Introduction
Acute myeloid leukemia (AML) is a type of leukemia
characterized by the rapid proliferation of blood-forming cells of
myeloid lineage and occurs with increasing frequency in elderly
patients (1). Chemotherapy,
conventionally a combination of anthracyclines and cytarabine (also
known as AraC), is used to treat AML patients, and the complete
remission rate (CR) is ~65–85% (1).
However, for patients older than 60 years of age, the CR rate is
lower, and they cannot endure a high intensity therapy due to the
toxicities of the chemotherapeutic agents such as myelosuppression.
Thus, there are unmet medical needs for AML patients, especially
for elderly patients aged over 60 years that require a low-toxicity
therapy.
Novel therapeutic approaches to treat AML are under
investigation such as proteasome inhibitors and DNA
methyltransferase inhibitors (2).
Fms-like tyrosine kinase 3 (FLT3), a member of the receptor
tyrosine kinase family, is a valuable target in the AML drug
development area. FLT3 is activated by the FLT3 ligand (FLT3LG)
upon binding and transduces a variety of signals, including the
proliferation, survival, and differentiation of hematopoietic
precursor cells (3). Constitutively
activating mutations of FLT3 were observed in AML patients. The
most common type of activating mutation is an internal tandem
duplication of the FLT3 juxtamembrane domain (FLT3-ITD) (4,5).
Mutations in the kinase domain (FLT3 D835) also have resulted in
the constitutive activation of FLT3 (6). FLT3-ITD mutation was reported to
constitutively activate downstream signal transducer and activator
of transcription 5 (STAT5) as well as induce the dysregulated cell
growth (7). FLT3 mutation is found
in approximately 1/3 of all AML patients, and it is associated with
a poor prognosis (8,9).
The FLT3 inhibitors evaluated in clinical trials for
AML therapy can be divided into three generations (10). Lestaurtinib, midostaurin, and
tandutinib are first generation inhibitors with modest efficacy.
Problems disclosed in first generation inhibitors (for example,
maintaining an effective plasma concentration) were resolved in the
second generation inhibitors, KW-2449 and quizartinib. The high
potency of KW-2449 and quizartinib made it possible to achieve an
effective plasma concentration in AML patients. The third
generation inhibitors (crenolanib and ASP2215) try to overcome the
resistance to FLT3 inhibitors observed in clinical trials.
Secondary mutations of FLT3 have emerged as resistant mechanisms,
and crenolanib has exhibited a potency against both FLT3-ITD and
FLT3-ITD with a secondary mutation (11). Currently, midostaurin is approved by
the FDA for the FLT3 mutation positive AML patients.
Continued discovery of novel chemical and
pharmacological information for FLT3 inhibitors is required. The
main reason for this is due to the emergence of resistant cancer
cells against the FLT3 inhibitor treatment (12). Indirubin derivatives as FLT3
inhibitors were previously reported by our group (13). Further optimization was carried out
(14), and a 5-carboxy indirubin
derivative (LDD-1076) exhibited the potent inhibition of FLT3
activity without any anti-proliferative activity. Corresponding
5-carboxy ester form of LDD-1076 (LDD-1075) have a strong growth
inhibitory activity with a relatively weak FLT3 inhibition. In this
study, LDD-1076 and LDD-1076 were characterized with an
antileukemic activity, and the bioconversion of LDD-1075 to
LDD-1076 was identified.
Materials and methods
Cell cultures
The MV4-11 human AML cell lines were purchased from
ATCC (Rockville, MD, USA). The MV4-11 cells harbors FLT3-ITD
mutation, which confers the constitutive kinase activity of FLT3.
The cells were cultured in an RPMI medium (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) supplemented with 10% fetal bovine serum
and 1% penicillin/streptomycin. The cultured cells were incubated
at 37°C with 5% CO2.
For the cytotoxicity assay, the cells were seeded in
a 96-well plate (15,000 cells per well) and incubated with the
indicated compound for 72 h. As a negative control, the cells were
treated with the vehicle only [i.e., dimethyl sulfoxide (DMSO)]. A
final concentration of 0.5% DMSO was used in all treatment groups
to exclude the effects of DMSO on the cell cytotoxicity. The cell
viability was measured by a tetrazolium-based assay with an
EZ-Cytox Cell Viability Assay kit (Daeil Lab Service Co., Ltd.,
Seoul, Korea). The half-maximal inhibitory concentration
(IC50) was calculated by nonlinear regression with the
Prism software, version 5.01 (GraphPad, La Jolla, CA, USA).
Chemical synthesis
The LDD-1075 and LDD-1076 compounds were chemically
synthesized in Professor Yong-Chul Kim's laboratory. The synthetic
scheme and analytical data of LDD-1075 and LDD-1076 are previously
described (14).
In vitro kinase assay
The inhibition of the FLT3 recombinant kinase
activity was measured using homogeneous, time-resolved fluorescence
(HTRF) assays. Recombinant proteins containing the FLT3 kinase
domain were purchased from Carna Biosciences (Kobe, Japan). The
optimal enzyme, ATP, and substrate concentrations were established
using an HTRF KinEASE kit (Cisbio, Codolet, France) according to
the manufacturer's instructions. The enzymes were mixed with
serially diluted compounds and peptide substrates in a kinase
reaction buffer [50 mM HEPES (pH 7.0), 500 µM ATP, 0.1 mM sodium
orthovanadate, 5 mM MgCl2, 1 µM DTT, 0.01% bovine serum
albumin (BSA), and 0.02% NaN3]. After adding the
reagents for detection, the TR-FRET signal was measured with a
Victor multilabel reader (Perkin Elmer, Waltham, MA, USA). The
IC50 was calculated by nonlinear regression with the
Prism software. Met, EGFR, FAK, Jak2, and Jak3 in vitro
kinase assays were also carried out using HTRF assay technology in
order to determine the selectivity against kinases.
Bioconversion of LDD-1075 to LDD-1076
in MV4-11 cell lysate
To determine whether LDD-1076 can be formed from
LDD-1075 in the MV4-11 cells, the whole cell lysates of MV4-11
cells were used. MV4-11 cells were suspended in PBS and disrupted
by three freeze/thaw cycles followed by sonication. The LDD-1075
compound was incubated with MV4-11 cell lysates at 37°C for 0, 15,
30, 60, and 90 min. The final incubation solutions contained 10 µM
LDD-1075, 1.2 mM NADPH, 1 mg/ml (total protein) cell lysates, and
100 mM phosphate buffer (pH 7.4). At each time point, the reaction
was terminated by removing itfrom the water bath and subsequently
adding100 µl of ice-cold acetonitrile containing 0.1 µg/ml of
internal standard (cilostazol) to a 50 µl aliquot of the reaction
mixture. The incubation solutions were then centrifuged, and the
concentration of LDD-1076 in the supernatants was analyzed by
LC-MS/MS.
Five microliters of the supernatant were injected
into the LC-MS/MS system. The LC-MS/MS system consisted of an
Agilent 1260® HPLC system (Agilent Technologies Inc.,
Hilden, Germany) and an Agilent 6460® triple-quadrupole
mass spectrometer (Agilent Technologies Inc., Singapore). The HPLC
mobile phases consisted of 0.1% formic acid (A) and 0.1% formic
acid in 90% acetonitrile (B). Chromatographic separation was
achieved on a reversed-phase Kinetex® C18
column (50×2.6 mm, 2.6 µm; Phenomenex, Torrance, CA, USA) using a
gradient elution at a flow rate of 0.3 ml/min. The total run time
was 4.5 min. Multiple reaction monitoring (MRM) detection in the
positive ion mode was employed using nitrogen as the collision gas
with a dwell time of 50 ms for each transition; the MRM transitions
monitored were m/z 322→260 for LDD-1076 and m/z
370→288 for the internal standard. The lower limit of the
quantitation of LDD-1076 in the reaction mixture was 10 nM. The
values of coefficients of correlation (R) were more than 0.998.
Flow cytometry analyses
MV4-11 cells were grown in 24-well plates (500,000
cells per well) and treated with LDD-1075 for 48 h. The cells were
fixed with 3.7% paraformaldehyde and treated with RNase A (50
µg/ml). The cells were then stained with propidium iodide (PI;
Sigma-Aldrich; Merck KGaA) and subjected to flow cytometry with an
Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA). The
data were analyzed by the BD Accuri C6 software (BD
Biosciences).
For the terminal deoxynucleotidyl transferase dUTP
nick end labeling (TUNEL) assay, the APO-DIRECT kit from BD
Biosciences was used (15). Cells
were grown in 6-well plates and treated with LDD-1075 for 24 h.
Cells were fixed with 2% formaldehyde for 30 min, washed twice in
PBS, and stored in 70% ethanol at −20°C until analysis. After
washing in wash buffer, cells were then incubated at 37°C for 60
min in a ratio of enzyme/FITC label solution, according to the
manufacturer's instructions. After a final wash in rinse buffer and
resuspension in PI/RNase solution, FACS analysis was performed.
Immunoblotting
Cells were lysed with an SDS lysis buffer (12 mM
Tris-Cl, pH 6.8, 5% glycerol, and 0.4% SDS) and 10~20 µg of protein
was subjected to electrophoresis on a 10% SDS-polyacrylamide gel
followed by western blotting. Antibodies against phospho-STAT5
(p-STAT5; cat. no. 9351, 1:1,000), PARP (cat. no. 9542, 1:1,000),
and cyclin D1 (cat. no. 2922, 1:1,000 were purchased from Cell
Signaling Technology (Danver, MA, USA). Antibody against STAT5
(cat. no. sc-835, 1:1,000) was from Santa Cruz (Santa Cruz, CA,
USA), and an antibody against β-actin was obtained from
Sigma-Aldrich; Merck KGaA (cat. no. A5441, 1:5,000).
Statistical analysis
The results were obtained from two or three
independent experiments. Data are expressed as the mean ± standard
error of the mean. Statistical significance was assessed using the
Student's t-test or one-way analysis of variance followed by
Tukey's post-hoc test using SPSS software for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of LDD-1075 and LDD-1076 on
the FLT3 kinase activity and cell cytotoxicity
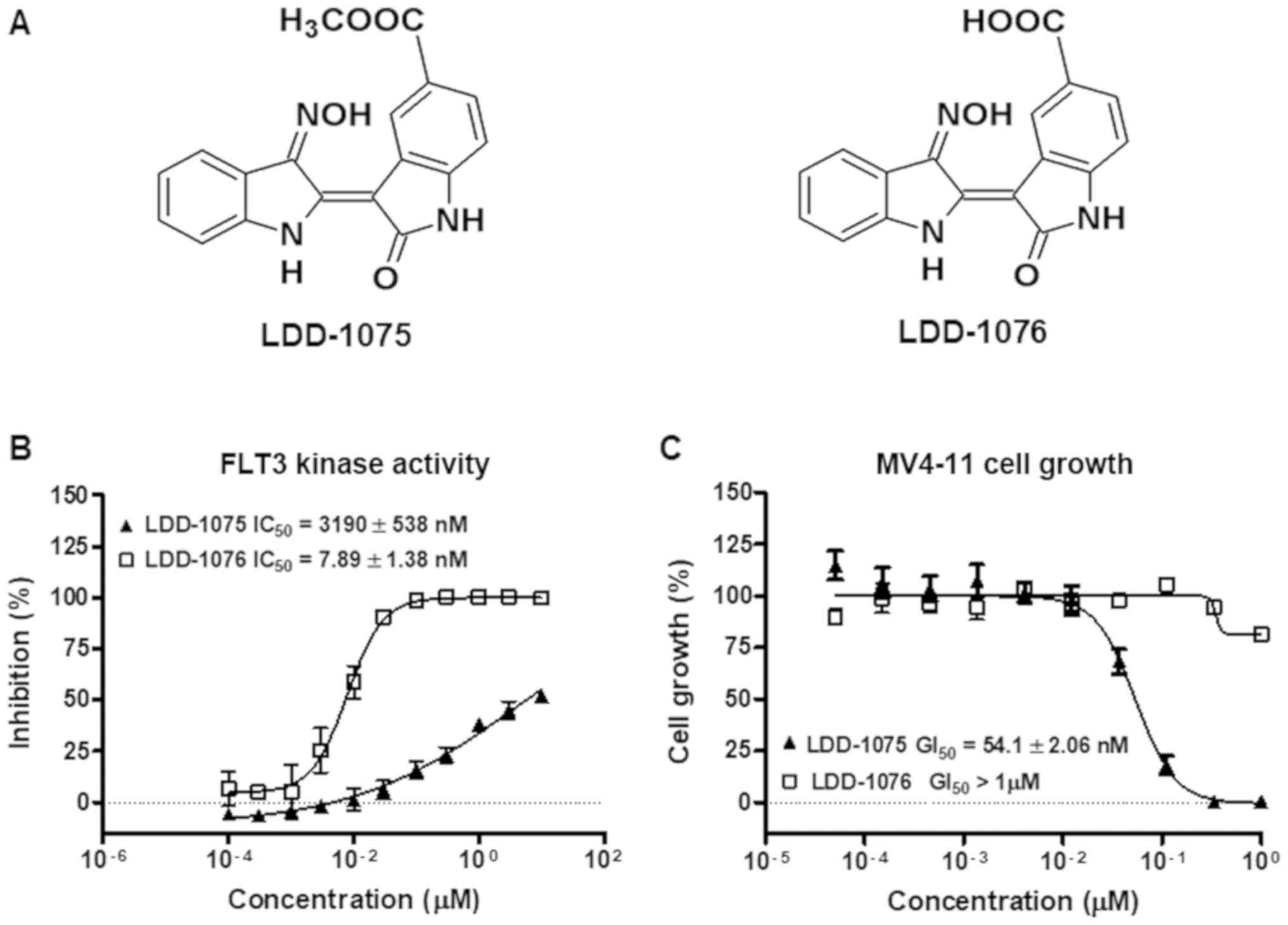
LDD-1075 and LDD-1076 are indirubin derivatives and
LDD-1075 is the ester form of LDD-1076 (Fig. 1A). LDD-1075 and LDD-1076 were
subjected to a FLT3 kinase activity assay in vitro. As shown
in Fig. 1B, the FLT3 kinase activity
inhibition was measured at the indicated concentrations of the LDD
compounds. LDD-1076 exhibited a potent activity with an
IC50 of 7.89±1.38 nM, while LDD-1075 showed a relatively
weak activity against FLT3 (IC50 3.19±0.538 µM). The
cytotoxic effect of the LDD compounds on the MV4-11 cells was
measured (Fig. 1C). The MV4-11 cell
line is AML cells with the constitutive active form of FLT3. In
contrast with the results of the kinase activity assay, LDD-1076
did not affect the MV4-11 cell growth. Interestingly, LDD-1075,
which exhibited a relatively low potency in the FLT3 kinase
inhibition, exhibited a strong cytotoxic effect against the MV4-11
cells (54.1±2.06 nM).
Table I shows the
kinase selectivity of the LDD-1075 and LDD-1076 compounds. LDD-1075
had an IC50 of more than 10 µM for the kinases tested.
LDD-1076 also showed negligible activity against Met, EGFR, and FAK
(IC50s >10 µM). Unexpectedly, Jak2 and Jak3 were
relatively strongly inhibited by LDD-1076 (IC50: 0.476
and 0.546 µM, respectively).
 | Table I.In vitro inhibition of LDD-1075
and LDD-1076 against activity of selected kinases. |
Table I.
In vitro inhibition of LDD-1075
and LDD-1076 against activity of selected kinases.
| Kinase | IC50, µM
LDD-1075 | LDD-1076 |
|---|
| Met | >10 | >10 |
| EGFR | >10 | >10 |
| FAK | >10 | >10 |
| Jak2 | >10 | 0.476 |
| Jak3 | >10 | 0.546 |
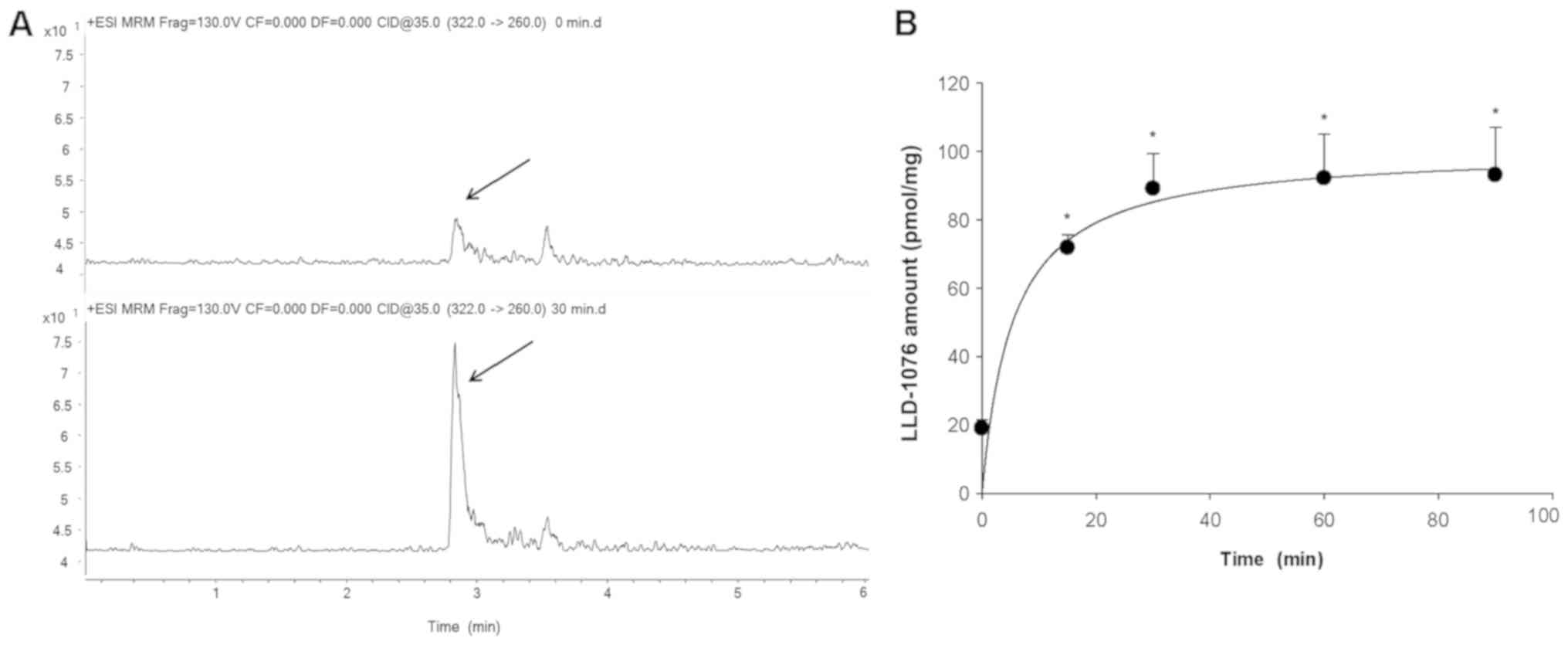
Bioconversion of LDD-1075 to LDD-1076
in MV4-11 cell lysates
The observation in Fig.
1B and C led to a hypothesis that LDD-1075 goes inside the
cells and hydrolyzes to LDD-1076. Generally, the ester form
structure compounds are more permeable to cell membranes than those
of the acid form and can be easily hydrolyzed to the acid form by
various enzymes. According to the hypothesis, LDD-1076 is expected
to not have cytotoxicity despite the high potency of the in
vitro FLT3 assay. This hypothesis also can explain the potent
cytotoxic activity of the LDD-1075 with its relatively low kinase
inhibitory activity. To investigate whether LDD-1076 can be formed
from LDD-1075 in the MV4-11 cells, the LDD-1075 was incubated with
MV4-11 cell lysates and analyzed by LC-MS/MS. The LDD-1076 was
detected after incubation of the LDD-1075 with MV4-11 cell lysates
(Fig. 2A), and the amounts increased
with the incubation time reaching a plateau at 30 min (Fig. 2B). This result suggests that LDD-1075
can be converted to LDD-1076 inside the MV4-11 cells, and LDD-1076
may partly contribute to the high cytotoxicity of LDD-1075 in the
MV4-11 cells.
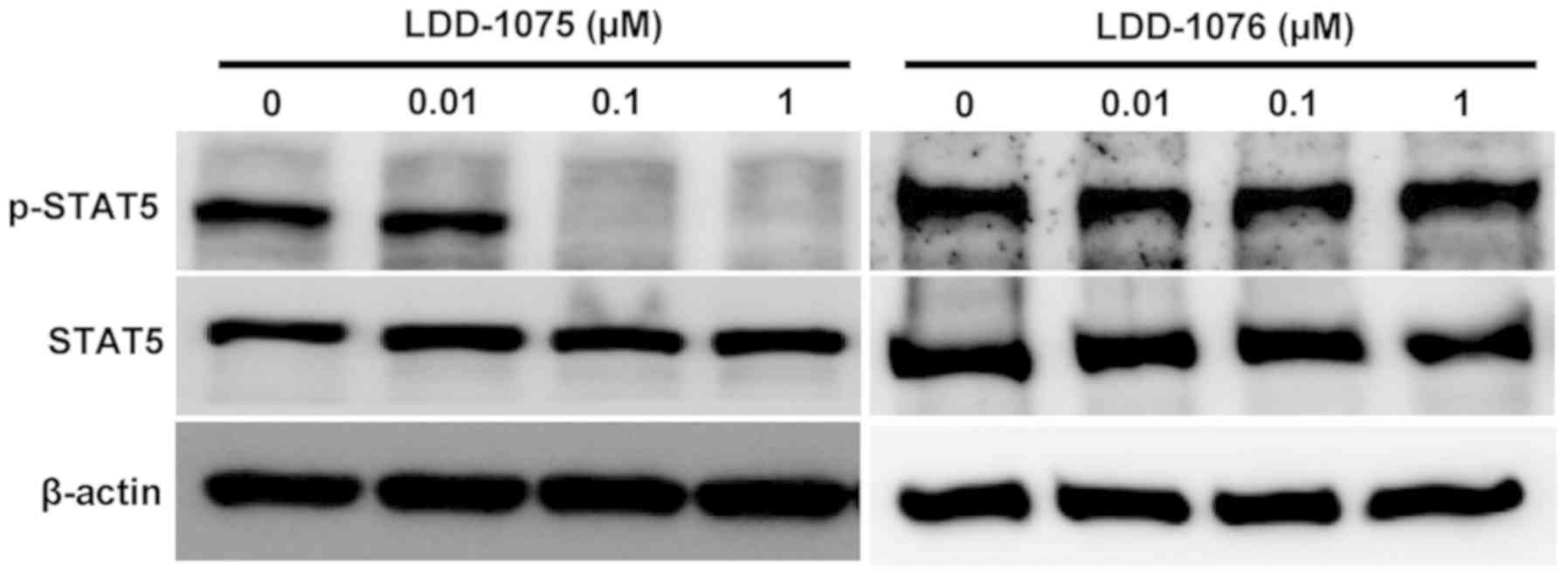
Inhibition of STAT5 phosphorylation by
LDD-1075
The effects of LDD-1075 on the downstream signaling
pathway were studied. The phosphorylated and activated FLT3
transduce signals to STAT5 resulting in an increase in the
phosphorylated form of STAT5. The STAT5 phosphorylation resulted in
the activation of STAT5 transcription factors. As shown in Fig. 3, the dose-dependent downregulation of
the phosphorylated form of STAT5 was observed by the LDD-1075
treatment. In contrast, the LDD-1076 did not affect the
phosphorylation of STAT5, which is consistent with the cytotoxicity
result shown in Fig. 1C. Presumably,
LDD-1076 cannot penetrate into cells. As a result, LDD-1076 cannot
evoke cellular events, while LDD-1075 can penetrate into cells and
be converted to the LDD-1076 inside the cells, resulting in FLT3
inhibition.
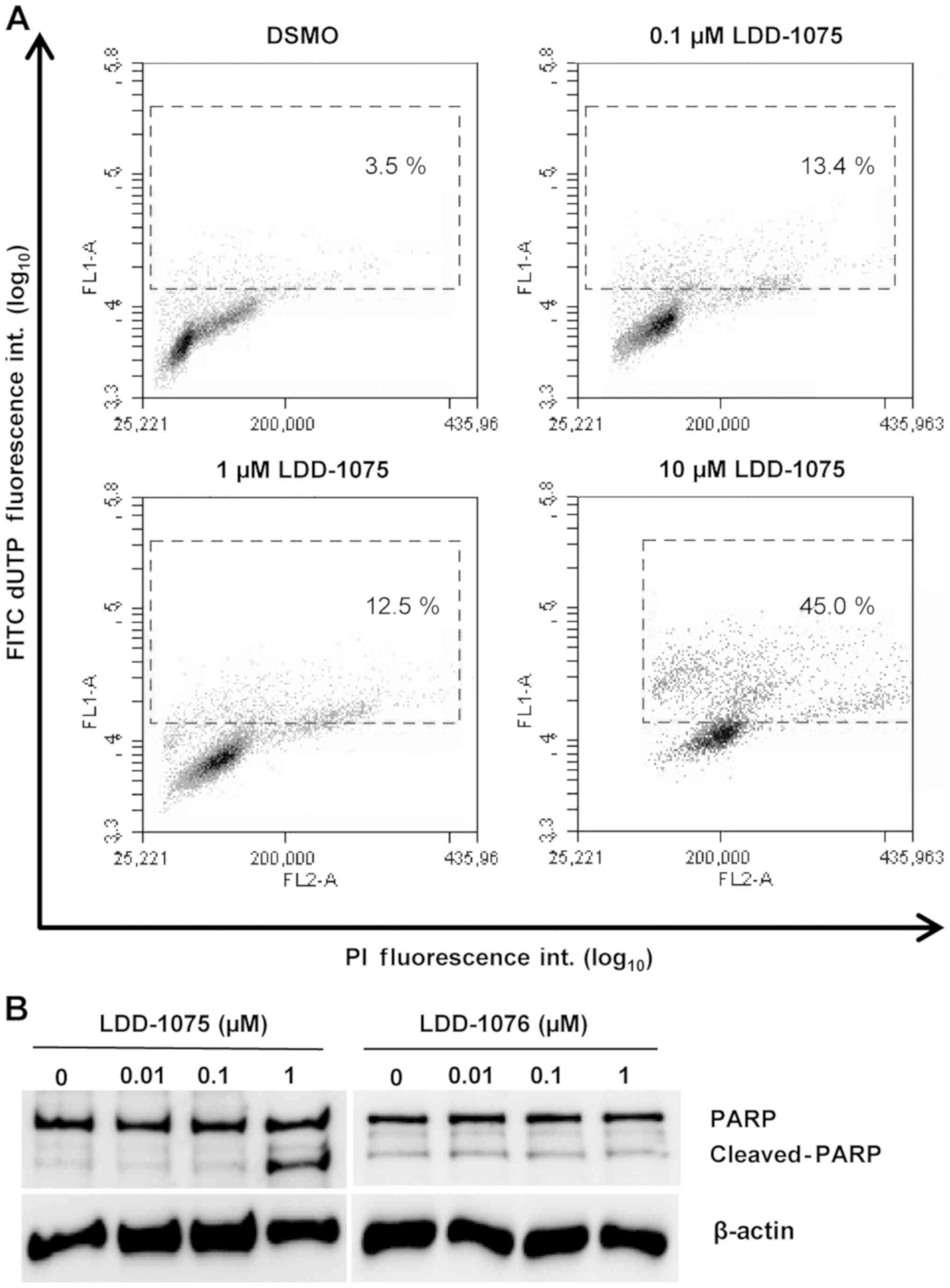
Induction of apoptosis by
LDD-1075
To investigate the effect of LDD-1075 on the
apoptosis of MV4-11 cells, the TUNEL assay was performed. Cells
were treated with 0.1, 1, and 10 µM of LDD-1075 along with the DMSO
treatment as a control for 24 h. Then, the cells were subjected to
the TUNEL assay and flow cytometry, as described in the Materials
and Methods section. As shown in Fig.
4A, the LDD-1075 treatment dose-dependently increased the
apoptotic population with highly FITC-labeled deoxyuridine
triphosphates (FITC-dUTP) and PI stained cells.
The PARP cleavage was measured using anti-PARP and
anti-cleaved PARP antibodies to confirm the apoptotic death of the
MV4-11 cells. As shown in Fig. 4B,
the treatment of LDD-1075 induced the PARP cleavage
dose-dependently. In contrast, the PARP cleavage was not observed
by the LDD-1076 treatment. These results suggest that the
LDD-1075-induced growth inhibition is mediated by an apoptotic cell
death.
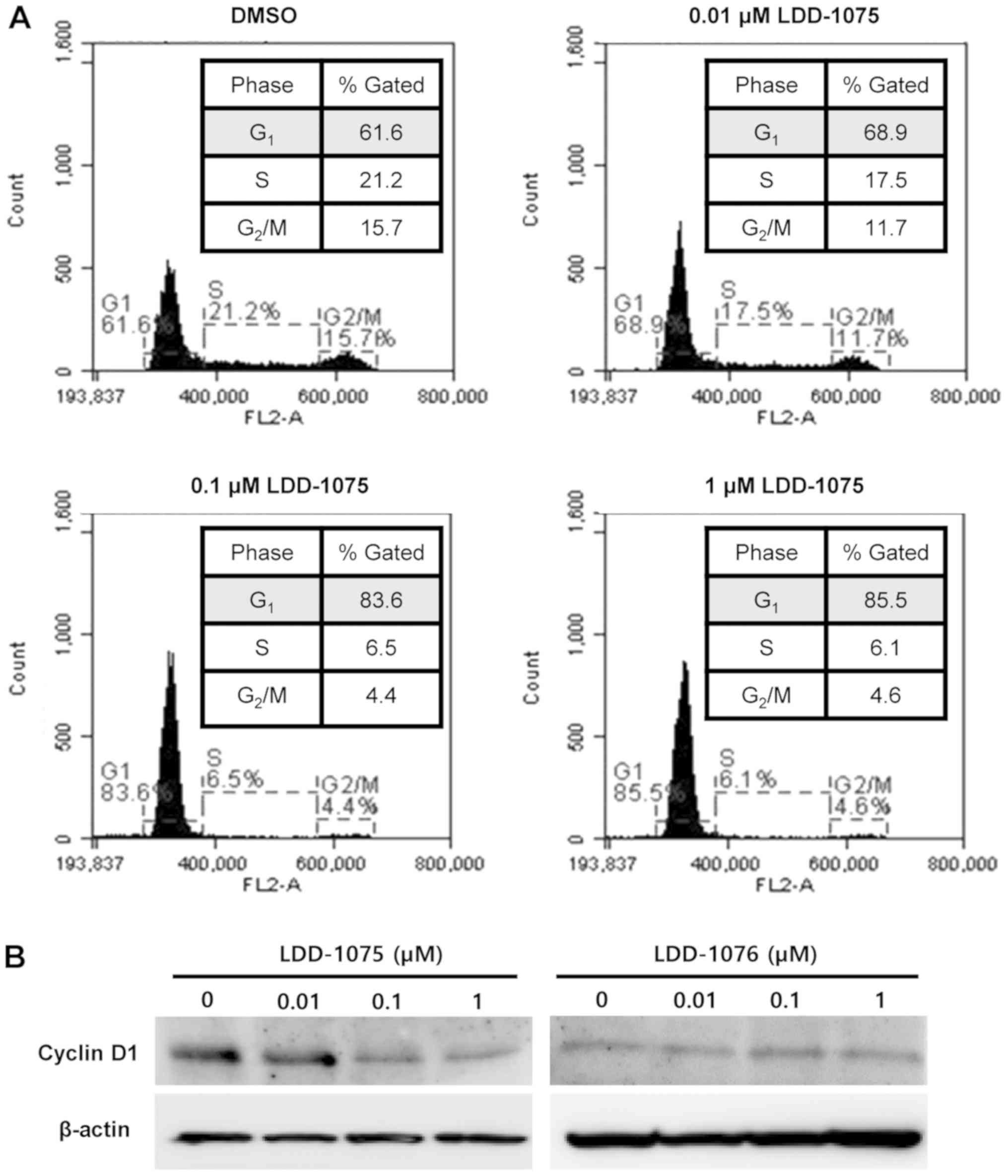
Induction of cell cycle arrest by
LDD-1075
The effect of LDD-1075 on the cell cycle was
investigated. MV4-11 cells were treated with 0.01, 0.1, and 1 µM of
the LDD-1075 compound along with the DMSO treatment as a control
for 24 h. The LDD-1075 treatment induced a cell cycle arrest at the
G1 phase dose-dependently (Fig. 5A).
The cell population at the G1 phase increased from 61.6% (DMSO) to
85.4% (1 µM LDD-1075). Treatment of 10 µM LDD-1075 resulted in the
increase of dead cell population extensively, which made it
difficult to observe cell cycle change. Thus, results up to 1 µM
LDD-1076 treatment was presented in Fig.
5A. Consistent with the G1 phase arrest, the cyclin D1
expression was reduced by the LDD-1075 treatment (Fig. 5B).
Discussion
Recognition of the antitumor effects of the
indirubin derivatives goes back to the traditional Chinese medicine
(TCM) used for to treat of chronic myeloid leukemia (16). A science-based approach that can
explain the anti-leukemic activity of the TCM led to the
characterization of the active ingredient of the traditional
Chinese preparation as indirubin (16,17).
Indirubin was shown to be effective in the clinical trials of
leukemic patients (18), and its
mechanism of action was investigated. DNA and protein synthesis
inhibition by indirubin was presented as a mechanism of the
anti-tumor effects (19,20). Later, indirubin was found to directly
inhibit the activity of cyclin-dependent kinases (CDKs) and
glycogen synthase kinase 3β (GSK3β) (21,22).
Many indirubin derivatives have been synthesized, to
develop anti-cancer drugs based on this information (13,23–25).
Depending on the substitutions of the indirubin derivatives, the
compounds have been shown to inhibit kinases other than CDKs and
GSK3β including, for example, the Jak family kinases (26), phosphorylase kinase (27), Aurora kinases (28), and dual-specificity tyrosine
phosphorylation-regulated kinase (DYRK) (29). Our lab has been investigating
indirubin derivatives as FLT3 inhibitors (13); thus, here, we report compounds with
an improved FLT3 inhibitory activity.
Small molecules with the ester form are commonly
used as prodrugs due to their better permeability through the
membrane. Our observation in Fig. 1B and
C led to a hypothesis that LDD-1075 may act like a prodrug
converting to LDD-1076 inside the cells. LDD-1075, the ester form
of the compound, exhibits a strong growth inhibition but a weak
kinase inhibition in vitro. LDD-1076, the acid from of the
compound, shows a strong kinase inhibition in vitro but
almost no cytotoxicity. The bioconversion of LDD-1075 to LDD-1076
was confirmed by incubating LDD-1075 with the cell lysates
(Fig. 2). This finding will be very
useful when the LDD-1075 compound is developed as a prospective
anti-cancer agent. LDD-1076 will be considered as the active form
converted from LDD-1075.
In conclusion, this study presented the LDD-1075
compound as a potent anti-tumor agent with a mechanism for FLT3
activity inhibition by way of bioconversion to LDD-1076. These
findings will extend our pharmacological understanding in the FLT3
research field.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National Research Foundation (grant no. 2018R1A2B6002081) funded by
the government of Korea (MEST).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KBY, HJL, HJC, JL and JC conducted the experiments
and analyzed the data. HJC, JDH, YCK and SYH contributed to the
study design. SYH was a major contributor in writing the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
DiPiro JT: Pharmacotherapy: A
pathophysiologic approach. McGraw-Hill Medical. (New York).
2016.
|
|
2
|
Tasian SK, Pollard JA and Aplenc R:
Molecular therapeutic approaches for pediatric acute myeloid
leukemia. Front Oncol. 4:552014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shurin MR, Esche C and Lotze MT: FLT3:
Receptor and ligand. Biology and potential clinical application.
Cytokine Growth Factor Rev. 9:37–48. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakao M, Yokota S, Iwai T, Kaneko H,
Horiike S, Kashima K, Sonoda Y, Fujimoto T and Misawa S: Internal
tandem duplication of the flt3 gene found in acute myeloid
leukemia. Leukemia. 10:1911–1918. 1996.PubMed/NCBI
|
|
5
|
Kiyoi H, Towatari M, Yokota S, Hamaguchi
M, Ohno R, Saito H and Naoe T: Internal tandem duplication of the
FLT3 gene is a novel modality of elongation mutation which causes
constitutive activation of the product. Leukemia. 12:1333–1337.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R,
Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C,
et al: Activating mutation of D835 within the activation loop of
FLT3 in human hematologic malignancies. Blood. 97:2434–2439. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hayakawa F, Towatari M, Kiyoi H, Tanimoto
M, Kitamura T, Saito H and Naoe T: Tandem-duplicated Flt3
constitutively activates STAT5 and MAP kinase and introduces
autonomous cell growth in IL-3-dependent cell lines. Oncogene.
19:624–631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schnittger S, Schoch C, Dugas M, Kern W,
Staib P, Wuchter C, Loffler H, Sauerland CM, Serve H, Buchner T, et
al: Analysis of FLT3 length mutations in 1003 patients with acute
myeloid leukemia: Correlation to cytogenetics, FAB subtype and
prognosis in the AMLCG study and usefulness as a marker for the
detection of minimal residual disease. Blood. 100:59–66. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thiede C, Steudel C, Mohr B, Schaich M,
Schakel U, Platzbecker U, Wermke M, Bornhauser M, Ritter M,
Neubauer A, et al: Analysis of FLT3-activating mutations in 979
patients with acute myelogenous leukemia: Association with FAB
subtypes and identification of subgroups with poor prognosis.
Blood. 99:4326–4335. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kiyoi H: Flt3 Inhibitors: Recent advances
and problems for clinical application. Nagoya J Med Sci. 77:7–17.
2015.PubMed/NCBI
|
|
11
|
Galanis A, Ma H, Rajkhowa T, Ramachandran
A, Small D, Cortes J and Levis M: Crenolanib is a potent inhibitor
of FLT3 with activity against resistance-conferring point mutants.
Blood. 123:94–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
El Fakih R, Rasheed W, Hawsawi Y,
Alsermani M and Hassanein M: Targeting FLT3 mutations in acute
myeloid leukemia. Cells. 7(pii): E42018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Choi SJ, Moon MJ, Lee SD, Choi SU, Han SY
and Kim YC: Indirubin derivatives as potent FLT3 inhibitors with
anti-proliferative activity of acute myeloid leukemic cells. Bioorg
Med Chem Lett. 20:2033–2037. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee HJ, Lee J, Jeong P, Choi J, Baek J,
Ahn SJ, Moon Y, Heo JD, Choi YH, Chin YW, et al: Discovery of a
FLT3 inhibitor LDD1937 as an anti-leukemic agent for acute myeloid
leukemia. Oncotarget. 9:924–936. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee J, Kang JS, Choi BY and Keum YS:
Sensitization of 5-fluorouracil-resistant SNUC5 colon cancer cells
to apoptosis by α-Mangostin. Biomol Ther (Seoul). 24:604–609. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiao Z, Hao Y, Liu B and Qian L: Indirubin
and meisoindigo in the treatment of chronic myelogenous leukemia in
China. Leuk Lymphoma. 43:1763–1768. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eisenbrand G, Hippe F, Jakobs S and
Muehlbeyer S: Molecular mechanisms of indirubin and its
derivatives: Novel anticancer molecules with their origin in
traditional Chinese phytomedicine. J Cancer Res Clin Oncol.
130:627–635. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gan WJ, Yang T, Wen S, Liu Y, Tan Z, Deng
C, Wu J and Liu M: Studies on the mechanism of indirubin action in
the treatment of chronic myelocytic leukemia (CML). II.
5′-Nucleotidase in the peripheral white blood cells of CML. Chin
Acad Med Sci Beijing. 6:611–613. 1985.
|
|
19
|
Zhang L, Wu GY, Qiu CC and Gao BH: Effect
of indirubin on DNA synthesis in vitro. Zhongguo Yi Xue Ke Xue Yuan
Xue Bao. 7:112–116. 1985.(In Chinese). PubMed/NCBI
|
|
20
|
Wu GY and Fang FD: Studies on the
mechanism of indirubin action in the treatment of chronic
granulocytic leukemia. II. Effects of indirubin on nucleic acid and
protein synthesis in animal transplantable tumor cells and normal
proliferating cells in vitro (author's transl)]. Zhongguo Yi Xue Ke
Xue Yuan Xue Bao. 2:83–87. 1980.(In Chinese). PubMed/NCBI
|
|
21
|
Hoessel R, Leclerc S, Endicott JA, Nobel
ME, Lawrie A, Tunnah P, Leost M, Damiens E, Marie D, Marko D, et
al: Indirubin, the active constituent of a Chinese antileukaemia
medicine, inhibits cyclin-dependent kinases. Nat Cell Biol.
1:60–67. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leclerc S, Garnier M, Hoessel R, Marko D,
Bibb JA, Snyder GL, Greengard P, Biernat J, Wu YZ, Mandelkow EM, et
al: Indirubins inhibit glycogen synthase kinase-3 beta and
CDK5/p25, two protein kinases involved in abnormal tau
phosphorylation in Alzheimer's disease. A property common to most
cyclin-dependent kinase inhibitors? J Biol Chem. 276:251–260.
2001.PubMed/NCBI
|
|
23
|
Li C, Go Y, Mao Z, Koyano K, Kai Y,
Kanehisa N, Zhu Q, Zhou Z and Wu S: The synthesis, antileukemic
activity and crystal structures of indirubin dervatives. Bull Chem
Soc Jpn. 69:1621–1627. 1996. View Article : Google Scholar
|
|
24
|
Moon MJ, Lee SK, Lee JW, Song WK, Kim SW,
Kim JI, Cho C, Choi SJ and Kim YC: Synthesis and structure-activity
relationships of novel indirubin derivatives as potent
anti-proliferative agents with CDK2 inhibitory activities. Bioorg
Med Chem. 14:237–246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Beauchard A, Ferandin Y, Frere S, Lozach
O, Blairvacq M, Meijer L, Thiery V and Besson T: Synthesis of novel
5-substituted indirubins as protein kinases inhibitors. Bioorg Med
Chem. 14:6434–6443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu L, Nam S, Tian Y, Yang F, Wu J, Wang
Y, Scuto A, Polychronopoulos P, Magiatis P, Skaltsounis L, et al:
6-Bromoindirubin-3′-oxime inhibits JAK/STAT3 signaling and induces
apoptosis of human melanoma cells. Cancer Res. 71:3972–3979. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Begum J, Skamnaki VT, Moffatt C, Bischler
N, Sarrou J, Skaltsounis AL, Leonidas DD, Oikonomakos NG and Hayes
JM: An evaluation of indirubin analogues as phosphorylase kinase
inhibitors. J Mol Graph Model. 61:231–242. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Myrianthopoulos V, Magiatis P, Ferandin Y,
Skaltsounis AL, Meijer L and Mikros E: An integrated computational
approach to the phenomenon of potent and selective inhibition of
Aurora kinases B and C by a series of 7-substituted indirubins. J
Med Chem. 50:4027–4037. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Myrianthopoulos V, Kritsanida M,
Gaboriaud-Kolar N, Magiatis P, Ferandin Y, Durieu E, Lozach O,
Cappel D, Soundararajan M, Filippakopoulos P, et al: Novel inverse
binding mode of indirubin derivatives yields improved selectivity
for DYRK kinases. ACS Med Chem Lett. 4:22–26. 2013. View Article : Google Scholar : PubMed/NCBI
|