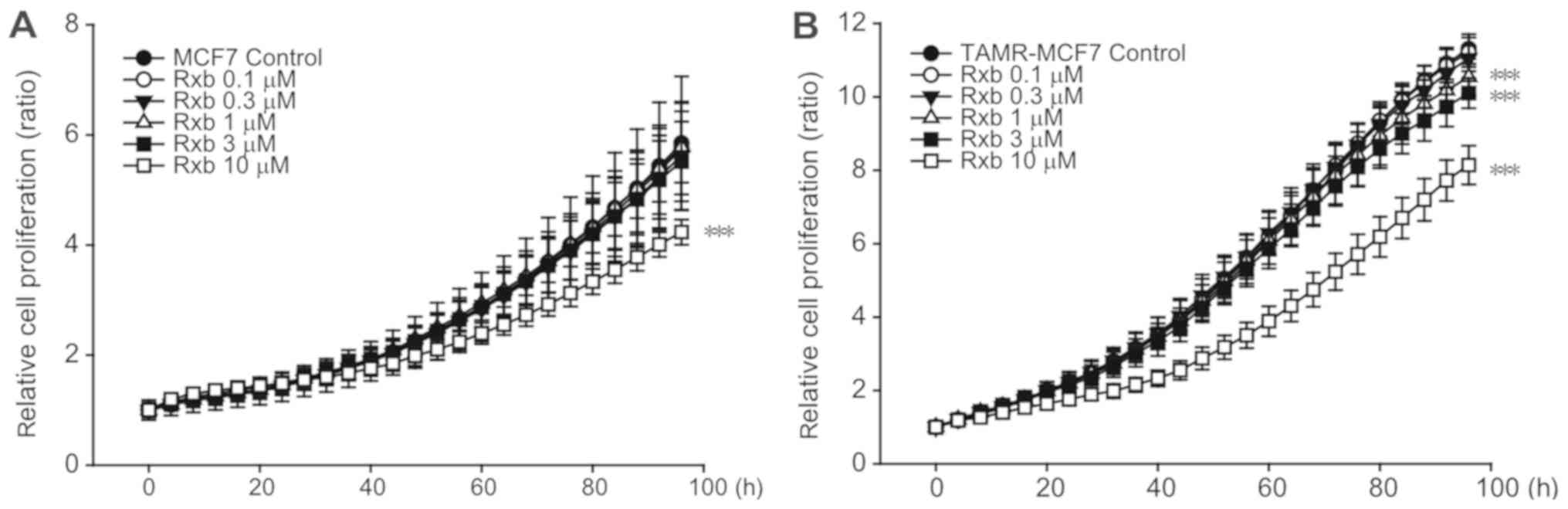
Ruxolitinib inhibits TAMR-MCF-7 cell
proliferation
To determine the effect of ruxolitinib on breast
cancer cell proliferation, real-time, live-cell monitoring and
analysis were performed using IncuCyte ZOOM. Ruxolitinib
significantly inhibited the growth of MCF-7 cells at a
concentration of 10 µM (Fig. 2A).
However, TAMR-MCF-7 cells showed enhanced sensitivity to
ruxolitinib (Fig. 2B). When
ruxolitinib at the concentration of 1 or 3 µM was exposed to
TAMR-MCF7 cells, the inhibiting effect of cell growth was slight
but significant. Also, this inhibitory effect was not observed in
MCF-7 cells treated with ruxolitinib at the same concentration
ranges. These data suggest that TAMR-MCF-7 cell proliferation is
partially dependent on the JAK2-STAT3 pathway and that ruxolitinib
preferentially suppresses TAM-resistant breast cancer cell
proliferation.
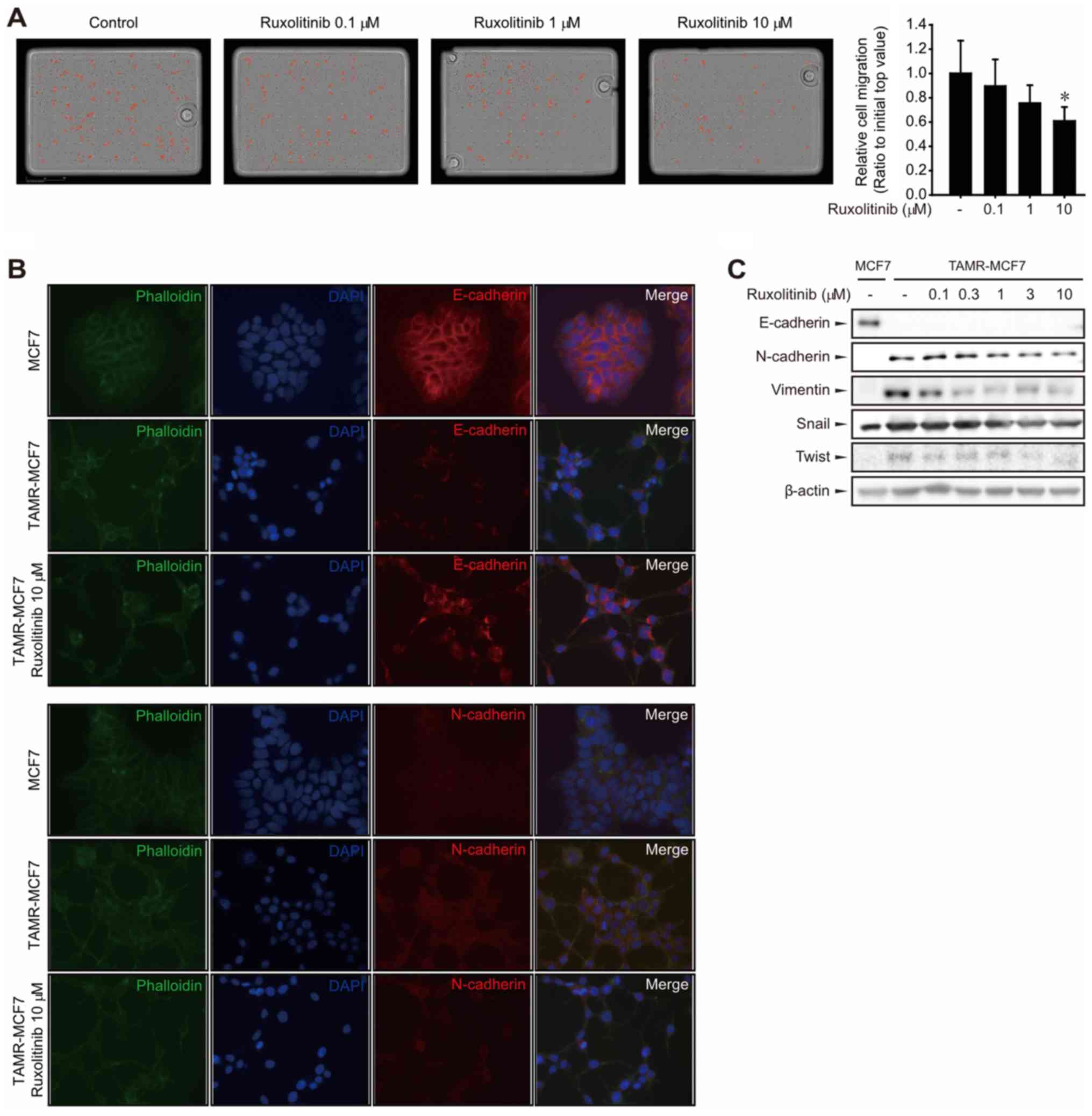
Ruxolitinib inhibits cell migration
and EMT progression in TAMR-MCF-7 cells
Because the JAK2-STAT3 pathway plays an important
role in cancer cell migration and invasion, we performed a
transwell migration assay to assess TAMR-MCF-7 cell migration. We
previously reported that TAMR-MCF-7 cells possess greater in
vitro migratory ability than MCF-7 cells (8). TAMR-MCF-7 cell migration was
significantly suppressed under treatment with 10 µM ruxolitinib
(Fig. 3A). Because TAMR-MCF-7 cells
acquire the migratory phenotype via EMT progression (7), we then examined whether JAK2-STAT3
inhibition by ruxolitinib affects the expression of EMT markers in
TAMR-MCF-7 cells. Representative biochemical markers of EMT include
loss of the epithelial adherence protein E-cadherin and
upregulation of the mesenchymal protein N-cadherin (9). Immunocytochemistry for E-cadherin,
N-cadherin, and phalloidin in MCF-7 and TAMR-MCF-7 cells showed
that E-cadherin downregulation and N-cadherin upregulation in
TAMR-MCF-7 cells were partially reversed by treatment with 10 µM
ruxolitinib (Fig. 3B). Western blot
analyses confirmed that higher expression of mesenchymal marker
proteins, such as N-cadherin, vimentin, snail, or twist, was
suppressed in TAMR-MCF-7 cells by ruxolitinib in a
concentration-dependent manner (Fig.
3C). Although E-Cadherin was slightly detected in TAMR-MCF-7
cells by immunocytochemistry, E-cadherin was not detectable by
immunoblottings in TAMR-MCF7 cells (Fig.
3C) (8). It may result from the
difference in detection sensitivity between immunoblotting and
immunocytochemistry. These results demonstrate that ruxolitinib may
inhibit cell migration in TAMR-MCF-7 cells, presumably by blocking
EMT.
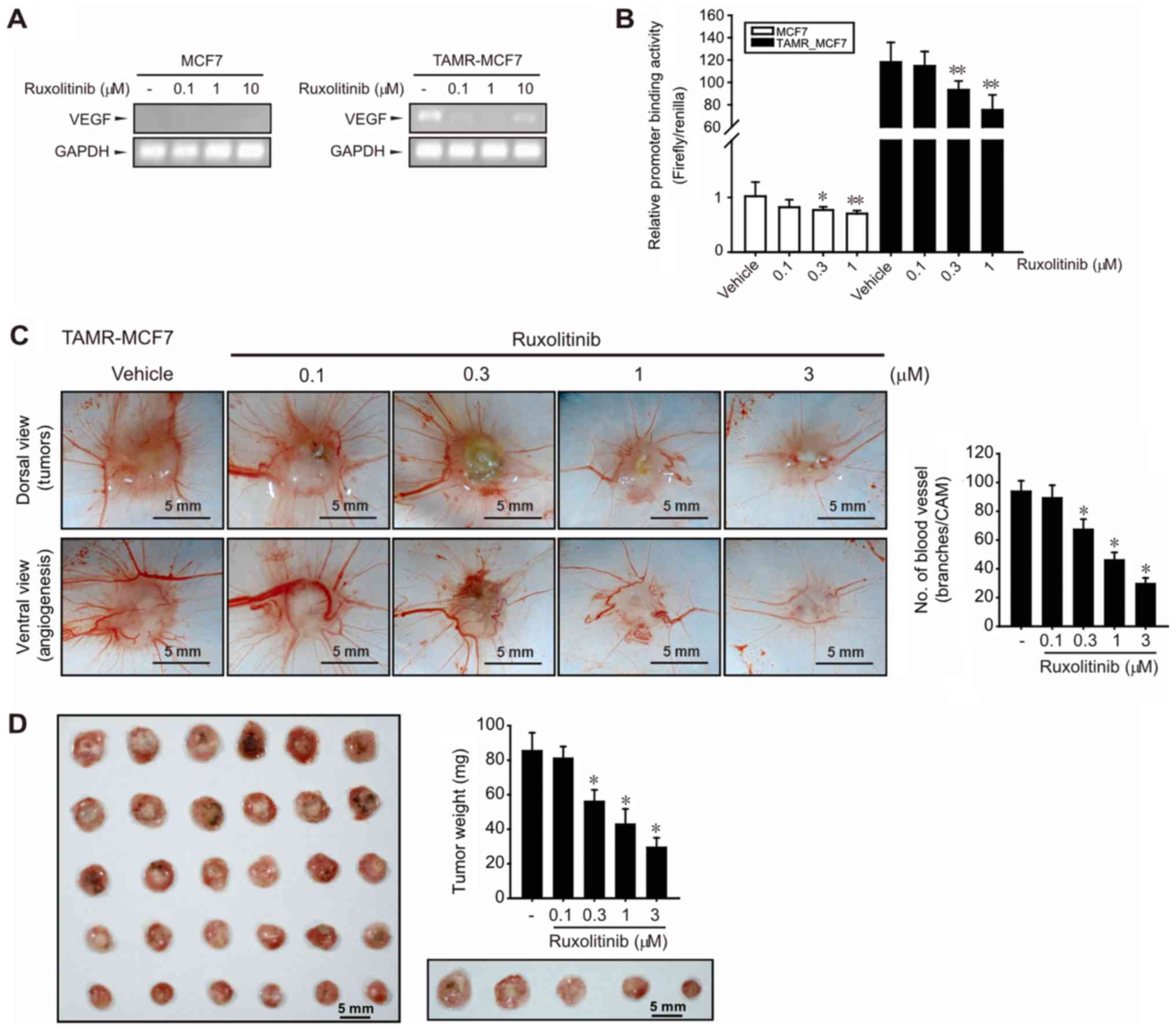
Ruxolitinib inhibits angiogenesis and
tumor growth
A clinical feature of TAM resistance in human breast
cancer is an increase in microvessel counts (31). We previously reported that angiogenic
potential was enhanced in TAMR-MCF-7 cells by VEGF upregulation
(10). In the current study, high
basal VEGF mRNA expression was observed in TAMR-MCF-7 cells, but
not in MCF-7 cells (Fig. 4A).
Consistent with the inhibitory effects of ruxolitinib on cell
migration and EMT progression of TAMR-MCF-7 cells, ruxolitinib
(0.1–10 µM) reduced VEGF mRNA levels in TAMR-MCF-7 cells (Fig. 4A). Moreover, a VEGF-luc reporter gene
assay further revealed that VEGF promoter binding activity was
significantly diminished by treatment with 0.3–1 µM ruxolitinib
(Fig. 4B). As ruxolitinib above 3 µM
causes a significant cell death in the reporter gene analysis
condition because of the lipid carrier-based membrane damage
(32), MCF-7 and TAMR-MCF-7 cells
were exposed to 0.3–1 µM ruxolitinib. These results suggest that
the overexpression of VEGF gene transcription in TAMR-MCF-7 cells
was partly a result of JAK2-STAT3 pathway activation. We then
performed chorioallantoic membrane (CAM) assays by placing
TAMR-MCF-7 cells on CAMs to verify the effect of ruxolitinib on
angiogenesis. Numbers of vessel branch points in TAMR-MCF-7 cells
were significantly decreased by ruxolitinib in a
concentration-dependent manner (Fig.
4C).
To estimate the inhibitory effect of ruxolitinib on
tumor growth, we analyzed the tumor weight of TAMR-MCF-7 cells on
CAMs. Implanted tumor growth was strongly suppressed by ruxolitinib
(0.3–3 µM) (Fig. 4D), which implies
that JAK2-STAT3 signaling is also critical to in vivo tumor
growth in TAM-resistant breast cancer.
Discussion
As a selective ER modulator, TAM is the most widely
used treatment for ER-positive breast cancer in both premenopausal
and postmenopausal patients (3).
Approximately 70% of breast cancer patients express ER; therefore,
ER is the major target for luminal breast cancer therapy (33). Nonetheless, intrinsic or acquired
resistance to hormonal therapy is the main challenge to clinicians,
with 50% of advanced metastatic breast cancer patients developing
resistance to TAM (34). Several
studies have suggested potential mechanisms underlying the
acquisition of TAM resistance, and therapeutic targets that may
cause drug sensitivity in TAM-resistant breast cancer.
Overexpression or formation of ER variants, coregulator switching,
abnormal microRNA expression, and genetic polymorphism have been
suggested as possible mechanisms for TAM resistance (34,35). In
addition, the dysregulation of intracellular signaling has been
seen during the acquisition of TAM resistance in several contexts,
including growth factor receptors [epidermal growth factor receptor
(EGFR/HER2), fibroblast growth factor receptor, type-I insulin-like
growth factor receptor (IGFIR)), phosphoinositide 3-kinase
(PI3K)-phosphatase and tensin homolog (PTEN)/AKT/mammalian target
of rapamycin (mTOR), and nuclear factor-κB (NF-κB) (36). In particular, the activation of
EGFR/HER2 or IGFR is a key molecular pathway implicated in TAM
resistance (4). However, clinical
trials combining anti-estrogens with EGFR inhibitors have provided
minimal, or no, clinical benefit to patients over anti-estrogens
alone (37,38). Because hormone resistance to breast
cancer typically appears to be caused by diverse factors,
identifying a new therapeutic target for the prevention of cancer
relapse under continued hormone therapy remains problematic.
Activation of the JAK-STAT pathway has been
considered an essential downstream signaling response to interferon
(IFN)-α, IFN-γ, and IL-6 (39).
Because the JAK-STAT pathway is also involved in the proliferation,
differentiation, migration, and apoptosis of cancer cells (40–42),
diverse inhibitors have been developed to target the JAK family
(43–46). Among these, tofacitinib (Xeljanz) and
ruxolitinib have been approved for the treatment of rheumatoid
arthritis, myelofibrosis, and polycythemia vera (PCV), and are now
in clinical trials for novel therapeutic applications, such as
colitis and various solid tumors (pancreatic cancer, breast cancer,
and non-small-cell lung cancer (NSCLC) (45). It has been reported that ruxolitinib
suppresses Hodgkin's lymphoma (HL) and primary mediastinal B-cell
lymphoma (PMBL) growth in vitro, and increases programmed
cell death against lymphoma cells (47). Ruxolitinib has also been shown to
significantly inhibit tumor growth and improve survival in HL and
PMBL xenograft mice (47). In the
current study, we showed for the first time that STAT3 activity was
highly increased in TAMR-MCF-7 cells, and that ruxolitinib
inhibited cell proliferation and tumor growth in TAM-resistant
breast cancer cells, though the inhibition intensity is marginal.
We believe that the functional role of JAK2, which can be inhibited
by ruxolitinib, is not mainly involved in the growth of TAMR-MCF7
cells. Lim et al (21)
recently demonstrated that ruxolitinib and calcitriol possessed
synergistic anticancer effect in MCF-7 cells. Although ruxolitinib
showed marginal inhibition rate in cell growth of MCF-7 cells,
synergistic inhibition of cell proliferation was observed by
ruxolitinib/calcitriol combination. Based on the finding, we
believe that ruxolitinib could be used as a regimen for combination
therapy in TAM-resistant breast cancer patients. In fact, JAK-STAT
has been reported to be constitutively active in a triple-negative
breast cancer (TNBC) cell line, MDA-MB-cells, and TNBC tumor
tissues (12,48–50), and
an oral JAK inhibitor suppresses tumor growth and metastasis of
TNBC cells in vitro and in vivo (51). In a terminated clinical study, single
administration of ruxolitinib showed no objective responses in a
treatment-refractory TNBC patient population (52). The authors indicated that the
discrepancy between clinical outcomes and on-target activity may be
due to intratumoral heterogeneity within individual TNBC biopsies
(50). Considering our cell-based
results, tamoxifen-resistant breast cancer patients may be able to
enroll in new clinical studies using ruxolitinib.
Increased metastasis and angiogenesis are the
representative clinical characteristics of TAM-resistant breast
cancer (31). We and other groups
have shown that EMT contributes to the enhanced migration ability
of TAM-resistant breast cancer cells, which may be required for
higher rates of metastasis (7–9). During
EMT progression, morphological changes to highly motile mesenchymal
cancer cells are associated with several molecular characteristics,
including increased levels of N-cadherin and vimentin, reduced
levels of E-cadherin and claudins, and the release of matrix
remodeling enzymes such as matrix metalloproteinases (52). Changes in the expression patterns of
these proteins have been shown to result from induction of
EMT-activating transcription factors, such as the snail family of
zinc finger transcription factors, basic helix-loop-helix factors,
and twist (53). In the current
study, protein expression of mesenchymal phenotype markers and
transcription factors upregulated in TAMR-MCF-7 cells was
diminished by ruxolitinib treatment. Consistent with our results,
JAK1/2 inhibition by ruxolitinib suppressed STAT3-mediated
expression of ZEB1 and snail as well as the emergence of a
mesenchymal phenotype in hepatocellular carcinoma (54). It has been also reported that the
JAK-STAT pathway is involved in VEGF gene transcription (55,56).
VEGF mRNA expression and gene transcription were highly upregulated
in TAMR-MCF-7 cells, but were suppressed by ruxolitinib. Consistent
with sqPCR results, a reporter gene assay using the VEGF-luc
promoter showed that ruxolitinib inhibited the activation of VEGF
gene transcription. It has been reported that protein expression
and secretion of VEGF mainly depend on the transcriptional
activation of the VEGF gene (10).
Considering our findings that ruxolitinib inhibits both the
VEGF-luc reporter activity and VEGF mRNA expression, VEGF protein
levels may be down-regulated by ruxolitinib in TAMR-MCF-7 cells.
The fact that we did not quantify the amount of secreted VEGF would
be a limitation of our study. CAM assay results clearly
demonstrated that ruxolitinib can suppress TAMR-MCF7-induced
angiogenesis, where this effect was mediated by VEGF. In
conclusion, ruxolitinib, a potent JAK-STAT pathway inhibitor,
blocked the EMT process and VEGF production, consequently
suppressing TAMR-MCF-7 cell migration and angiogenesis. These
results hold great promise in improving the clinical outcomes of
TAM-resistant breast cancer patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Research Foundation of Korea (NRF) grant (grant nos.
NRF-2018R1A2B2003590 and 2017R1A4A1015860).
Availability of data and materials
The dataset generated and/or analysed during this
study are available from the corresponding author on reasonable
request.
Author's contributions
KWK and JAK designed the research. JWK, JG and JEK
performed the research. JWK and JEK contributed to the western blot
analysis, cell culture, sqPCR, luciferase assay and
immunocytochemistry assay. JG contributed to the CAM assay and data
analysis.
Ethics approval and consent to
participate
The experimental studies were approved by the
Institutional Animal Care and Use Committee of Yeungnam University
(Gyeongsangbuk, Korea).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TAM
|
tamoxifen
|
|
CAM
|
chorioallantoic membrane
|
|
DAPI
|
4′6-diamidino-2-phenylindole
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
EDTA
|
ethylenediamine tetraacetic acid
|
|
EGFR
|
epidermal growth factor
|
|
EMT
|
epithelial to mesenchymal
transition
|
|
ER
|
estrogen receptor
|
|
FBS
|
fetal bovine serum
|
|
FGFR
|
fibroblast growth factor receptor
|
|
IGFR
|
insulin-like growth factor
|
|
IL-6
|
interleukin-6
|
|
JAK
|
Janus kinase
|
|
MMP2
|
matrix metalloproteinase-2
|
|
MMP9
|
matrix metalloproteinase-9
|
|
MPN
|
myeloproliferative neoplasm
|
|
PBS
|
phosphate-buffered saline
|
|
PDGFR
|
platelet-derived growth factor
receptor
|
|
RT
|
room temperature
|
|
SDS
|
sodium dodecyl sulfate
|
|
STAT
|
signal transducer and activator of
transcription
|
|
VEGF
|
vascular endothelial growth factor
|
References
|
1
|
Mueller SO, Clark JA, Myers PH and Korach
KS: Mammary gland development in adult mice requires epithelial and
stromal estrogen receptor alpha. Endocrinology. 143:2357–2365.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Petrangeli E, Lubrano C, Ortolani F,
Ravenna L, Vacca A, Sciacchitano S, Frati L and Gulino A: Estrogen
receptors: New perspectives in breast cancer management. J Steroid
Biochem Mol Biol. 49:327–331. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rose C, Thorpe SM, Andersen KW, Pedersen
BV, Mouridsen HT, Blichert-Toft M and Rasmussen BB: Beneficial
effect of adjuvant tamoxifen therapy in primary breast cancer
patients with high oestrogen receptor values. Lancet. 1:16–19.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ali S and Coombes RC: Endocrine-responsive
breast cancer and strategies for combating resistance. Nat Rev
Cancer. 2:101–112. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Choi HK, Yang JW, Roh SH, Han CY and Kang
KW: Induction of multidrug resistance associated protein 2 in
tamoxifen-resistant breast cancer cells. Endocr Relat Cancer.
14:293–303. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clarke R, Thompson EW, Leonessa F, Lippman
J, McGarvey M, Frandsen TL and Brünner N: Hormone resistance,
invasiveness and metastatic potential in breast cancer. Breast
Cancer Res Treat. 24:227–239. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim MR, Choi HK, Cho KB, Kim HS and Kang
KW: Involvement of Pin1 induction in epithelial-mesenchymal
transition of tamoxifen-resistant breast cancer cells. Cancer Sci.
100:1834–1841. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bui QT, Im JH, Jeong SB, Kim YM, Lim SC,
Kim B and Kang KW: Essential role of Notch4/STAT3 signaling in
epithelial-mesenchymal transition of tamoxifen-resistant human
breast cancer. Cancer Lett. 390:115–125. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Christiansen JJ and Rajasekaran AK:
Reassessing epithelial to mesenchymal transition as a prerequisite
for carcinoma invasion and metastasis. Cancer Res. 66:8319–8326.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim MR, Choi HS, Yang JW, Park BC, Kim JA
and Kang KW: Enhancement of vascular endothelial growth
factor-mediated angiogenesis in tamoxifen-resistant breast cancer
cells: Role of Pin1 overexpression. Mol Cancer Ther. 8:2163–2171.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bollrath J, Phesse TJ, von Burstin VA,
Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T,
Canli O, Schwitalla S, et al: gp130-mediated Stat3 activation in
enterocytes regulates cell survival and cell-cycle progression
during colitis-associated tumorigenesis. Cancer Cell. 15:91–102.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berishaj M, Gao SP, Ahmed S, Leslie K,
Al-Ahmadie H, Gerald WL, Bornmann W and Bromberg JF: Stat3 is
tyrosine-phosphorylated through the interleukin-6/glycoprotein
130/Janus kinase pathway in breast cancer. Breast Cancer Res.
9:R322007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vignais ML, Sadowski HB, Watling D, Rogers
NC and Gilman M: Platelet-derived growth factor induces
phosphorylation of multiple JAK family kinases and STAT proteins.
Mol Cell Biol. 16:1759–1769. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vainchenker W and Constantinescu SN:
JAK/STAT signaling in hematological malignancies. Oncogene.
32:2601–2613. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu H and Jove R: The STATs of cancer-new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Furqan M, Mukhi N, Lee B and Liu D:
Dysregulation of JAK-STAT pathway in hematological malignancies and
JAK inhibitors for clinical application. Biomark Res. 1:52013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu K, Gao H, Wang Q, Wang L, Zhang B, Han
Z, Chen X, Han M and Gao M: Hispidulin suppresses cell growth and
metastasis by targeting PIM1 through JAK2/STAT3 signaling in
colorectal cancer. Cancer Sci. 109:1369–1381. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X,
Li J, Li C, Yan M, Zhu Z, et al: IL-6 secreted by cancer-associated
fibroblasts promotes epithelial-mesenchymal transition and
metastasis of gastric cancer via JAK2/STAT3 signaling pathway.
Oncotarget. 8:20741–20750. 2017.PubMed/NCBI
|
|
19
|
Wei Z, Jiang X, Qiao H, Zhai B, Zhang L,
Zhang Q, Wu Y, Jiang H and Sun X: STAT3 interacts with Skp2/p27/p21
pathway to regulate the motility and invasion of gastric cancer
cells. Cell Signal. 25:931–938. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Quintás-Cardama A, Vaddi K, Liu P,
Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, et
al: Preclinical characterization of the selective JAK1/2 inhibitor
INCB018424: Therapeutic implications for the treatment of
myeloproliferative neoplasms. Blood. 115:3109–3117. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lim ST, Jeon YW, Gwak H, Kim SY and Suh
YJ: Synergistic anticancer effects of ruxolitinib and calcitriol in
estrogen receptor-positive, human epidermal growth factor receptor
2-positive breast cancer cells. Mol Med Rep. 17:5581–5588.
2018.PubMed/NCBI
|
|
22
|
Tavallai M, Booth L, Roberts JL, McGuire
WP, Poklepovic A and Dent P: Ruxolitinib synergizes with DMF to
kill via BIM+BAD-induced mitochondrial dysfunction and via reduced
SOD2/TRX expression and ROS. Oncotarget. 7:17290–17300. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tavallai M, Booth L, Roberts JL,
Poklepovic A and Dent P: Rationally repurposing ruxolitinib (Jakafi
(®)) as a solid tumor therapeutic. Front Oncol.
6:1422016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Knowlden JM, Hutcheson IR, Jones HE,
Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE and Nicholson
RI: Elevated levels of epidermal growth factor receptor/c-erbB2
heterodimers mediate an autocrine growth regulatory pathway in
tamoxifen-resistant MCF-7 cells. Endocrinology. 144:1032–1044.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Auerbach R, Kubai L, Knighton D and
Folkman J: A simple procedure for the long-term cultivation of
chicken embryos. Dev Biol. 41:391–394. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee JS, Kang Y, Kim JT, Thapa D, Lee ES
and Kim JA: The anti-angiogenic and anti-tumor activity of
synthetic phenylpropenone derivatives is mediated through the
inhibition of receptor tyrosine kinases. Eur J Pharmacol.
677:22–30. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Religa P, Cao R, Religa D, Xue Y,
Bogdanovic N, Westaway D, Marti HH, Winblad B and Cao Y: VEGF
significantly restores impaired memory behavior in Alzheimer's mice
by improvement of vascular survival. Sci Rep. 3:20532013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Al Zaid Siddiquee K and Turkson J: STAT3
as a target for inducing apoptosis in solid and hematological
tumors. Cell Res. 18:254–267. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Siveen KS, Sikka S, Surana R, Dai X, Zhang
J, Kumar AP, Tan BK, Sethi G and Bishayee A: Targeting the STAT3
signaling pathway in cancer: Role of synthetic and natural
inhibitors. Biochim Biophys Acta. 1845:136–154. 2014.PubMed/NCBI
|
|
31
|
Marson LP, Kurian KM, Miller WR and Dixon
JM: The effect of tamoxifen on breast tumour vascularity. Breast
Cancer Res Treat. 66:9–15. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kongkaneramit L, Sarisuta N, Azad N, Lu Y,
Iyer AK, Wang L and Rojanasakul Y: Dependence of reactive oxygen
species and FLICE inhibitory protein on lipofectamine-induced
apoptosis in human lung epithelial cells. J Pharmacol Exp Ther.
325:969–977. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yager JD and Davidson NE: Estrogen
carcinogenesis in breast cancer. N Engl J Med. 354:270–282. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garcia-Becerra R, Santos N, Diaz L and
Camacho J: Mechanisms of resistance to endocrine therapy in breast
cancer: Focus on signaling pathways, miRNAs and genetically based
resistance. Int J Mol Sci. 14:108–145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Goetz MP, Rae JM, Suman VJ, Safgren SL,
Ames MM, Visscher DW, Reynolds C, Couch FJ, Lingle WL, Flockhart
DA, et al: Pharmacogenetics of tamoxifen biotransformation is
associated with clinical outcomes of efficacy and hot flashes. J
Clin Oncol. 23:9312–9318. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hosford SR and Miller TW: Clinical
potential of novel therapeutic targets in breast cancer: CDK4/6,
Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways.
Pharmgenomics Pers Med. 7:203–215. 2014.PubMed/NCBI
|
|
37
|
Smith IE, Walsh G, Skene A, Llombart A,
Mayordomo JI, Detre S, Salter J, Clark E, Magill P and Dowsett M: A
phase II placebo-controlled trial of neoadjuvant anastrozole alone
or with gefitinib in early breast cancer. J Clin Oncol.
25:3816–3822. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Johnston SR, Martin LA, Leary A, Head J
and Dowsett M: Clinical strategies for rationale combinations of
aromatase inhibitors with novel therapies for breast cancer. J
Steroid Biochem Mol Biol. 106:180–186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Matthes T, Manfroi B and Huard B:
Revisiting IL-6 antagonism in multiple myeloma. Crit Rev Oncol
Hematol. 105:1–4. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Darnell JE Jr, Kerr IM and Stark GR:
Jak-STAT pathways and transcriptional activation in response to
IFNs and other extracellular signaling proteins. Science.
264:1415–1421. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Taga T, Hibi M, Hirata Y, Yamasaki K,
Yasukawa K, Matsuda T, Hirano T and Kishimoto T: Interleukin-6
triggers the association of its receptor with a possible signal
transducer, gp130. Cell. 58:573–581. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
O'Shea JJ, Schwartz DM, Villarino AV,
Gadina M, McInnes IB and Laurence A: The JAK-STAT pathway: impact
on human disease and therapeutic intervention. Annu Rev Med.
66:311–328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Aparicio-Siegmund S, Sommer J, Monhasery
N, Schwanbeck R, Keil E, Finkenstädt D, Pfeffer K, Rose-John S,
Scheller J and Garbers C: Inhibition of protein kinase II (CK2)
prevents induced signal transducer and activator of transcription
(STAT) 1/3 and constitutive STAT3 activation. Oncotarget.
5:2131–2148. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Buchert M, Burns CJ and Ernst M: Targeting
JAK kinase in solid tumors: Emerging opportunities and challenges.
Oncogene. 35:939–951. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang S, Luo C, Gu Q, Xu Q, Wang G, Sun H,
Qian Z, Tan Y, Qin Y, Shen Y, et al: Activating JAK1 mutation may
predict the sensitivity of JAK-STAT inhibition in hepatocellular
carcinoma. Oncotarget. 7:5461–5469. 2016.PubMed/NCBI
|
|
47
|
Lee S, Shah T, Yin C, Hochberg J, Ayello
J, Morris E, van de Ven C and Cairo MS: Ruxolitinib significantly
enhances in vitro apoptosis in Hodgkin lymphoma and primary
mediastinal B-cell lymphoma and survival in a lymphoma xenograft
murine model. Oncotarget. 9:9776–9788. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Buettner R, Mora LB and Jove R: Activated
STAT signaling in human tumors provides novel molecular targets for
therapeutic intervention. Clin Cancer Res. 8:945–954.
2002.PubMed/NCBI
|
|
49
|
Bharti AC, Donato N and Aggarwal BB:
Curcumin (diferuloylmethane) inhibits constitutive and
IL-6-inducible STAT3 phosphorylation in human multiple myeloma
cells. J Immunol. 171:3863–3871. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Stover DG, Gil Del Alcazar CR, Brock J,
Guo H, Overmoyer B, Balko J, Xu Q, Bardia A, Tolaney SM, Gelman R,
et al: Phase II study of ruxolitinib, a selective JAK1/2 inhibitor,
in patients with metastatic triple-negative breast cancer. NPJ
Breast Cancer. 4:102018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lee HJ, Seo NJ, Jeong SJ, Park Y, Jung DB,
Koh W, Lee HJ, Lee EO, Ahn KS, Ahn KS, et al: Oral administration
of penta-O-galloyl-β-D-glucose suppresses triple-negative breast
cancer xenograft growth and metastasis in strong association with
JAK1-STAT3 inhibition. Carcinogenesis. 32:804–811. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Garg M: Epithelial plasticity and cancer
stem cells: Major mechanisms of cancer pathogenesis and therapy
resistance. World J Stem Cells. 9:118–126. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sánchez-Tilló E, Liu Y, de Barrios O,
Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A
and Postigo A: EMT-activating transcription factors in cancer:
Beyond EMT and tumor invasiveness. Cell Mol Life Sci. 69:3429–3456.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Smigiel JM, Parameswaran N and Jackson MW:
Potent EMT and CSC phenotypes are induced by Oncostatin-M in
pancreatic cancer. Mol Cancer Res. 15:478–488. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang SW and Sun YM: The IL-6/JAK/STAT3
pathway: Potential therapeutic strategies in treating colorectal
cancer (Review). Int J Oncol. 44:1032–1040. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Huang Y, de Reyniès A, de Leval L, Ghazi
B, Martin-Garcia N, Travert M, Bosq J, Brière J, Petit B, Thomas E,
et al: Gene expression profiling identifies emerging oncogenic
pathways operating in extranodal NK/T-cell lymphoma, nasal type.
Blood. 115:1226–1237. 2010. View Article : Google Scholar : PubMed/NCBI
|