Introduction
Circulating tumor DNA (ctDNA), determined in the
cell-free fraction of blood, represents a variable and generally
small fraction of the total circulating DNA (1). Plasma-derived ctDNA, determined in the
cell-free fraction of blood in patients with cancer, has been
recognized as a potential non-invasive biomarker for tumor tissue
biopsies (2–4). Next generation sequencing (NGS) studies
on ctDNA have revealed that ctDNA is a potential marker associated
with various human cancer types (2–4). In
2010, to enhance the clinical management of patients with cancer,
Leary et al (2) introduced
the concept of using ctDNA for the development of personalized
biomarkers to provide an exquisitely sensitive and broadly
applicable approach. Van der Vaart et al (3) used a parallel tagged sequencing method
to sequence circulating DNA obtained from healthy controls as well
as patients with cancer (12 patients with prostate cancer). Chan
et al (5) reported the use of
shotgun massive parallel sequencing to obtain a non-invasive,
genome-wide view of somatic copy number alterations and
cancer-associated mutations in four patients with hepatocellular
carcinoma (HCC) and a patient with synchronous breast and ovarian
cancer, demonstrating the use of ctDNA as a powerful tool for
cancer detection, and its potential role as a powerful tool for
elucidating important tumoral characteristics, cancer monitoring
and research. In a study containing 30 females with metastatic
breast cancer who were receiving systemic therapy, Dawson et
al (4) compared the radiographic
imaging of tumors with the assay of CA 15-3, circulating tumor
cells and ctDNA. The results demonstrated that ctDNA is an
inherently specific, informative and highly sensitive biomarker of
metastatic breast cancer. The development of ctDNA is rapid, which
indicates notable potentialities and feasibility of using it to
monitor tumor dynamics in various solid cancer types. However, the
majority of the methods used to test ctDNA are expensive (3,4).
Liver cancer is one of the most common cancer types
and cause of cancer-associated mortalities in China (6). Targeted therapies have been achieved by
addressing the specific molecular drivers of a patient, which is
safer and more efficacious (1,7,8). However, ctDNA with specific tumor
mutations has not been extensively investigated or analyzed in
patients with liver cancer. The majority of the research used whole
genome sequencing to investigate ctDNA (2–5). It has
been reported that there are >6,000 genes associated with liver
cancer, but the majority of which occurred rarely
(liverome.kobic.re.kr/index.php). As for the specific nature of
ctDNA, sequencing of the sample in depth is required to retrieve
the genetic information. Therefore, if the region is too large, it
would be a waste of resources, because only the regions associated
with liver cancer are required. Furthermore, the majority of the
liver cancer-associated genes are not suitable to be markers as the
majority of these genes are rarely observed in patients with liver
cancer (liverome.kobic.re.kr/index.php). Therefore, these regions
specific to liver cancer should be determined. In the present
study, liver cancer samples from several databases were used to
develop a set of regions specific to liver cancer. Those selected
regions were then determined to be suitable for ctDNA analysis. Due
to the small size of the designed regions, samples could be
sequenced deeply, which would help to make ctDNA a potential marker
for cancer monitoring. To the best of our knowledge, this is the
first research aiming to develop and design the liver cancer
regions for the analysis of ctDNA. Furthermore, the present study
could provide valuable information for other cancer chip design
types.
In conclusion, in order to detect somatic mutations
and design a specific method to quantify ctDNA in liver tumors, a
selected region covering multiple classes of somatic mutations that
may be identified in patients with liver tumors was designed, and
its performance was evaluated in 3 patients with liver cancer. The
results provided a set of personalized cancer-specific markers, and
evaluated the prognosis of therapy.
Materials and methods
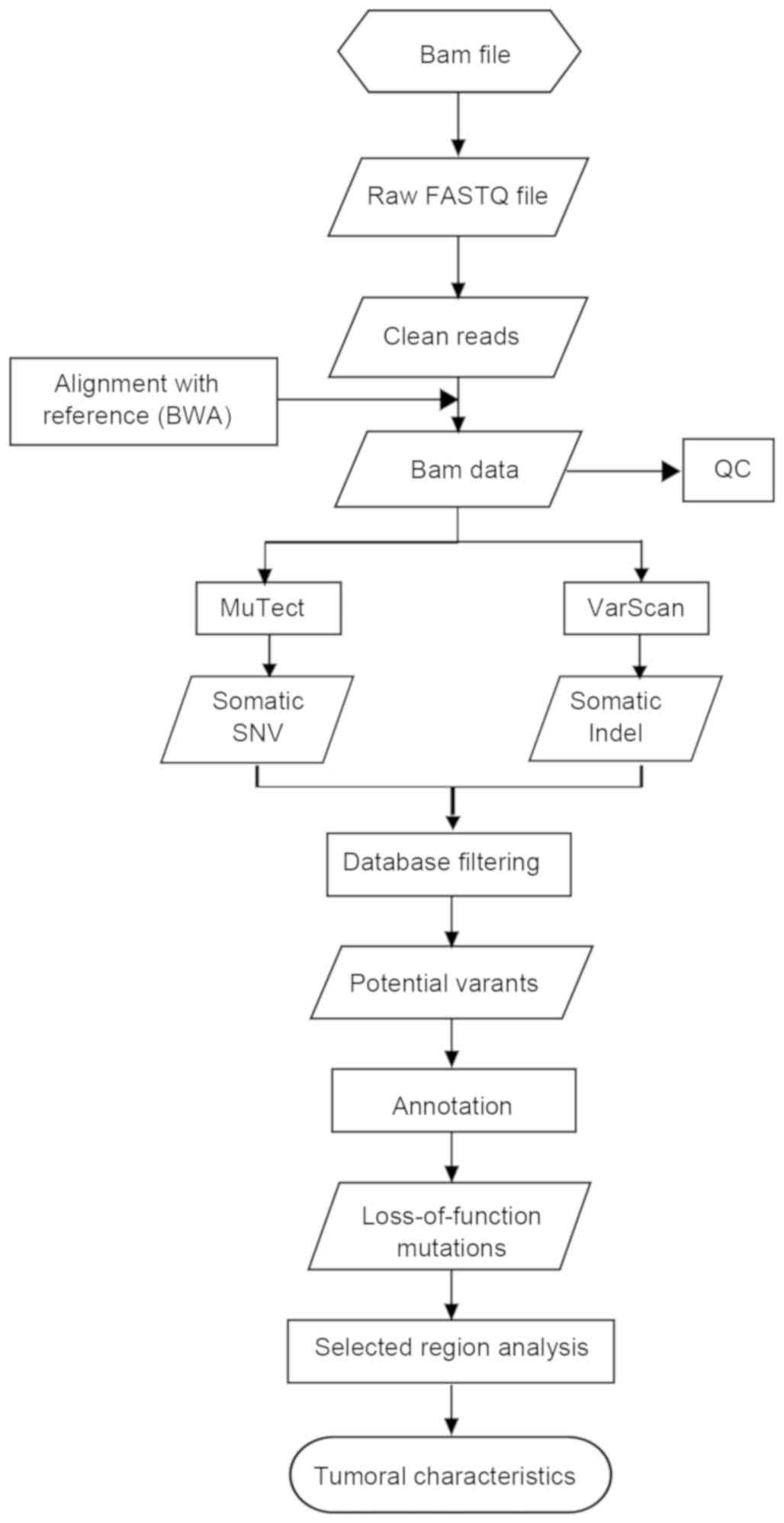
Data analysis pipeline
In the present study, a data analysis pipeline,
including data filtration, alignment, variants detection and
results annotation for whole genome data and data in the selected
regions, was established (Fig. 1).
The sequencing data (bam file) were provided by Professor Yuk-Ming
Dennis Lo from The Chinese University of Hong Kong (Hong Kong,
China). The bam file was sorted, and bedtools (bedtools version
2.25.0) was used to change the bam file into an fq file, via
filtering of certain reads with the same read ID, for further
analysis. The pipeline started from the clean reads, as follows:
Firstly, the clean reads with a length of 50 bps were mapped to the
human reference genome (hg19) from the University of California,
Santa Cruz database (hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/)
using BWA (Burrows Wheeler Aligner; bio-bwa.sourceforge.net/); secondly, following removal
of polymerase chain reaction-derived duplications using Picard
(broadinstitute.github.io/picard/) and realigning by GATK
(https://software.broadinstitute.org/gatk/documentation/tooldocs/current/),
the bam results were then used to determine variant detection.
Somatic SNVs calling were performed using MuTect (software.broadinstitute.org/cancer/cga/mutect).
To reduce false-positives, stringent criteria were used in the
present study, as follows: Firstly, a mutation was kept only when
it was completely absent in the result blood sample; secondly, a
mutation was kept only when the sequencing depth was >20-fold.
This threshold was applied to lower the false-positive detection
rate. Somatic Indel calling was performed using Varscan software
(varscan.sourceforge.net/). Detailed
filtering parameters followed the best practice guidelines
(varscan.sourceforge.net/using-varscan.html).
Local realignment around Indels was also included. The identified
somatic variants were directly annotated by the Catalogue of
Somatic Mutations in Cancer (COSMIC, http://cancer.sanger.ac.uk/cosmic/) database, dbSNP,
Hapmap, 1000 Genome and dbNSFP using our own PERL scripts if the
alterations have been reported to be disease-causing mutations or
targets for therapy.
Design of a liver cancer-associated
selector
In the present study, the focus was on liver cancer;
therefore, specific regions, including the coding sequences (CDSs),
covering recurrent alterations in potential recurrent mutated genes
were designed. Using the method by Newman et al (9) a novel algorithm to determine the liver
cancer-associated regions was developed. Firstly, a list of genes
covering recurrent alterations in potential driver genes were
selected using the top 100 genes listed in COSMIC (10). Subsequently, regions of CDS
containing recurrent single nucleotide variants (SNVs) were
selected based on the mutation frequency of the CDS region (cut-off
value, 5; Fig. 2) to form potential
liver cancer-associated regions. Following this, to maximize the
number of mutations per patient while minimizing the region, a
novel algorithm was applied using whole-exome sequencing (WES) data
from 243 patients (the LINC-JP_Liver_Cancer-NCC_JP project by
December 6, 2014) with liver cancer with cancer-associated SNVs and
Indels profiled by International Cancer Genome Consortium
(https://icgc.org/). Of the 243 patients included in
the study, 74.49% (181/243) were male, and the age of the 243
patients at diagnosis ranged from 23–85 years. Detailed information
of the patients can be found in the ICGC database (https://icgc.org/icgc/cgp/66/420/824).
Additionally, a number of genomic regions harboring liver cancer
driver genes known to be associated with liver cancer therapy were
included. The present study and the protocols used were approved by
the Institutional Ethics Committee of BGI (Shenzhen, China).
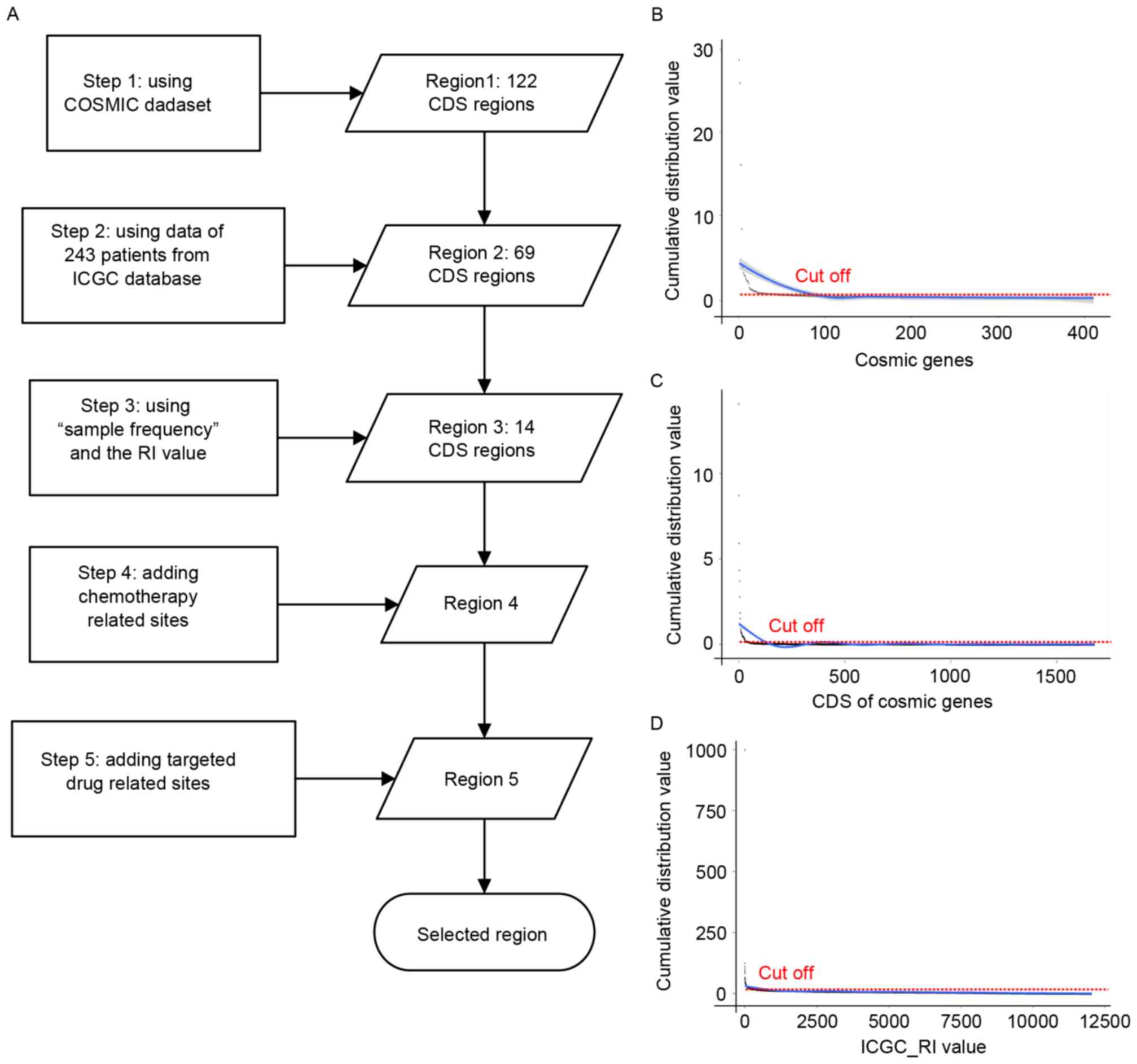
The main steps and cut off parameters are as follows
(Fig. 2A): i) Step 1: Region 1. To
select the CDS region (initial region 1) covering recurrent
alterations in known driver genes, the data of patients with liver
cancer from COSMIC was analyzed. Subsequently ‘mutation frequency’
in a specific gene region in patients with liver cancer was used to
select the initial seed genes. The cut-off value was 12 (Fig. 2B), and 100 genes from the COSMIC
patients with liver cancer were obtained. Subsequently, ‘mutation
frequency’ of the CDS region (Fig.
2C; cut-off value was 5) was used to select the initial seed
regions and a region with 141 CDS regions was obtained. The final
region 1 was generated from the intersection of the seed genes and
the CDS regions. Finally, region 1 with 122 CDS regions was
selected (data not shown);
ii) Step 2: Region 2. To maximize the number of
mutations per patient while minimizing the region, a novel
algorithm was used using WES data from 243 patients (the
LINC-JP_Liver_Cancer-NCC_JP project) with cancer-associated SNVs
and Indels profiled by International Cancer Genome Consortium
(icgc.org/). Firstly, patients with a mutation in
region 1 were removed. A total of 142 samples were removed, which
indicated a high liver cancer-associated nature of region 1. The
remaining samples were retained for further analysis. Secondly, the
‘sample frequency’ of all the CDSs in the remaining patients was
calculated. A cut off value of 4 was used in this step (Fig. 2D). Thirdly, the remaining CDS region,
which would be selected only if at least one new patient was
identified, was analyzed. This was repeated until no further region
met these criteria. In this step, region 2 (69 CDS regions) was
selected. Additionally, 91 more samples were covered in this step,
and a total of 233 samples (233/243) were covered in regions 1 and
2.
iii) Step 3: Region 3. In this step, ‘sample
frequency’ and the recurrence index (RI) value were used. Firstly,
a sample cutoff value of 2 and RI cutoff value of 10 was used to
generate a seed list of 257 candidate CDS regions. The cutoff value
aforementioned is not a stringent filtering parameter. Therefore,
the ‘mutation frequency’ of the aforementioned seed CDS regions was
calculated. A cutoff value of 3 was used, which means that only the
CDS regions with at least 3 mutations were obtained. Region 3 was
obtained in this step, which contains 14 CDS regions in total
(region 3).
iv) Step 4: Add chemotherapy-associated site. Single
nucleotide polymorphisms, which were reported to be associated with
chemotherapy (region 4) were included.
v) Step 5: Add targeted drug-associated sites. By
the time of submission of this paper, there has only been one drug
demonstrated to treat liver cancer, Sorafenib, which blocks the
RAF/mitogen-activated protein kinase kinase/extracellular
signal-regulated kinase pathway (11). A number of potential targeted genes
reported to be targetable to some drugs (region 5) were also
included.
Selected regions performance
assessment
To investigate the specificity of the selected
genomic regions selected, WES data from patients with liver
cancer-associated SNVs and Indels profiled by TCGA (by January 10,
2015) were used for assessment. The aim was to evaluate the
fraction of the 192 patients containing at least one
loss-of-function mutation in the selected regions.
Evaluation of somatic mutation
detection in 3 patients with liver cancer
To investigate the performance of the designed
regions, 3 patients with liver cancer were recruited, who all
received surgery [formalin-fixed paraffin embedded (FFPE) sample
and blood sample sequencing data prior to surgery, and ctDNA
sequencing data prior to and following surgery]. The samples were
collected in the Prince of Wales Hospital (Hong Kong, China), as
previously described (5). Written
informed consent was obtained from all the participants at the time
of sample collection. The sequencing data was provided by Professor
Yuk-Ming Dennis Lo from The Chinese University of Hong Kong. The
following pipeline was used to identify patient specific mutations,
and then evaluate the performance of the designed regions in
monitoring cancer in the 3 patients. The bioinformatics pipeline is
depicted in Fig. 1.
Statistical analysis
To assess statistical significance, random selectors
were selected using the same size to the present selected region.
The performance of random selectors and liver cancer-associated
selectors were compared (Z-test), and P-values were calculated
accordingly.
Results
Liver cancer-associated selector
The following algorithm was used to design the liver
cancer-associated selector, aiming to collect all the liver
cancer-associated genes and regions. In the whole process, two
values were primarily introduced to maximize the association of the
selected regions and liver cancer, whilst minimizing the length of
the designed regions: One was ‘mutation frequency’, which is
defined as the number of SNVs/Indels that occur within a given
cohort of patients; the other one was ‘sample frequency’, which is
defined as the number of the samples (carrying specific mutations)
in a specific CDS region. The number of patients with mutations/CDS
length in kb [known as RI (9)] was
used to measure patient-level recurrence frequency at the CDS
level. This RI could normalize gene or CDS size.
Following all of the aforementioned steps, a
specific region of 211 kb, which included 159 genes, was
obtained.
Reliability and accuracy evaluation of
the selector
To investigate the specificity of the aforementioned
selected genomic regions, WES data from 192 patients with liver
cancer-associated SNVs and Indels profiled by TCGA were used for
assessment. Detailed information of the patients can be found in
TCGA database (by January 10, 2015). A total of 107 patients
(107/192, 55%) contained at least one loss-of-function mutation in
the selected region. This independent cohort contained a mean of
284.4 mutations following the removal of 5 samples, which contained
>100,000 mutations each and would influence the sample
frequency. The RI of the selected region in the remaining samples
was 1, which means the number of patients with loss-of-function
mutations/CDS length in kb was >1. To assess the statistical
significance, a 211 kb region was randomly selected for comparison.
A mean RI of 0.0123 was determined in the randomly selected region
(P<1.0×10-5 for the difference between random selectors and
liver cancer-associated selector; Z-test), which validated a high
specificity of the selected genomic regions to liver cancer.
Analysis of the 3 patients with liver
cancer
To demonstrate the use of the designed region in
elucidating notable tumoral characteristics, and the potential role
of ctDNA as a powerful biomarker, the WGS (whole genome sequencing)
data of 3 patients with HCC (blood and FFPE pre-resection samples,
and pre- and post-resection plasma samples), which were recruited
for another study (5), were
analyzed. The analysis was started from the bam file provided by
Professor Yuk-Ming Dennis Lo from The Chinese University of Hong
Kong. The bam file was sorted first, and then bedtools was used to
change the bam file into a fq file for further analysis. During
this process, some reads with the same read ID were removed.
Subsequently, the aforementioned pipeline was used for analysis.
The sequencing results are detailed in Table I.
 | Table I.Whole genome and selected region
performance results. |
Table I.
Whole genome and selected region
performance results.
| Sample | Type | Mapped reads | Duplicate rate
(%) | Whole genome coverage
(%) | Coverage of selected
regions (%) | WGS sequencing
depth | Sequencing depth of
selected regions |
|---|
| case013W | Blood | 1,004,693,827 | 1.89 | 92.58 | 98.59 | 25.49 | 42.63 |
| T013 | FFPE | 1,082,465,782 | 4.09 | 86.08 | 98.04 | 26.86 | 58.93 |
| Plsm013_pre | Plasma | 951,847,727 | 2.34 | 92.05 | 98.08 | 16.04 | 18.73 |
| Plsm013_post | Plasma | 1,074,184,598 | 2.02 | 92.20 | 98.48 | 18.16 | 22.03 |
| case023W | Blood | 971,811,353 | 2.00 | 92.57 | 98.59 | 24.64 | 41.16 |
| T023 | FFPE | 1,128,719,103 | 2.04 | 92.57 | 98.40 | 28.62 | 49.26 |
| Plsm023_pre | Plasma | 881,754,515 | 1.77 | 91.52 | 98.24 | 14.99 | 21.35 |
| Plsm023_post | Plasma | 987,003,992 | 2.42 | 92.02 | 98.38 | 16.66 | 23.36 |
| case027W | Blood | 1,810,697,288 | 3.97 | 92.83 | 98.58 | 44.98 | 76.61 |
| T027 | FFPE | 1,064,991,038 | 4.20 | 74.04 | 93.20 | 26.41 | 97.33 |
| Plsm027_pre | Plasma | 928,705,384 | 2.88 | 92.24 | 97.66 | 15.56 | 16.66 |
| Plsm027_post | Plasma | 1,065,556,168 | 2.10 | 92.32 | 98.29 | 18.00 | 20.91 |
For all the samples included in the present study, a
mean of 1,079,369,231 reads were mapped to regions of the hg19
genome, resulting in a 23-fold average depth (Table I). The mean coverage of the 3 blood
samples (all sample types) was >91.5%, except FFPE sample T013
and T027. The mean sequencing depth of the blood sample, FFPE
samples and plasma sample was 31.7, 27.3 and 16.6, respectively,
which demonstrated a relatively low sequencing depth of the plasma
sample. The data in the selected region was then removed from the
WGS data for further analysis. The mean coverage of the 3 patients
(all sample types) in the selected region was ~98%, except for
sample T27, while the mean sequencing depth of the blood sample,
FFPE samples and plasma sample of the selected region was 53.5,
68.5 and 20.5, respectively.
No Indels were identified in the selected regions;
therefore, the primary focus was on the somatic SNVs. The total
number of detected SNVs was detailed in Table II. There was a mean of 3,097
mutations detected in the whole genome region of all the tissue
samples. The mutations determined in the plasma prior to and
following surgery are almost equal, which made it difficult to
annotate and generate the association between those mutations and
the cancer states. While the mutations located in the selected
region were relative small and liver-cancer associated. The
comparison between whole genome data and data of the selected
region was aimed to reveal its potential role as a powerful tool
for cancer detection and monitoring. As detailed in Table II, the mean detected mutations of
the FFPE samples and plasma sample of the designed selected region
was 71.3 and 5.5, respectively.
| Table II.Analysis results of somatic SNVs. |
Table II.
Analysis results of somatic SNVs.
| Patient | Gender | Age, years | Sample pair | Somatic SNVs | Somatic SNVs in
selected region |
|---|
| Patient 1 | Male | 77 | case013W-T013 | 3,500 | 75 |
|
|
|
|
case013W-Plsm013_pre | 274 | 6 |
|
|
|
|
case013W-Plsm013_post | 330 | 7 |
| Patient 2 | Male | 58 | case023W-T023 | 3,060 | 86 |
|
|
|
|
case023W-Plsm023_pre | 345 | 10 |
|
|
|
|
case023W-Plsm023_post | 306 | 6 |
| Patient 3 | Male | 39 | case027W-T027 | 2,732 | 53 |
|
|
|
|
case027W-Plsm027_pre | 98 | 3 |
|
|
|
|
case027W-Plsm027_post | 114 | 1 |
Monitoring of serial ctDNA levels of
the liver cancer- associated selector
The specific pattern(s) of ctDNA involved in the
disease states identified by targeted NGS sequencing may provide a
set of biomarker/therapeutic that could monitor tumor dynamics with
high accuracy, which may be used as a more precise diagnostic tool
to predict disease risk and treatment responses.
The performance of circulating biomarkers from the
selected regions in the 3 patients with HCC prior to and following
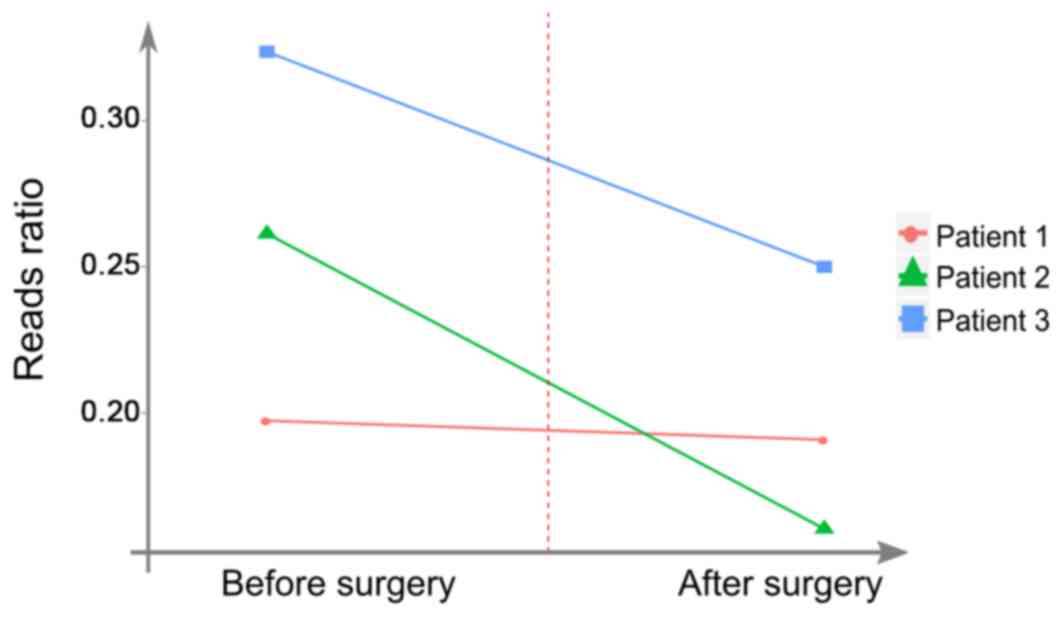
surgery was analyzed (Fig. 3). This
was performed to evaluate whether the fluctuations of ctDNA in the
selected regions can reflect the dynamics of the disease. The
significantly detectable levels of ctDNA data from the selected
region may be used to determine tumor volumes and clinical
responses to therapy.
In the present study, the analysis of the reads
ratio in the selected region was performed to monitor the effect of
surgery. The reads ratio was defined as the fraction of mutated
reads in all detected reads. The reads ratio in patients 2 and 3
was notably reduced following surgery (Fig. 3). Those two patients exhibited
similar dynamic patterns, which may reflect tumor burden in plasma
samples. However, further experiments are required to be performed
to support this result.
There were 6 SNVs exhibited in the pre-surgery
sample of patient 1. A total of three mutations were not identified
in the post-surgery sample of patient 1, including: TERT c.C1310G
and c.G555C, and MET c.T1621G, which may partially reflect the
clinical effect of surgical treatment. However, three mutations
(ALK c.A2242G, NOTCH1 c.A4111C and FLCN c.T884G) in the pre-surgery
sample of patient 1 were also exhibited in the post-surgery sample.
These minimal residuals may indicate possible progression of occult
microscopic disease. These data may highlight the promise of ctDNA
analysis from the specific selector region for identifying patients
with residual disease following treatment.
Somatic mutation reads upregulated/downregulated,
according to the treatment sensitivity of the patients, are
considered to be novel biomarkers. In the present study, the
results in the selected region demonstrated that the present method
provided an optimal method, including the design of specific liver
cancer-associated selector, and then the monitoring of treatment
response, which could provide a more accurate, sensitive and
economic method, compared with whole genome sequencing. If the data
indicates a strong association between ctDNA level and therapy
sensitivity of the patients, it will provide evidence of the role
of ctDNA in cancer monitoring, and may further validate previous
investigations.
Discussion
Continuing ineffective therapies and unnecessary
side effects are common limitations of cancer treatment (12), and thus far there is no effective
method to monitor treatment response, and to determine the benefit
of novel therapeutics. Generally, the use of serial imaging,
including radiographic measurements, in assessing treatment
response and detecting changes in tumor burden, frequently fails.
For example, patients with non-small cell lung cancer undergoing
definitive radiotherapy frequently have surveillance computed
tomography (CT)/positron emission tomography (PET)-CT scans that
are difficult to interpret due to fibrotic changes and
radiation-induced inflammatory in the lung and surrounding tissues
(13). Thus, there is an urgent
requirement for more sensitive and specific biomarkers to measure
tumor burden. Along with the development of NGS, the research of
ctDNA has become a hot spot in this field. It can be used as a
potential marker to predict disease risk, patient outcomes or
response to treatment (2–4). However, the reports published thus far
have a number of limitations. Firstly, it is difficult to detect
low frequency mutations; secondly, the acute monitoring of ctDNA
requires the detection of somatic mutations in tumor-tissue
samples, which are required to be performed in an invasive way; and
finally, evaluation of the ctDNA levels is different in reported
articles. For example, certain studies used absolute levels of
ctDNA or fraction of mutant allele in plasma to represent the ctDNA
levels (4,14), while other studies used the mean
allele frequency of the major clone to represent the ctDNA levels
(15). This inconsistency makes it
difficult to use ctDNA as a biomarker to monitor tumor dynamics in
patients with various solid types of cancer.
To solve those problems, the aim of the present
study was to develop a novel method. Firstly, based on
considerations of high sequencing depth, a selector only containing
liver-associated genes was designed; and secondly, the aim was to
establish a more precise target in analyzing ctDNA in cancer
monitoring. In the present study, patient specific mutation sites
were analyzed instead of the whole ctDNA level through targeted NGS
sampling at different times. This could make ctDNA an inherently
specific, informative and highly sensitive biomarker for liver
cancer.
There are >6,000 genes reported to be liver
cancer associated (liverome.kobic.re.kr/index.php). To the best of
our knowledge, no available database provides a selector specific
for liver cancer. Only a minority of tumor types can be defined
using a small number of recurrent mutations at predefined
positions. If the region is too large (such as WGS/WES), the cost
of sequencing would increase substantially. The cost greatly
influences the clinical application of ctDNA detection, meaning
that the design of the specific selector is vital in the present
study. First, it dictates which mutations can be detected with high
probability for a patient with a particular cancer type; and
secondly, the selector size directly impacts the cost and depth of
sequence coverage. For example, the smaller the selector is, the
deeper the sequencing depth could be achieved.
In the present study, a novel algorithm was used in
a liver cancer-associated selector design. It was considered that
‘mutation frequency’ and ‘sample frequency’ should be considered at
the same time. In the first step of process of designing a liver
cancer-associated selector, genes and CDS were taken into
consideration to generate region 1, due to it being considered that
genes are functional elements, which could be used as the first
filter, and CDS regions could be used to minimize the regions
associated with liver cancer mutations. To determine the regions
most associated with liver cancer, the algorithm used was more
stringent in the first 3 steps, particularly in the third step,
where 3 values were used to calculate and filter.
The mean sequencing depth of the blood sample, FFPE
samples and plasma sample of the selected region was 53.5, 68.5 and
20.5, respectively, which demonstrated a relatively low sequencing
depth of the plasma samples. The coverage of FFPE sample T013 and
T027 was 86.08 and 74.04%, respectively, which may be a result of
degradation of the samples. Only the associations of SNV and
disease progression were discussed in the present study. The whole
genome CNV status has been investigated in previous research
(5). The key point in the present
study is the sensitivity of designed selector.
A number of researchers reported that the absolute
levels of ctDNA in pretreatment plasma were significantly
correlated with tumor volume, as measured by CT and PET imaging
(2–4). Therefore, they used total volume of
ctDNA as a marker. However, in a number of cases, it has also been
observed that certain mutations dominated the plasma (4,14,16). As
a result, it was considered that mutations in potential driver
genes or actionable mutations could better reflect the tumor
dynamics. Analysis of the total reads in the selected region was
conducted to monitor the effect of surgery. The reads ratio in
patients 2 and 3 is notably reduced following surgery (Fig. 3), which demonstrated similar dynamic
patterns in plasma. To support this result, more experiments are
required to be performed. However, the ratio of the ctDNA remained
detectable in the plasma following surgery, which is the reason why
further investigation is required.
Along with the development and improvement of NGS,
the research of ctDNA has become a hot spot in this field. The
monitoring of ctDNA levels requires the identification of somatic
mutations in patients with cancer. However, due to the high cost of
sequencing, the characteristic of ctDNA slowed down the application
of ctDNA in the clinical setting. The present study provided an
optimal method to design a cost-effective alternative for ctDNA
analysis, and targeted deep sequencing can be readily expanded to
include other genes known to be recurrently mutated in a specific
cancer type. Therefore, Chinese HCC samples will be collected for
testing in future work.
Acknowledgements
The authors would like to thank Professor Yuk-Ming
Dennis Lo from The Chinese University of Hong Kong (Hong Kong,
China) for providing access to the sequencing data of the patients
with hepatocellular carcinoma.
Funding
The present study was supported by National Natural
Science Foundation of China (grant no. 81502593).
Availability of data and materials
The datasets generated and/or analyzed during the
present study are available in the European Genome-Phenome Archive
(EGA; www.ebi.ac.uk/ega/), which is hosted by
the European Bioinformatics Institute (EBI), under accession number
EGAS00001000370.
Authors' contributions
YS, RM, HT, HW, JX, GW and JW designed the research
and wrote the first draft of the article. JL, XG, YM, YY, XW, FM
and MN contributed to revising the manuscript critically. YS, HT,
HW and MN developed the algorism. YS, RM, JX, HT, HW, MN, XG, YM,
YY, XW, FM and JL performed the data analysis. All authors approved
the final version to be published.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of BGI-Shenzhen (Shenzhen, China). Written informed
consent was obtained from all the participants at the time of
sample collection.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ignatiadis M and Dawson SJ: Circulating
tumor cells and circulating tumor DNA for precision medicine: Dream
or reality? Ann Oncol. 25:2304–2313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leary RJ, Kinde I, Diehl F, Schmidt K,
Clouser C, Duncan C, Antipova A, Lee C, McKernan K, De La Vega FM,
et al: Development of personalized tumor biomarkers using massively
parallel sequencing. Sci Transl Med. 2:20ra142010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van der Vaart M, Semenov DV, Kuligina EV,
Richter VA and Pretorius PJ: Characterisation of circulating DNA by
parallel tagged sequencing on the 454 platform. Clin Chim Acta.
409:21–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dawson SJ, Tsui DW, Murtaza M, Biggs H,
Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B,
et al: Analysis of circulating tumor DNA to monitor metastatic
breast cancer. N Engl J Med. 368:1199–1209. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chan KC, Jiang P, Zheng YW, Liao GJ, Sun
H, Wong J, Siu SS, Chan WC, Chan SL, Chan AT, et al: Cancer genome
scanning in plasma: Detection of tumor-associated copy number
aberrations, single-nucleotide variants, and tumoral heterogeneity
by massively parallel sequencing. Clin Chem. 59:211–224. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen JG and Zhang SW: Liver cancer
epidemic in China: Past, present and future. Semin Cancer Biol.
21:59–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mabert K, Cojoc M, Peitzsch C, Kurth I,
Souchelnytskyi S and Dubrovska A: Cancer biomarker discovery:
Current status and future perspectives. Int J Radiat Biol.
90:659–677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pantel K and Alix-Panabieres C: Real-time
liquid biopsy in cancer patients: Fact or fiction? Cancer Res.
73:6384–6388. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Newman AM, Bratman SV, To J, Wynne JF,
Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, et
al: An ultrasensitive method for quantitating circulating tumor DNA
with broad patient coverage. Nat Med. 20:548–554. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Forbes SA, Tang G, Bindal N, Bamford S,
Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, et al: COSMIC
(the Catalogue of Somatic Mutations in Cancer): A resource to
investigate acquired mutations in human cancer. Nucleic Acids Res
38 (Database Issue). D652–D657. 2010. View Article : Google Scholar
|
|
11
|
Lang L: FDA approves sorafenib for
patients with inoperable liver cancer. Gastroenterology.
134:3792008. View Article : Google Scholar
|
|
12
|
Kumar B, Singh S, Skvortsova I and Kumar
V: Promising targets in anti-cancer drug development: Recent
updates. Curr Med Chem. 24:4729–4752. 2017.PubMed/NCBI
|
|
13
|
Jahangiri P, Pournazari K, Torigian DA,
Werner TJ, Swisher-McClure S, Simone CB II and Alavi A: A
prospective study of the feasibility of FDG-PET/CT imaging to
quantify radiation-induced lung inflammation in locally advanced
non-small cell lung cancer patients receiving proton or photon
radiotherapy. Eur J Nucl Med Mol Imaging. 46:206–216. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Forshew T, Murtaza M, Parkinson C, Gale D,
Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley
D, et al: Noninvasive identification and monitoring of cancer
mutations by targeted deep sequencing of plasma DNA. Sci Transl
Med. 4:136ra1682012. View Article : Google Scholar
|
|
15
|
Murtaza M, Dawson SJ, Pogrebniak K, Rueda
OM, Provenzano E, Grant J, Chin SF, Tsui DW, Marass F, Gale D, et
al: Multifocal clonal evolution characterized using circulating
tumour DNA in a case of metastatic breast cancer. Nat Commun.
6:87602015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kidess E, Heirich K, Wiggin M, Vysotskaia
V, Visser BC, Marziali A, Wiedenmann B, Norton JA, Lee M, Jeffrey
SS and Poultsides GA: Mutation profiling of tumor DNA from plasma
and tumor tissue of colorectal cancer patients with a novel,
high-sensitivity multiplexed mutation detection platform.
Oncotarget. 6:2549–2561. 2015. View Article : Google Scholar : PubMed/NCBI
|