Introduction
Hepatocellular carcinoma (HCC) is the main
pathological type of liver cancer. The incidence of HCC in
developed countries has significantly increased in the past few
decades (1). Due to its complexity,
heterogeneity and high recurrence following surgical resection, HCC
ranks the second or third major cause of cancer-associated
mortalities in the world (2,3). The fact that the diagnosis of HCC
primarily depends on the serologic alterations that occur in
advanced HCC restricts the therapeutic options for HCC (4). Thus, the identification of specific
molecular targets for early diagnosis and timely treatment for HCC
is imperative (5).
With the extensive application of gene chip
technologies, abundant expression profile information and screening
of differentially expressed genes (DEGs), biomarkers in tumor
tissues could be detected efficiently by integrating publicly
available datasets (3,6,7). Several
studies based on the integrated analysis of microarray data have
provided valuable insights to the underlying molecular mechanisms
of diseases (7–10). For instance, Gan et al
(10) reported that microRNA
(miR)-145-5p was associated with lymph node metastasis in non-small
cell lung cancer through revealing that the expression levels of
miR-145-5p were significantly lower in these tissues compared with
those in healthy tissues following a meta-analysis and integrated
analysis of microarray data. Guo et al (7) performed an integrated bioinformatics
analysis of four colorectal cancer (CRC) expression profiles in
Gene Expression Omnibus (GEO) and identified 31 key candidate genes
in CRC. Similarly, Liang et al (3) and Zhang et al (11) analyzed a publicly available data of
HCC in GEO and The Cancer Genome Atlas (TCGA) database, and
explored the diagnostic role of miR-133a-3p and miR-224-5p in HCC,
respectively. These findings provide novel and valuable directions
for further research into various cancer types, including HCC. It
was proposed that a thorough re-analysis using integrated
bioinformatics methods combined with the newest datasets would be
innovative, and may provide novel additional insights into the
underlying molecular mechanisms of HCC.
The GEO database is a public database of microarray
profile founded by the National Center for Biotechnology
Information and provides access to high-throughput screening of
abnormally expressed genes in cancerous tissues (7). Numerous studies have explored
microarray data profiling (8,9).
However, although reliable molecular biomarkers have been
clinically observed in patients with HCC, the underlying molecular
mechanisms of HCC remain to be clarified.
The present study identified a number of DEGs in HCC
tissues by integrated analysis of the two newest datasets (GSE76427
and GSE84402) from the GEO database and HCC samples in TCGA
(12,13). Functional enrichment was conducted on
key candidate genes with the DEGs threshold of | log2
(fold change) |≥1.5 in the GEO data or fold change ≥10 in the TCGA
data and P≤0.001. GSE14520 and GSE3500 (14,15), and
the TCGA dataset with expression profiles of cancer and adjacent
non-cancerous tissues were used for validation of candidate genes.
In addition, overall survival (OS) analysis of candidate genes was
performed to analyze the potential of using such genes as
prognostic biomarkers of HCC. The present study may provide novel
insights into the molecular mechanism of HCC and may serve as a
reference for clinical studies in HCC.
Materials and methods
Microarray data information
The gene expression profiles of GSE76427, GSE84402,
GSE14520 and GSE3500 were downloaded from the GEO database
(https://www.ncbi.nlm.nih.gov/), and gene
expression data for HCC and adjacent non-cancerous tissue were
obtained. The newest datasets available (GSE76427 and GSE84402)
were based on the platforms GPL10558 (Illumina HumanHT-12 v4.0
Expression BeadChip; Illumina, Inc., San Diego, CA, USA) and GPL570
(Affymetrix Human Genome U133 Plus 2.0 Array; Affymetrix; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), respectively. GSE76427
data included 115 HCC primary tumor tissues and 52 adjacent
non-tumor tissues (submission date, 30th December 2015) (12). GSE84402 included 14 HCC tissues and
14 adjacent non-tumor tissues (submission date, 14th July 2016)
(13). GSE14520 was based on the
GPL571 (Affymetrix Human Genome U133A 2.0 Array; Affymetrix; Thermo
Fisher Scientific, Inc.) and GPL3921 platforms (Affymetrix HT Human
Genome U133A Array; Affymetrix; Thermo Fisher Scientific, Inc.).
GSE14520 included 66 tumor and paired non-tumor samples (submission
date, 22nd January 2009) (16).
GSE3500 data was based on 13 platforms (GPL2648, GPL2649, GPL2831,
GPL2868, GPL2906, GPL2935, GPL2938, GPL2948, GPL3007, GPL3008,
GPL3009, GPL3010 and GPL3011), and included 102 primary HCC tissues
(from 82 patients), 74 non-tumor tissues (from 72 patients), seven
benign tumor tissues (three adenoma and four focal nodular
hyperplasia), 10 metastatic cancer tissues, and 10 HCC cell lines
(14).
Another expression profile of liver cancer was
obtained from the TCGA database by Cancer RNA-seq Nexus (CRN)
online (http://syslab4.nchu.edu.tw/) using
University of California, Santa Cruz Refseq Gene Array, which
consisted of 84 HCC tissues and 42 adjacent non-tumor tissues.
GSE76427, GSE84402 and TCGA data were used for the identification
of DEGs. The GSE14520 and GSE3500 datasets and TCGA data were used
for the validation of expression profiles. The details and patient
information of the datasets in GEO and TCGA are listed in Table I.
 | Table I.Detailed information of the datasets
in GEO and TCGA patients. |
Table I.
Detailed information of the datasets
in GEO and TCGA patients.
| GEO ID | GSE14520 | GSE3500 | GSE76427 | GSE84402 | TCGA |
|---|
| Total no. of
patients | 22 | 82 | Not mentioned | 14 | 371 |
| Total no. of
samples | 132 | 193 | 167 | 14 | 373 |
| No. of non-tumor
samples | 66 (paired
non-tumor samples) | 74 (non-tumor
samples) | 52 (adjacent
non-tumor tissues) | 14 (>5 cm
laterally from the edge of the cancerous) region | 42 (healthy
tissues) |
| No. of primary HCC
samples | 66 (tumor
samples) | 102 (tumor samples
from HBV+ patients) | 115 (primary tumor
tissues) | 14 (percentage of
tumor cells >70%) | 84 (tumor
samples) |
| Tumor types | Hepatocellular
carcinoma | Hepatocellular
carcinoma | Hepatocellular
carcinoma | Hepatocellular
carcinoma | Hepatocellular
carcinoma |
| Grading of tumors
(TNM stage) | No subdivision | No subdivision | No subdivision | No subdivision | Stage II |
| Pathological
grade | No subdivision | No subdivision | No subdivision | No subdivision | No subdivision |
| Comment | N.A. | N.A. | Percentage of HCC
patients with HBV infection and cirrhosis were 46% and 54%. | N.A. | Using CRN (Cancer
RNA-Seq Nexus tool) for analysis. |
Identification of overexpressed DEGs
in HCC tissues
The raw data from the downloaded datasets GSE76427
and GSE84402 as well as TCGA data were analyzed following
integrated transformation and correlation analysis using Funrich
(version 3.3; http://www.funrich.org/) and Morpheus
software (https://software.broadinstitute.org/morpheus). Data
processing was conducted by two professional analysts. DEGs between
human HCC tissues and paired non-tumor or healthy tissues were
defined using the Student's t-test with a cut-off criterion of
P<0.001 and log2 (fold change) |≥1.5 in GEO data or
fold change |≥10 in TCGA data. Upon overlapping with Funrich
software, candidate genes with highly overexpressed levels in HCC
tissues were identified in the three datasets.
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of
candidates
Functional enrichment analysis of GO and KEGG
pathways was parsed using the online tool Database for Annotation
Visualization and Integrated Discovery (https://david.ncifcrf.gov/), which contains integrated
gene visualization. Funrich software was also utilized to identify
functional enrichment under a threshold of P<0.05.
Construction of protein-protein
interaction (PPI) network and heatmap analysis
DEGs-encoded proteins and PPI network information
data were obtained using the Search Tool for the Retrieval of
Interacting Genes (STRING) database (http://string-db.org). Cytoscape software (version
3.7.0; http://cytoscape.org/) was used for
visualization of the interactions among the candidate DEGs. Heatmap
hot spots with gradient from red to blue color, which were
generated from the TCGA dataset, were used to show the relative
expression level of key candidate genes in HCC.
Validation of the aberrant expression
of UBE2C in HCC based on GEO and TCGA datasets
The expression profiles of candidate genes in DEGs
were validated using another two GEO data (GSE14520 and GSE3500)
with the online tool Oncomine (https://www.oncomine.org/resource/login.html), which
functions as a profiler for intuitive expression of the GEO
database. An independent sample Student's t-test was used to
compare the DEG levels between HCC and non-cancerous tissues. The
online tool Cancer Cell Line Encyclopedia (https://portals.broadinstitute.org/ccle) and
Firebrowse (http://firebrowse.org/) were employed
to evaluate the relative transcription level of candidate genes
among human malignancies and different human HCC cell lines
according to the data obtained from the GEO and TCGA databases.
Survival association of candidate
genes with clinicopathological parameters of patients with HCC
Survival analysis of patients with high expression
of candidate DEGs in TCGA HCC data was performed using Kaplan-Meier
estimator survival curves with several clinicopathological
characteristics taken into consideration, including sex and
pathological stage. Data was obtained from cBioPortal (http://www.cbioportal.org) and Kaplan-Meier Plotter
(http://kmplot.com/analysis/) was used
for analysis. A threshold of P<0.05 was used to set the cut-off
criterion.
Results
Identification of DEGs and
overexpressed candidate genes in human HCC tissues
To date, the GEO database contains the most
comprehensive public microarray/gene data resources, while TCGA
processes the largest quantity of cancer genes. The gene expression
profile of HCC and adjacent non-cancerous tissues was downloaded
from GEO (GSE76427 and GSE84402) and TCGA. Under the inclusion
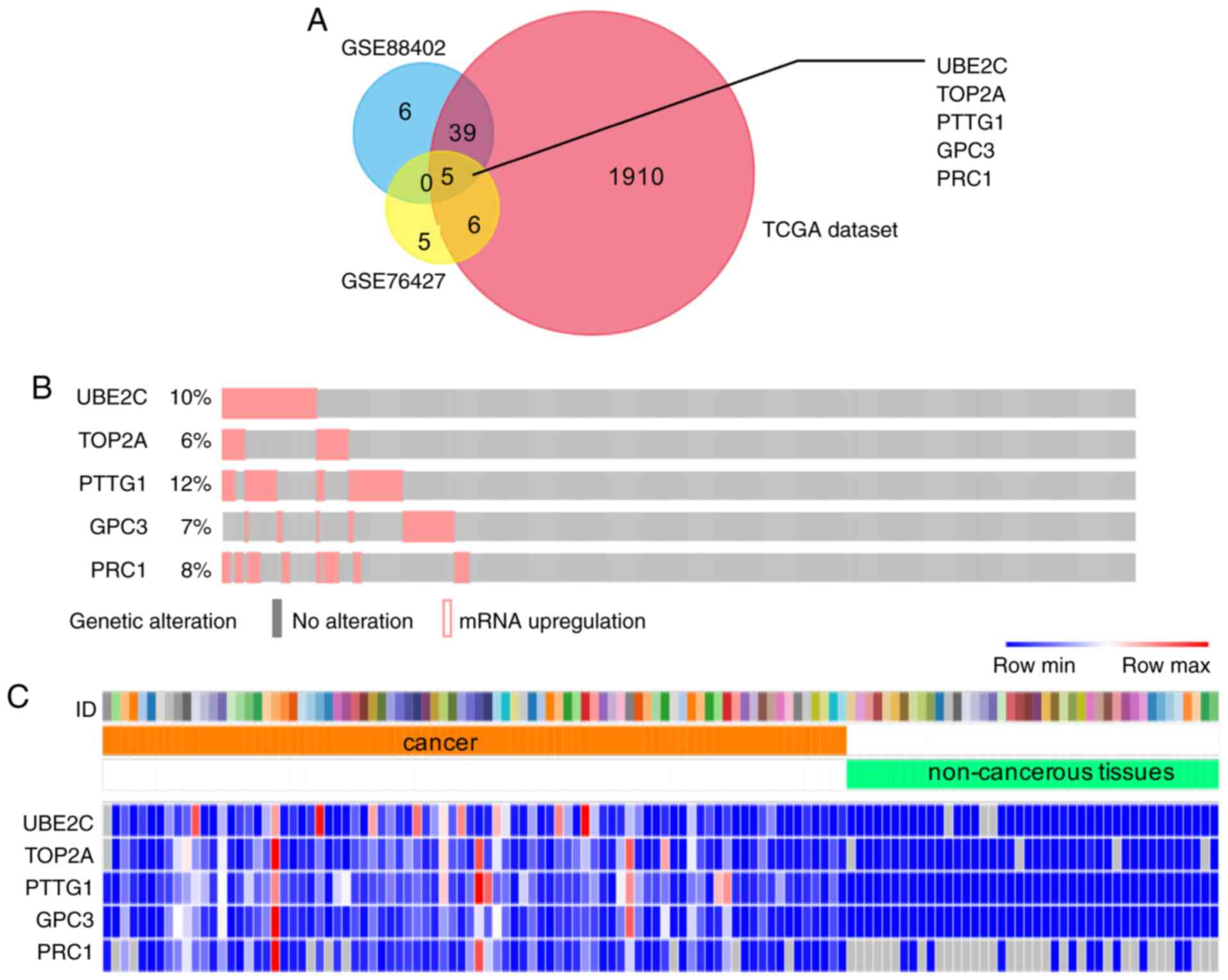
criteria, a total of 1,650 and 1,960 DEGs were identified from
GSE76427, GSE84402 and TCGA data, respectively. Using the FunRich
software, five upregulated genes were identified
[ubiquitin-conjugating enzyme 2C (UBE2C), topoisomerase II α
(TOP2A), pituitary tumor transforming gene 1 (PTTG1), glypican-3
(GPC3) and polycomb-repressive complex 1 (PRC1)], which were
overlapped in three groups and were considered as candidate genes
for selection of HCC biomarkers (Fig.
1A). The OncoPrint created from 373 HCC tissues in the TCGA
database using available data from the cBioPortal website indicated
that 27% (100/373) of clinical cases exhibited gene upregulation in
patients with HCC (Fig. 1B). A
heatmap of these five DEGs in the TCGA data was visualized using
STRING and Morpheus software (Fig.
1C), which partly revealed the significant discrepancy between
human HCC and healthy tissues.
GO and KEGG pathway enrichment
analysis for DEGs
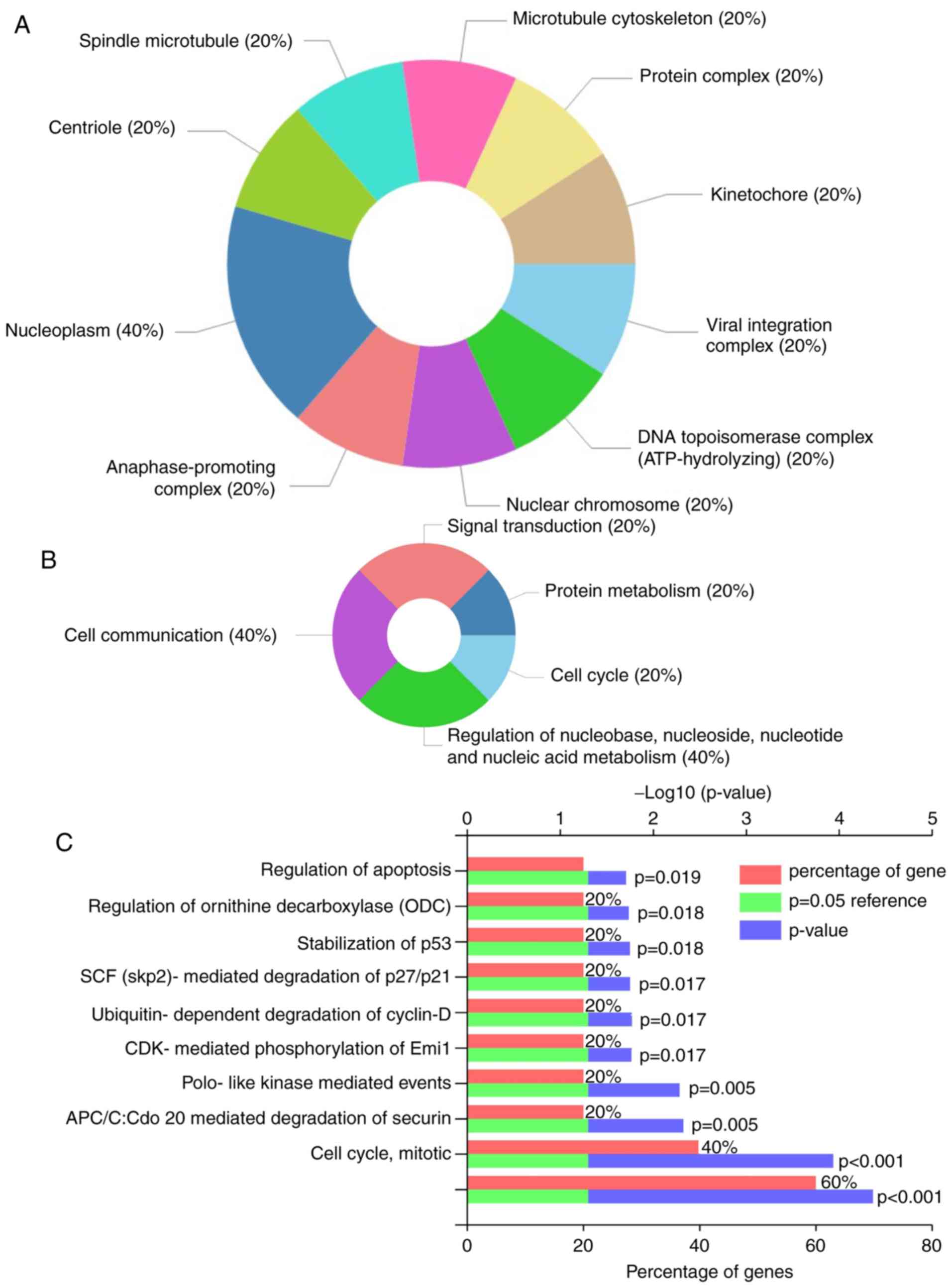
The five overlapped upregulated candidated genes
were subjected to functional enrichment analysis via GO. It was
observed that they were primarily enriched in: i) Cellular
components, including nucleoplasm, nuclear chromosome and
kinetochore and DNA topoisomerase complex (ATP-hydrolyzing)
(Fig. 2A); and ii) biological
processes, including cell communication, signal transduction and
cell cycle (Fig. 2B). Candidate
genes were associated with various functions, including apoptosis
regulation, stabilization of p53 and CDK-mediated phosphorylation,
and the removal of cell division cycle 6 (Fig. 2C). These results suggested that these
five candidate genes serve important biological roles during HCC
development.
KEGG pathway enrichment analysis revealed that PTTG1
was enriched in two pathways, including ‘Oocyte meiosis’ and ‘Cell
cycle’ (P<0.05) (data not shown).
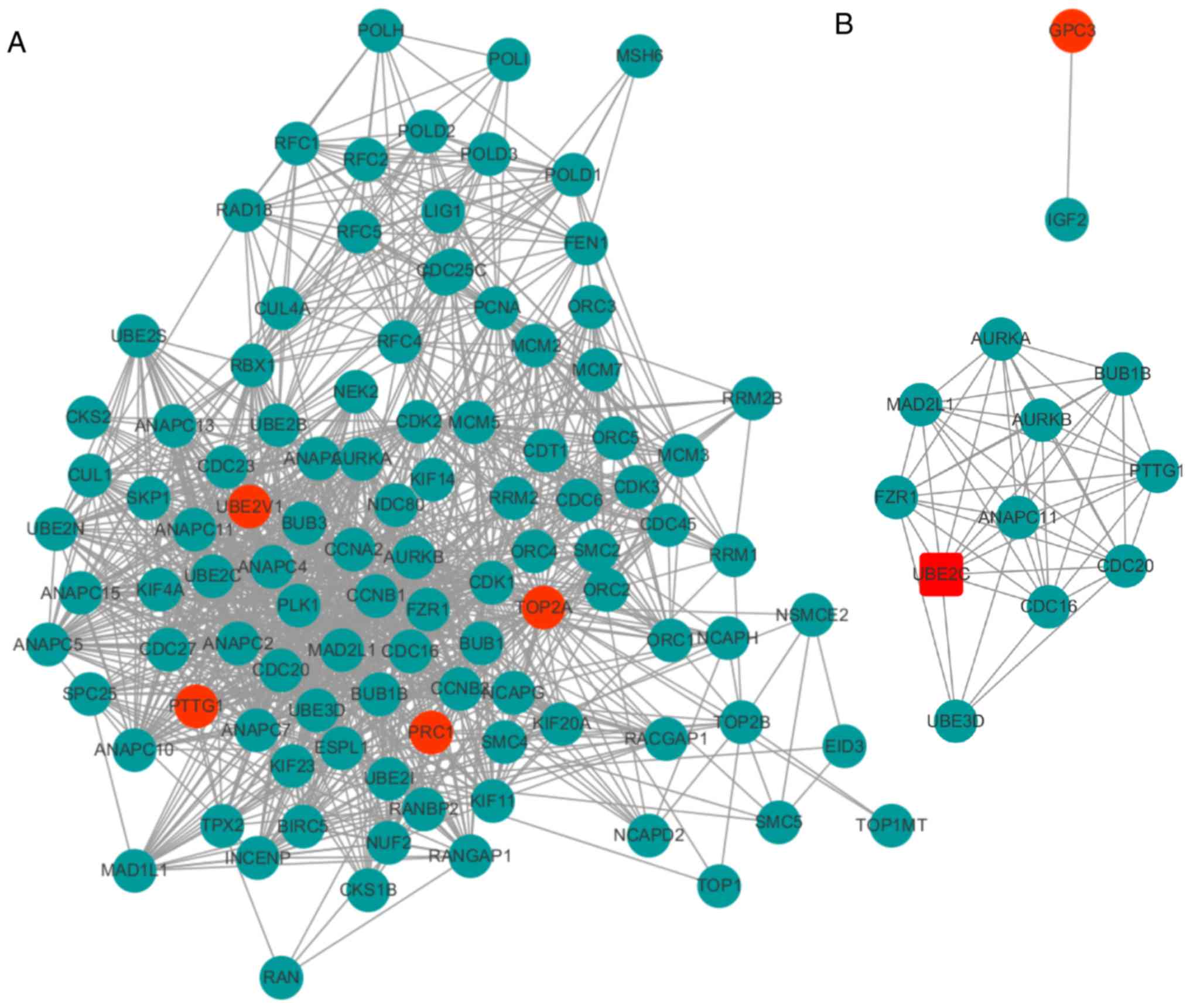
Construction of PPI network
Five DEGs and their interactions were included in a
PPI network, in which there were 105 nodes (proteins) and 1,036
edges (interactions) (Fig. 3A). In
the integrated PPI network, a module consisting of UBE2C and PTTG1,
was identified, which included 11 nodes and 50 edges (Fig. 3B). Based on the expression levels of
these five genes, it was observed that UBE2C exhibited relatively
higher expression levels compared with other genes in HCC tissues
from TCGA. Thus, UBE2C was considered a candidate biomarker for
HCC.
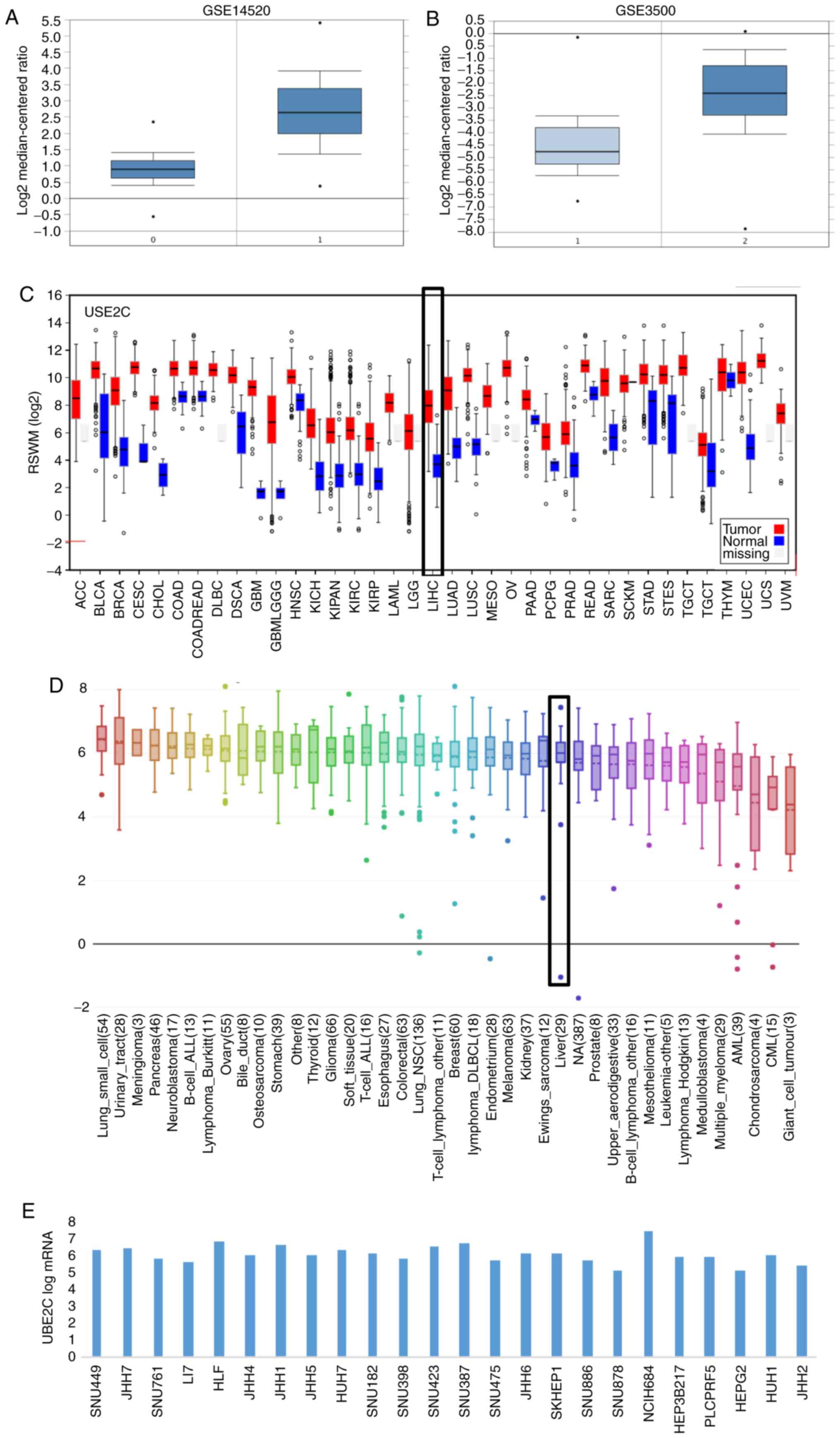
Validation of UBE2C overexpression in
HCC tissues and cell lines
To verify the overexpression of UBE2C in HCC
tissues, the GSE14520 and GSE3500 data were downloaded, and the
expression profile of UBE2C was analyzed. As expected, the
significant overexpression of UBE2C in HCC tissues compared with
adjacent non-cancerous tissues was validated in the datasets
GSE14520 and GSE3500 (Fig. 4A and
B), as well as in other various human malignant tumors,
compared with healthy or adjacent non-cancerous tissues in data
from the GEO and TCGA databases (Fig. 4C
and D). High expression levels of UBE2C in different human
liver cell lines was further demonstrated (Fig. 4E). These results revealed that UBE2C
serves important roles in the development of HCC and other cancer
types, and may function as an oncogene in tumorigenesis.
Kaplan-Meier analysis of DEGs and
UBE2C in HCC tissues
Kaplan-Meier analysis was conducted for five DEGs
(UBE2C, TOP2A, PTTG1, GPC3 and PRC1). Survival analysis indicted
that patients with overexpression of these five DEGs exhibited
significantly significantly shorter survival times (P=0.0127)
compared with patients with relatively low expression levels
(Fig. 5A). Analysis of UBE2C in
cohorts with different clinical characteristics revealed that
patients with higher UBE2C expression exhibited short survival
times [hazard ratio (HR)=1.44; confidence interval (CI)=1.02–2.04;
log-rank P=0.037] (Fig. 5B), and
patients in stage III HCC with high UBE2C expression had relatively
short survival times compared with patients in stage III HCC with
low UBE2C expression (HR=1.84; CI=1.01–3.34; log-rank P=0.043)
(Fig. 5C-F). Stage IV was ignored
due to low sample size (n<10). Overexpression of UBE2C may serve
as a novel indicator of reduced survival time in patients with
diagnosed HCC. Thus, UBE2C may be used as a prognostic biomarker
for HCC.
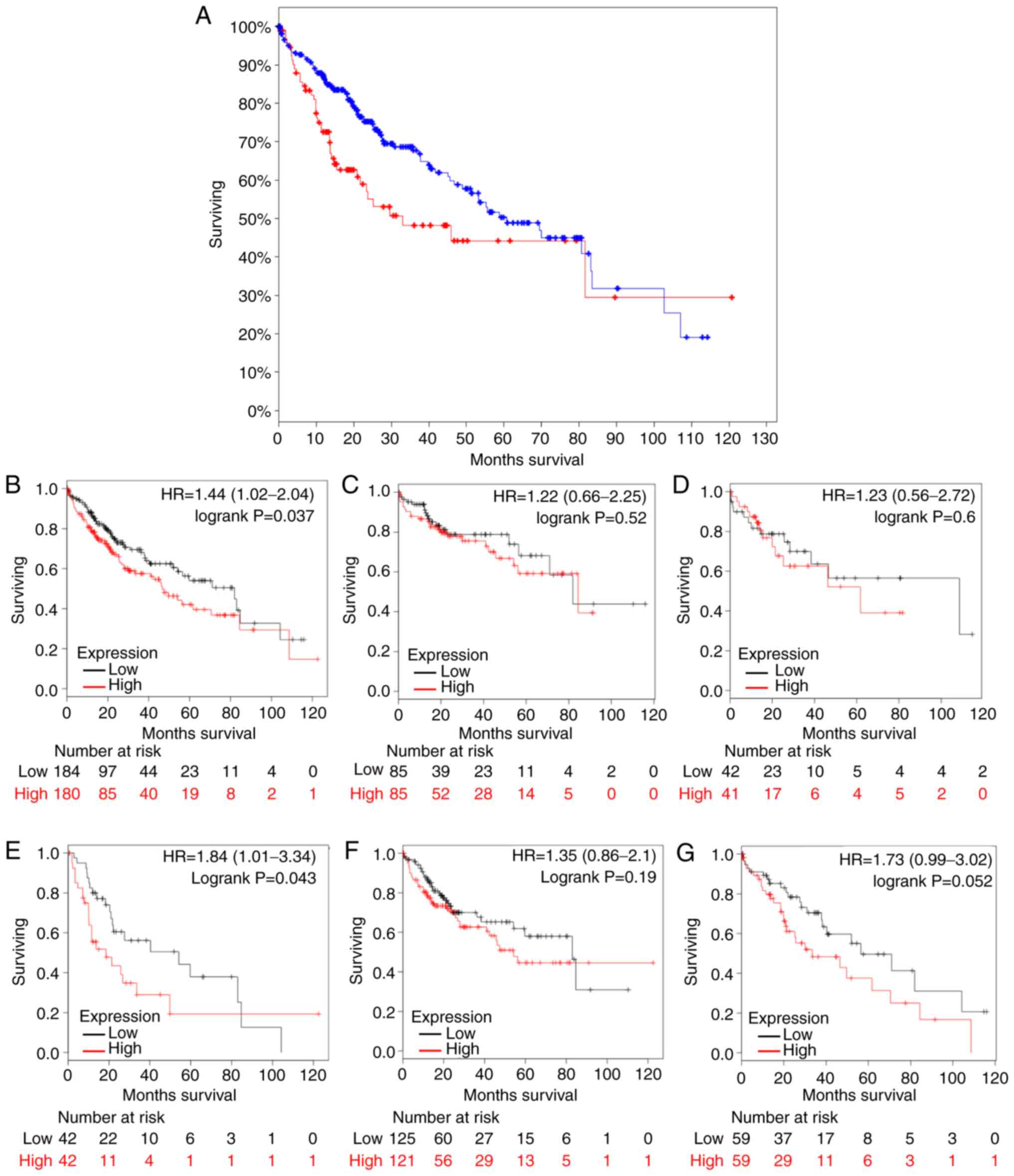 | Figure 5.Kaplan-Meier estimator analysis of
candidate genes in patients with HCC. (A) Kaplan-Meier plots
patients with HCC with five differentially expressed genes (UBE2C,
topoisomerase II α, pituitary tumor transforming gene 1, glypican-3
and polycomb-repressive complex 1) overexpression with data
recruited from cBioPortal (P=0.0127). The blue line represents HCC
patients with relatively low UBE2C, TOP2A, PTTG1, GPC3 and PRC1
mRNA expression, and the red line represents HCC patients with high
expression of the aforementioned genes. Log-rank analysis revealed
that patients with HCC and high mRNA expression exhibited
significant short survival times. (B-G) Kaplan-Meier plots of
different clinicopathological features associated with UBE2C
overexpression. The black line represents UBE2C low expression and
the red line represents UBE2C overexpression. The X and Y axes
indicate survival rate and overall survival time (days),
respectively. (B) All patients. (C) Patients in clinical stage I.
(D) Patients in clinical stage II. (E) Patients in clinical stage
III. (F) Male and (G) female patients with HCC. Log-rank analysis
revealed that patients with HCC in clinical stage III with high
UBE2C mRNA expression exhibited significantly lower survival times
compared with patients with low UBE2C mRNA expression. Log rank
P-value and 95% confidence intervals of the hazard ratio were used
for statistical analysis. HCC, hepatocellular carcinoma; mRNA,
messenger RNA; UBE2C, ubiquitin-conjugating enzyme 2C. |
Discussion
The overall survival of patients with HCC is
markedly short, and morbidity is increasing in developed and
developing contries. In USA, 22,000 newly diagnosed cases occurred
annually, and 18,000 patients succumbed to HCC (4). Approximately 1,000,000 patients in the
world are diagnosed with HCC each year (17). Numerous studies and clinical trails
had attempted to uncover the molecular mechanisms of HCC (18–20). The
present study integrated and thoroughly re-analyzed three datasets
of HCC. UBE2C and four other overexpressed DEGs (TOP2A, PTTG1, GPC3
and PRC1) were identified to be associated with HCC. Enrichment
analysis revealed that these genes were important for HCC
development via different signaling pahtways. Expression validation
analysis using GEO and TCGA data demonstrated that these genes were
associated with HCC pathogenesis, and survival analysis revaled
that the overexpression of these five genes, particularly UBE2C,
was important for survival of patients with HCC.
Formerly known as UBCH10, UBE2C is essential for
ubiquitination and inactivation of protein activity (21). Normal expression of UBE2C guarantees
normal physiology functions, such as cell cycle progression and
programmed cell death (22,23). However, high expression levels of
UBE2C frequently leads to the destruction of essential proteins,
thus disrupting the function of mitotic cyclins, spindle checkpoint
control and euploidy status of cells (24,25).
Numerous studies have identified the overexpression of UBE2C in
several types of cancer, such as human cervical carcinoma, bladder
cancer and breast cancer (21,26,27). It
has been previously reported that UBE2C demonstrated potential in
becoming a promising cancer biomarker (28). Inhibition of UBE2C, by contrast,
suppressed tumor cell proliferation, inhibited tumorigenesis and
sensitized cancer cells to radiation (21,27,28).
Zhang et al (29) reported
that the inhibtion of oncogenic miR-17/20a suppressed gastic cancer
cell proflieration by downregulating UBE2C. These studies, as well
as the finding that UBE2C was upregualted in HCC tiusses compared
with controls in the present study, suggest that the overexpression
of UBE2C may be associated with the tumorigenesis of HCC.
A previous study revealed the molecular structure of
anaphase-promoting complex/cyclosome (APC/C)-coactivator complexes
under cryo-electron microscopy (30). The union of APC/C complexes with
UBE2C promotes ubiquitination, whereas early mitotic inhibitor-1
(Emi1) constrains this effect. Deubiquitination enzymes are
activated as soon as UBE2C is separated from APC/C, but the process
is slow, with unclear molecular patterns (31). In the present study, GO analysis
revealed that the upregulated UBE2C, TOP2A, PTTG1, GPC3 and PRC1
genes were primarily involved in cell cycle, cell communication and
protein metabolism biological processes. Paclitaxel (PTX) is a
widely used microtubule-poisoning drug in anti-neoplastic
strategies, which triggers cell death by activating the spindle
assembly checkpoint (SAC) (32).
Cancer cells senstive to PTX do not achieve SAC, and undergo
mitosis by degradation of APC/C and cyclin B, which is called
mitotic catastrophe, thus leading to mitotic slippage or cell
apoptosis (33). High expression of
UBE2C has been demonstrated to override SAC (24). Since opposite characteristics are
observed for UBE2C and Emi1 when interacting with APC/C, the exact
role of Emi, as well as its expression levels and significance in
PTX-treated HCC, require further study.
In summary, the present study confirmed that the
high-expression level of UBE2C as well as other four genes (TOP2A,
PTTG1, GPC3 and PRC1) was associated with poor overall survival of
patients with HCC. Overexpression of UBE2C may serve as a novel
indicator of short survival in patients with HCC. Furher studies
should foucus on the potential use of UBE2C as a poor prognostic
factor for HCC. Whether UBE2C overexpression serves a specific role
in the growth of HCC, its inhibtion may block the growth of HCC
in vivo or in vitro.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Postgraduate
Research & Practice Innovation Program of Jiangsu Province
(grant no. KYCX18_1462), National Natural Science Foundation of
China (grant no. 81700392), Nanjing Health Youth Talent Training
Project in 13th Five-Year (grant no. QRX17113) and Natural Science
Foundation of Jiangsu Province Youth Fund Project (grant no.
BK20150106).
Availability of data and materials
The datasets analyzed in the present study are all
available on NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi).
Authors' contributions
ZW, YL, XS and BX designed the study. ZW and SQ
wrote the manuscript. XL, QL, JH, JZ, ZHW, SQ and AS performed the
bioinformatics analysis. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Neumann H, Longo D, Fauci A, Kasper D,
Hauser S, Jameson J and Loscalzo J: Harrison's Principles of
Internal Medicine. 2011.
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liang HW, Yang X, Wen DY, Gao L, Zhang XY,
Ye ZH, Luo J, Li ZY, He Y, Pang YY and Chen G: Utility of
miR-133a-3p as a diagnostic indicator for hepatocellular carcinoma:
An investigation combined with GEO, TCGA, meta-analysis and
bioinformatics. Mol Med Rep. 17:14692018.PubMed/NCBI
|
|
4
|
Yang N, Ekanem NR, Sakyi CA and Ray SD:
Hepatocellular carcinoma and microRNA: New perspectives on
therapeutics and diagnostics. Adv Drug Deliv Rev. 81:62–74. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kulasingam V and Diamandis EP: Strategies
for discovering novel cancer biomarkers through utilization of
emerging technologies. Nat Clin Pract Oncol. 5:588–599. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo Y, Bao Y, Ma M and Yang W:
Identification of key candidate genes and pathways in colorectal
cancer by integrated bioinformatical analysis. Int J Mol Sci.
18:E7222017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang L, Gao L, Zou XP, Huang ML, Chen G,
Li JJ and Cai XY: Diagnostic significance and potential function of
miR-338-5p in hepatocellular carcinoma: A bioinformatics study with
microarray and RNA sequencing data. Mol Med Rep. 17:2297–2312.
2018.PubMed/NCBI
|
|
9
|
Zeng T, Wang D, Chen J, Chen K, Yu G, Chen
Q, Liu Y, Yan S, Zhu L, Zhou H, et al: AF119895 regulates NXF3
expression to promote migration and invasion of hepatocellular
carcinoma through an interaction with miR-6508-3p. Exp Cell Res.
363:129–139. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gan TQ, Xie ZC, Tang RX, Zhang TT, Li DY,
Li ZY and Chen G: Clinical value of miR-145-5p in NSCLC and
potential molecular mechanism exploration: A retrospective study
based on GEO, qRT-PCR, and TCGA data. Tumour Biol.
39:10104283176916832017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Huang L, Liang H, Zhang R, Chen
G, Pang Y and Feng Z: Clinical value and potential targets of
miR-224-5p in hepatocellular carcinoma validated by a TCGA- and
GEO- based study. Int J Clin Exp Pathol. 10:9970–9989. 2017.
|
|
12
|
Grinchuk OV, Yenamandra SP, Iyer R, Singh
M, Lee HK, Lim KH, Chow PK and Kuznetsov VA: Tumor-adjacent tissue
co-expression profile analysis reveals pro-oncogenic ribosomal gene
signature for prognosis of resectable hepatocellular carcinoma. Mol
Oncol. 12:89–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang H, Huo X, Yang XR, He J, Cheng L,
Wang N, Deng X, Jin H, Wang N, Wang C, et al: STAT3-mediated
upregulation of lncRNA HOXD-AS1 as a ceRNA facilitates liver cancer
metastasis by regulating SOX4. Mol Cancer. 16:1362017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen X, Cheung ST, So S, Fan ST, Barry C,
Higgins J, Lai KM, Ji J, Dudoit S, Ng IO, et al: Gene expression
patterns in human liver cancers. Mol Biol Cell. 13:1929–1939. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roessler S, Long EL, Budhu A, Chen Y, Zhao
X, Ji J, Walker R, Jia HL, Ye QH, Qin LX, et al: Integrative
genomic identification of genes on 8p associated with
hepatocellular carcinoma progression and patient survival.
Gastroenterology. 142:957–966. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Roessler S, Jia HL, Budhu A, Forgues M, Ye
QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX and Wang XW: A
unique metastasis gene signature enables prediction of tumor
relapse in early-stage hepatocellular carcinoma patients. Cancer
Res. 70:10202–10212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dhanasekaran R, Limaye A and Cabrera R:
Hepatocellular carcinoma: Current trends in worldwide epidemiology,
risk factors, diagnosis, and therapeutics. Hepat Med. 4:19–37.
2012.PubMed/NCBI
|
|
18
|
Guo X, Wang Z, Zhang J, Xu Q, Hou G, Yang
Y, Dong C, Liu G, Liang C, Liu L, et al: Upregulated KPNA2 promotes
hepatocellular carcinoma progression and indicates prognostic
significance across human cancer types. Acta Biochim Biophys Sin
(Shanghai). 51:285–292. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tong H, Liu X, Li T, Qiu W, Peng C, Shen B
and Zhu Z: INTS8 accelerates the epithelial-to-mesenchymal
transition in hepatocellular carcinoma by upregulating the TGF-beta
signaling pathway. Cancer Manag Res. 11:1869–1879. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aziz K, Limzerwala JF, Sturmlechner I,
Hurley E, Zhang C, Jeganathan KB, Nelson G, Bronk S, Velasco RF,
van Deursen EJ, et al: Ccne1 overexpression causes chromosome
instability in liver cells and liver tumor development in Mice.
Gastroenterology. 2019. View Article : Google Scholar
|
|
21
|
Bose MV, Gopisetty G, Selvaluxmy G and
Rajkumar T: Dominant negative Ubiquitin-conjugating enzyme E2C
sensitizes cervical cancer cells to radiation. Int J Radiat Biol.
88:629–634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu YC: Ubiquitin ligases and the immune
response. Annu Rev Immunol. 22:81–127. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Williamson A, Wickliffe KE, Mellone BG,
Song L, Karpen GH and Rape M: Identification of a Physiological E2
module for the human anaphase-promoting complex. Proc Natl Acad
USA. 106:18213–11821. 2009. View Article : Google Scholar
|
|
24
|
Reddy SK, Rape M, Margansky WA and
Kirschner MW: Ubiquitination by the anaphase-promoting complex
drives spindle checkpoint inactivation. Nature. 446:921–926. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
van Ree JH, Jeganathan KB, Malureanu L and
van Deursen JM: Overexpression of the E2 ubiquitin-conjugating
enzyme UbcH10 causes chromosome missegregation and tumor formation.
J Cell Biol. 188:83. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Morikawa T, Kawai T, Abe H, Kume H, Homma
Y and Fukayama M: UBE2C is a marker of unfavorable prognosis in
bladder cancer after radical cystectomy. Int J Clin Exp Pathol.
6:1367–1374. 2013.PubMed/NCBI
|
|
27
|
Rawat A, Gopal G, Selvaluxmy G and
Rajkumar T: Inhibition of ubiquitin conjugating enzyme UBE2C
reduces proliferation and sensitizes breast cancer cells to
radiation, doxorubicin, tamoxifen and letrozole. Cell Oncol
(Dordr). 36:459–467. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hao Z, Zhang H and Cowell J:
Ubiquitin-conjugating enzyme UBE2C: Molecular biology, role in
tumorigenesis, and potential as a biomarker. Tumor Biol.
33:723–730. 2012. View Article : Google Scholar
|
|
29
|
Zhang Y, Han T, Wei G and Wang Y:
Inhibition of microRNA-17/20a suppresses cell proliferation in
gastric cancer by modulating UBE2C expression. Oncol Rep.
33:2529–2536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang L, Zhang Z, Yang J, McLaughlin SH
and Barford D: Atomic structure of the APC/C and its mechanism of
protein ubiquitination. Nature. 522:450–454. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xie C, Powell C, Yao M, Wu J and Dong Q:
Molecules in focus: Ubiquitin-conjugating enzyme E2C: A potential
cancer biomarker. Int J Biochem Cell Biol. 47:113–118. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rowinsky EK and Donehower RC: Paclitaxel
(taxol). N Engl J Med. 332:1004–1014. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gascoigne KE and Taylor SS: Article:
Cancer cells display profound intra- and interline variation
following prolonged exposure to antimitotic drugs. Cancer Cell.
14:111–123. 2008. View Article : Google Scholar : PubMed/NCBI
|