Introduction
Chromosomal translocations have been detected in a
variety of cancers. Certain types of translocation are known to
cause specific cancers. Translocations include several types of
fusion genes, for example, fusion genes in which a chimeric open
reading frame produces a chimeric protein after inter-intronic
recombination between two different genes, and promoter/enhancer
translocation upstream of a different gene, with subsequent control
of expression by the translocated promoter/enhancer (1,2).
Among fusion genes involved in hematopoietic
malignancies, the first to be cloned was the BCR/ABL fusion
gene from the Philadelphia chromosome, t(9;22), associated with
chronic myelogenous leukemia, although dozens of translocated genes
were already identified (3).
Multiple myeloma (MM) results from malignantly transformed plasma
cells and is reportedly associated with specific translocations,
such as t(11;14), t(4;14), and t(14;16) (4–6). As a
common feature, the enhancer region of the immunoglobulin heavy
chain (IgH) gene is rearranged to the target gene;
transcriptional activation of the gene in a B lymphocyte-specific
manner results in its functioning as an oncogene. The most
frequently detected translocation in MM is rearrangement between
the constant region of the IgH gene on the long arm of
chromosome 14 and the 5′-upstream region of the cyclin D1
(CCND1) gene on the long arm of chromosome 11. Similar
translocations were initially identified in the locus of B-cell
CLL/lymphoma 1 (Bcl1) in t(11;14)(q13;q32) (7,8).
CCND1 is a key molecule of the cell cycle control machinery,
involved in regulating the G1/S transition process (9). Cyclin activity is controlled by
transcription level. In the middle of the G1 phase, transcriptional
activation of the cyclin D1 gene results in an increase in
protein production, which then complexes with and induces the
kinase activity of cdk4 and cdk6, in turn stimulating cell cycle
progression. CCND1 gene juxtaposed to the downstream of
IgH constant region shows accelerated expression of CCND1
proteins by the effect of IgH super-enhancer in B
lymphocyte-specific manner, therefore, t(11;14)-harboring cells
indicate increased cell proliferation (10).
Sequencing of the junction point of t(11; 14)(q13;
q32) in B-cell malignancies revealed a difference between B-cell
lymphoma and MM (4,11,12). Two
types of gene rearrangement mechanisms are involved in chromosomal
translocations associated with B-cell malignancies: one that
involves VDJ recombination mediated by recombination activating
gene (RAG)1/2, and another that involves
activation-induced cytidine deaminase (AID) in
immunoglobulin class switching processes (3). Translocation in MM is induced by
AID (13,14). The resulting differences in the
origin of translocation-containing malignant cells in various
B-cell differentiation stages cause differences in clinical
condition (4,15).
Genome editing systems are now widely utilized in
many research fields (16,17). Among these systems, the
Streptococcus pyogenes type II clustered, regularly
interspaced, short palindromic repeats (CRISPR)-Cas9 system enables
efficient sequence-specific editing, enhancing the homologous
recombination-mediated template replacement of target genes
(18–20). Software that provides for effective
visual target estimation of CRISPR/Cas9 in the genome database in
combination with an evaluation score enhances the convenience of
the system (21). Recent reports
have described the induction of chromosomal translocations by
simultaneous cutting of two specific DNA sequences using
CRISPR-Cas9 (22–27) to engineer translocations that produce
chimeric proteins from the fusion genes. Translocation of
promotor/enhancer regions such as the B-cell lymphoma t(11;14) has
not been reported, however.
In this study, we developed a CRISPR/Cas9-mediated
genome editing system to induce the MM-specific chromosomal
translocation t(11;14)(q13;q32) in cultured cells and confirmed the
induction of a translocation identical to the experimental design.
In addition, we analyzed the DNA sequence of the translocation
junction, the gene expression and growth characteristics of
t(11;14)-positive cells.
Materials and methods
Materials
Cell culture reagents, Dulbecco's modified Eagle's
medium (DMEM) and penicillin/streptomycin, were obtained from
Nakalai Tesque (Kyoto, Japan). Plasmids used in this study,
lentiCRISPRv2 (#98290), pCAG-EGxxFP (#50716) (28), pX330 Cetn1/1 (#50718), and
pCAG-EGxxFP-Cetn1 (#50717), were obtained from Addgene (www.addgene.org). lentiCRISPRv2 puro was a gift from
Brett Stringer. Synthetic oligonucleotides were obtained from
Eurofin Genomics (Tokyo, Japan). DNA iso, RNAiso, Guide-it mutation
detection kit, and pMD20-T TA cloning vector were purchased from
Takara bio (Kyoto, Japan). Thunderbird® SYBR qPCR Mix,
KOD-PLUS, and ReverTraAce were obtained from Toyobo (Tokyo, Japan).
PEImax 40000 was obtained from Polyscience Inc. (Warrington, PA,
USA).
Cell culture, transfection, and virus
packaging
293 and 293T (SV40 T antigen introduced 293, 293T)
cells originated from Japanese Collection of Research Bioresources
Cell Bank were cultured in Dulbecco's modified Eagle medium
containing 10% fetal calf serum and incubated in a humidified
chamber at 37°C with 95% air and 5% CO2. These cells
were confirmed to be mycoplasma free. Plasmid transfection was
carried out using PEImax 40000, following the manufacturer's
protocol. For lentivirus packaging, plasmids were introduced into
293T cells, with subsequent recovery of the culture supernatant.
Virus particles were recovered by centrifugation of the supernatant
following addition of PEG6000 and NaCl (29).
CRISPR target localization
CRISPRscan (www.crisprscan.org/) (21) was used to localize the 20-bp target
sequences of the IgH Eµ region and CCND1 gene
5′-upstream region on the UCSC genome browser (http://www.ucsc.genome.edu/). Candidate gRNAs without
potential off-targets were selected using Cutting Frequency
Determination score (30) indicated
by CRISPRscan.
Plasmid construction
Double-stranded oligonucleotides for the guide RNA
of IgH Eµ (5′-CACCGGACTGGCCTAGCGGAGGCTC-3′ and
5′-AAACGAGCCTCCGCTAGGCCAGTCC-3′ for candidate A,
5′-CACCGGAGAACATACCAAGCCCCAC-3′ and 5′-AAACGTGGGGCTTGGTATGTTCTCC-3′
for candidate B) and CCND1
(5′-CACCGGGGGTAGGAAGCCTCGGCTGTGG-3′ and
5′-AAACCCACAGCCGAGGCTTCCTACCCCC-3′ for candidate A,
5′-CACCGGTGGCGAGGTGGGACCGCGG-3′ and 5′-AAACCCGCGGTCCCACCTCGCCACC-3′
for candidate B) were ligated into lentiCRISPRv2 vector digested
with BsmB1. For construction of pCAG-EGxxFP vectors to
monitor gRNA activity, IgH or CCND1 gene target
sequences were PCR-amplified from 293T cell genomic DNA using
5′-TTAGACAAGGGCGATGCCAG-3′ and 5′-TCAAGACCACTTTTCAACTACTCAC-3′ for
IgH candidate A, 5′-TCATTACCACCCTCCACTACCT-3′ and
5′-CCACTAGAAGGGGAACTGGTC-3′ for IgH candidate B,
5′-CACATGCCCGAAGTCAAACC-3′ and 5′-ATCACCGAGATCAGAAGGCT-3′ for
CCND1 candidate A, and 5′-CTTCTCACGAGCTGCCTTTG-3′ and
5′-GCTCATCACACAGCTTGACG-3′ for CCND1 candidate B with
KOD-Plus DNA polymerase. After cloning into pMD20-T for DNA
sequence confirmation, the target sequences were cloned into the
pCAG-EGxxFP cleavage site. For the IgH-CCND1
tandem-lentiCRISPRv2 vector, the U6-IgH gRNA B expression
cassette cloned into pMD20-T was PCR-amplified from IgH-B
lentiCRISPRv2 using 5′-GCAGAGATCCAGTTTGGTTAAT-3′ and
5′-ACCTAGCTAGCGtATTCAAAAA-3′ with KOD-Plus DNA polymerase and then
cloned into CCND1 B lentiCRISPRv2.
Homology-directed repair
(HDR)-mediated EGxxFP repair monitoring of gRNA activity
IgH or CCND1 gRNA activity was
monitored according to a previous report (28). Briefly, IgH (or CCND1,
IgH-CCND1 tandem) lentiCRISPRv2 and pCAG-EGxxFP IgH (or
CCND1) vectors were co-transfected into 293T cells. Two days
later, EGFP fluorescence was observed using a fluorescence
microscope (Axio Vert.A1, Zeiss, Oberkochen, Germany) and a flow
cytometer (S3e cell sorter, Bio-Rad, Hercules, CA, USA). The
fluorescence of co-transfected cells was compared to that of the
positive control, pX330-Cetn1/1 and pCAG-EGxxFP Cetn1 combination,
and the negative control, empty lentiCRISPRv2 and pCAG-EGxxFP. In
flow cytometric analyses, mean fluorescence intensity of
GFP-positive live cells was obtained using forward/side
scatter-gating and FL1 window.
Confirmation of CRISPR genome editing
activity
The genome editing activity of the IgH-CCND1
CRISPR vectors was confirmed by infecting 293T and 293 cells with
lentiCRISPRv2 virus. These cells were selected as previous reports
successfully established artificial translocation using CRISPR/Cas9
(22,25). Cells were infected with virus
suspension and cultured for 14 days with puromycin. Colonies were
picked, and DNA was recovered using a DNAiso kit according to the
manufacturer's protocol. The genome regions of gRNA target sites
were then PCR-amplified from puromycin-selected cells and parent
cells using the primers used in EGxxFP vector construction. A
Guide-IT mutation detection kit was used to detect mutations
introduced into the genomic DNA of CRISPR target sites. Purified
PCR products of selected and parental origin were mixed
equivalently, denatured, reannealed, incubated with Guide-it
nuclease, and electrophoresed to detect mismatch-directed cleavage.
In addition, to confirm genome editing at DNA sequence level,
PCR-amplified fragments were recombined into the pMD20-T TA cloning
vector, and inserted DNA was sequenced by Fasmac Co. (Kanagawa,
Japan).
Confirmation of translocation
PCR confirmation of translocation was performed
using the IgH/CCND1 translocation specific primer
pair (5′-AAGGGTGCGATGATGACCTAC-3′ for IgH side and
5′-AGCTGTTCTTGTAGTGGTGCC-3′ for CCND1 side) with
Thunderbird® SYBR qPCR Mix and a Light Cycler Nano
(Roche Diagnostics, Basel, Switzerland). For calibration of DNA
content in quantitative PCR (Q-PCR), primers for ACTB gene
(5′-AGAAAATCTGGCACCACACC-3′ and 5′-AACGGCAGAAGAGAGAACCA-3′) were
used for reference. PCR condition was, denaturation of template:
94°C 120 sec, DNA amplification cycles: 50 cycles of (94°C 10
sec-62°C 10 sec-72°C 20 sec), melting temperature measurement: 72°C
to 94°C at 0.1°C/sec. The presence of translocation-positive clones
was assessed based on the melting temperature of PCR-amplified DNA
compared with the positive control DNA. To confirm the efficiency
of IgH/CCND1 CRISPR/Cas9-induced translocation,
translocation-specific Q-PCR was performed using DNA of IgH/CCND1
lentiCRISPRv2-infected cells. Positive control DNA was amplified
from genomic DNA 5′-TCATTACCACCCTCCACTAC-3′ and
5′-TTTGCTAGCCACTGGCATCGCCCT-3′ for the IgH side and
5′-GCTCATCACACAGCTTGACG-3′ and 5′-TTTGCTAGCGCGGTGGGGTCTTGTGTTG-3′
for the CCND1 side, and the two PCR-amplified fragments were
recombined into a translocation-mimic PCR template
(5′-IgH-CCND1−3′). PCR-amplified DNA of candidate positive
cells was cloned into TA cloning vector, and inserted DNA was
sequenced. DNA sequence alignment was performed using ApE
(http://jorgensen.biology.utah.edu/wayned/ape/).
RT-qPCR
RNA was isolated from sub-confluent cultures of 293T
and t(11;14) positive cells two days after inoculation using RNAiso
reagent according to the manufacturer's instructions. cDNAs were
synthesized from total RNA using random primers and RevTraAce
reverse transcriptase. Quantitative PCR (Q-PCR) was performed using
the primers 5′-GACCCCGCACGATTTCATTG-3′ and
5′-CTCTGGAGAGGAAGCGTGTG-3′ for CCND1 and
5′-ACACTTCTGCTCGTTGCCTT-3′ and 5′-ACACAAATGCTCCTCTCACC-3′ for
MYEOV as the target and 5′-CACCAGGGCTGCTTTTAACTCT-3′ and
5′-TGGGATTTCCATTGATGACAAG-3′ for GAPDH as the reference with
Thunderbird qPCR mix and a LightCycler Nano (n=3) using the same
condition to DNA Q-PCR described above. Ratio of 2−Δ∆Cq
values were calculated from the values obtained by LightCyclerNano
Software (31).
Growth curve analysis
A total of 5×104 cells were inoculated
into 35-mm dishes and cultured. The number of viable attached cells
was determined using a dye-exclusion assay with trypan blue
(n=3).
Statistical analysis
Quantitative data were presented as mean ± standard
deviation obtained from three independent replicates. Statistical
analyses were performed using Welch's t-test on Microsoft Excel
version 14 on Windows 7. P<0.05 was considered to indicate a
statistically significant difference.
Results
Construction of IgH/cyclinD1-specific
CRISPR/Cas9
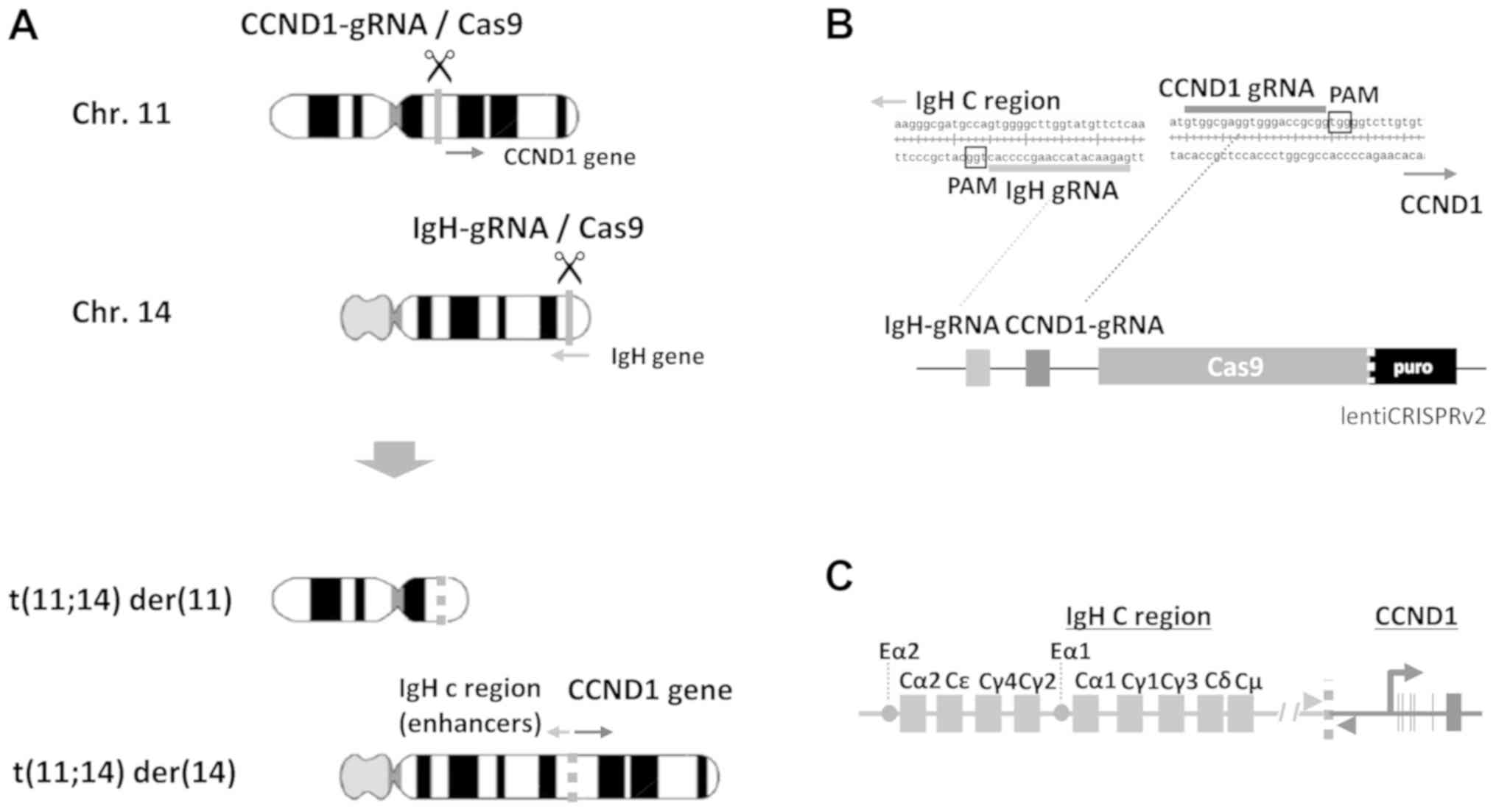
According to the scheme to obtain artificial
induction of t(11;14) by cutting CCND1 upstream on
chromosome (chr) 11 and IgH Eµ and Iµ regions on chr
14 (Fig. 1A), we designed two
efficient gRNA sequences to express in one CRISPR/Cas9 vector
(Fig. 1B) as described below. In
order to mimic t(11;14) in MM (Fig.
1C), we searched target sequences of CRISPR/Cas9 around the
previously reported junction sequence (32) using CRISPR Scan. As Ronchetti et
al (32) were unable to identify
clear clustering of junction sites of CCDN1, we searched the
targets from 10 to 20 kb upstream of the protein-coding sequence.
For IgH genes, Eµ and Iµ regions were
searched. Candidate gRNA sequences predicted to exhibit high
editing activity with minimal off-target activity were obtained.
Four gRNA target sequences were selected for IgH and
CCND1 genes, and synthetic oligonucleotides of these gRNA
candidates were recombined into lentiCRISPRv2.
The potency of gRNA sequences was monitored using
pCAG-EGxxFP. Both gRNA-coding lentiCRISPRv2 (IgH-A, -B,
CCND1-A or -B) and corresponding gRNA target
sequence-containing pCAG-EGxxFP (IgH-A, -B, CCND1-A
or -B) were introduced into 293T cells, and editing activity was
observed by recovery of EGFP fluorescence after HDR (Fig. 2A). EGFP fluorescence of cells into
which the candidates were introduced was compared to that of
positive control cells into which mouse Cetn1/1 pX330 and
pCAG-EGxxFP Cetn1 were introduced, and candidates exhibiting
fluorescence similar to that of Cetn1/1 cells were chosen (Fig. 2B). By comparing two gRNA candidates A
and B for IgH or CCND1, selected gRNA sequences were
5′-GAGAACATACCAAGCCCCACTGG−3′ (IgH-B) for
IgH, Chr14 genome position 105861042-105861064 in Genome
Reference Consortium Human 38 (GRCh38), and
5′-GTGGCGAGGTGGGACCGCGGTGG−3′ (CCND1-B) for
CCND1, Chr11 genome position 69627757-69627779 in GRCh38
(underlined sequences indicate protospacer adjacent motif,
PAM).
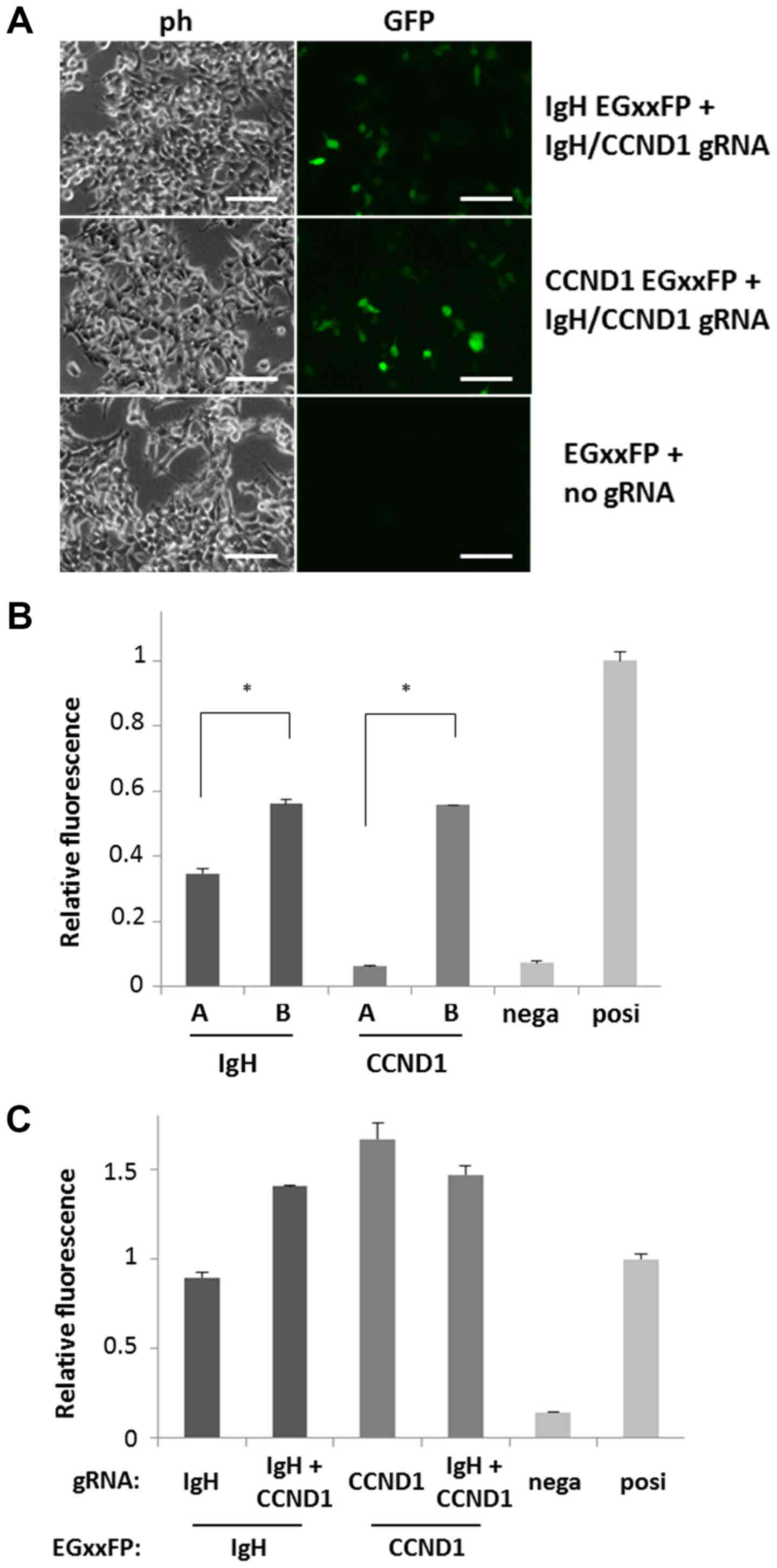 | Figure 2.Confirmation of DNA cutting activity
of the gRNA candidates. (A) 293T cells were introduced with target
DNA (IgH, CCND1 or empty)-containing EGxxFP and gRNA
(IgH and CCND1 or empty) expressing lentiCRISPRv2
vectors. Cells with high fluorescence indicate efficient gRNA
activity to cut target DNA region. Scale bars, 50 µm. (B) gRNA
activity are compared among IgH and CCND1 gRNA
candidates-coding lentiCRISPRv2. Bars A and B indicate the negative
control (empty lentiCRISPRv2) and positive control (pX330
Cetn1), respectively. Every gRNA vector was co-transfected
with the gRNA target DNA-containing EGxxFP vector, and the
intensity of cell fluorescence was analyzed by flow cytometry.
Relative fluorescence compared to positive control is indicated
(n=3). IgH-B and CCND1-B exhibited stronger
fluorescence compared to IgH-A and CCND1-A,
respectively. *P<0.001, as indicated. (C) Activity of single
(IgH-B or CCND1-B) gRNA and dual (IgH-B and
CCND1-B) gRNA expressing vectors were compared by
co-transfecting with the gRNA target DNA-containing EGxxFP vectors
(indicates as EGxxFP:). Negative and positive controls are the same
as those in 2B. IgH, immunoglobulin heavy chain; CCND1, cyclin D1;
ph, phase contrast micrograph; GFP, green fluorescent protein;
nega, negative; posi, positive. |
To induce chromosomal translocation with a single
vector capable of editing both IgH and CCND1, the
U6-CCND1 gRNA portion of CCND1-B lentiCRISPRv2 was
PCR-amplified and cloned downstream of the U6-IgH gRNA of
IgH-B lentiCRISPRv2 (Fig.
1B). The tandem gRNA (IgH-CCND1) lentiCRISPRv2 vector
exhibited activity similar to that of EGxxFP fluorescence recovery
compared with the IgH or CCND1 single-target vectors
using IgH-B or CCND1-B EGxxFP vectors (Fig. 2C). There were some differences of
activities between single and tandem gRNA vectors because of
unknown reasons. However, we could detect similar activities to
that of the positive control reproducibly using these vectors.
Induction of chromosomal
translocation
The dual gRNA lentiCRISPRv2 vector was packaged into
a lentivirus to observe its translocation inducing activity, and
then 293T cells were infected with the virus. Puromycin resistant
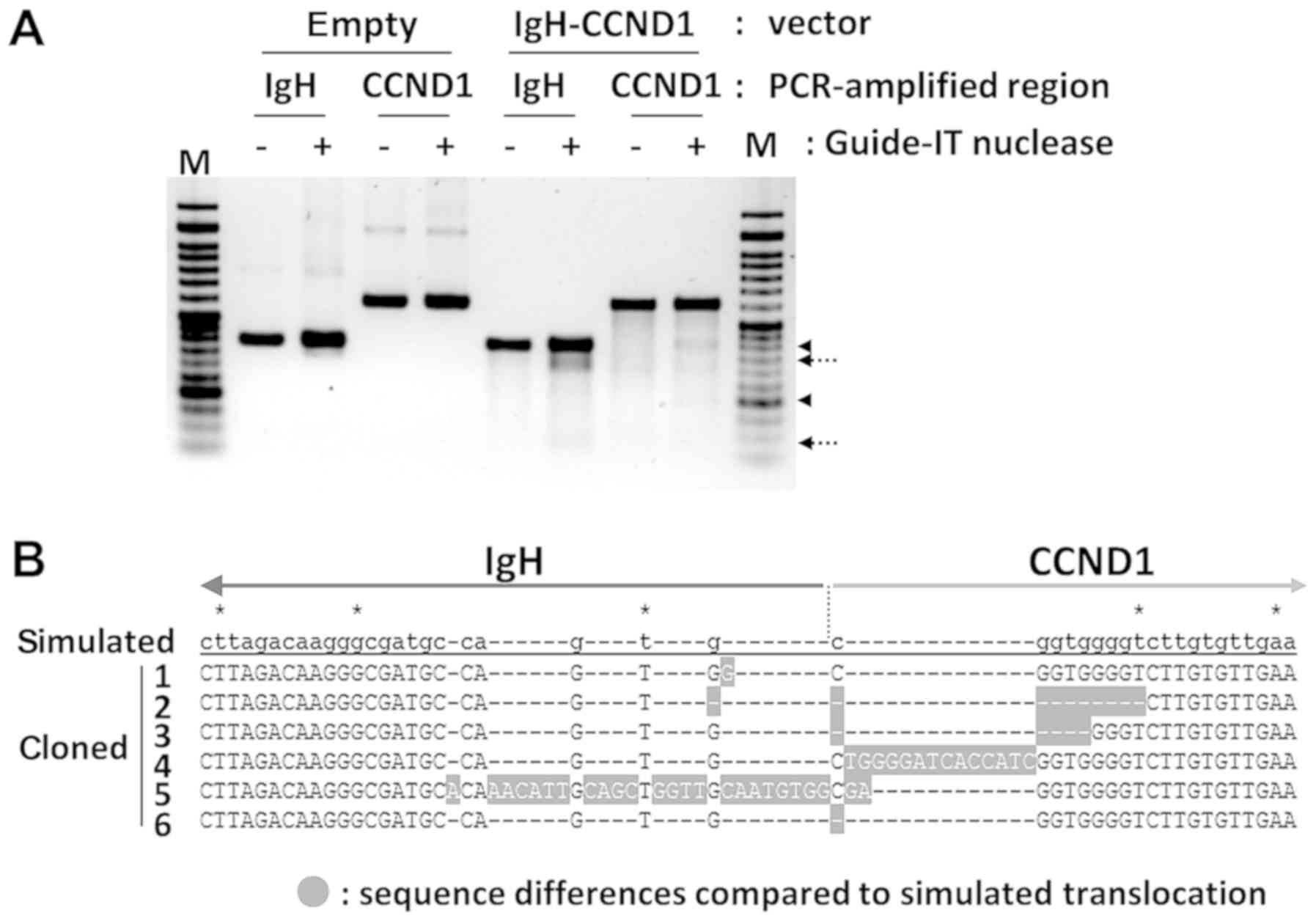
cells were isolated after seven days. Genome editing activity was
confirmed using a mismatch cleavage assay with PCR-amplified
IgH and CCND1 genome regions from the DNA of
puromycin-resistant cells. Mismatch cleavages by Guide-IT nuclease
were indicated in vector-containing cells, demonstrating that
Cas9-mediated double-strand breaks and repair by non-homologous end
joining (NHEJ) had occurred (Fig.
3A). Translocation-specific PCR was then performed to confirm
the presence of the designed translocation in puromycin-resistant
cell DNA. Q-PCR analysis using translocation control DNA as the
reference revealed a ratio of translocation-positive cells of
1/2,400 in 293T cells, whereas the ratio was 1/70 in 293 cells.
Then, DNA bands of the expected length in translocation-positive
cells were isolated and cloned into the TA vector, and the DNA
sequence of several clones was determined. The observation of
characteristic insertion and deletion of several base pairs in the
DNA sequencing results for six clones showed that NHEJ had occurred
(Fig. 3B). In addition, IgH
and CCND1 gRNA target regions in the non-translocated allele
were also PCR-amplified and cloned. DNA sequence of four clones
each for IgH and CCND1 had small deletions showing
that genome editing occurred even in the non-translocated alleles
(data not shown).
To clone translocation-positive 293T cells, Q-PCR
was first used to identify wells containing positive cells in
48-well plates inoculated with 200 cells/well. Positive wells were
then reseeded at 20 cells/well in 24-well plates, with subsequent
identification of wells containing positive cells by Q-PCR as
described above. Positive wells were inoculated into dishes at 100
cells/dish, and single colonies were then picked and expanded. We
ultimately succeeded in isolating translocation-positive 293T cells
using this serial dilution and PCR confirmation approach (data not
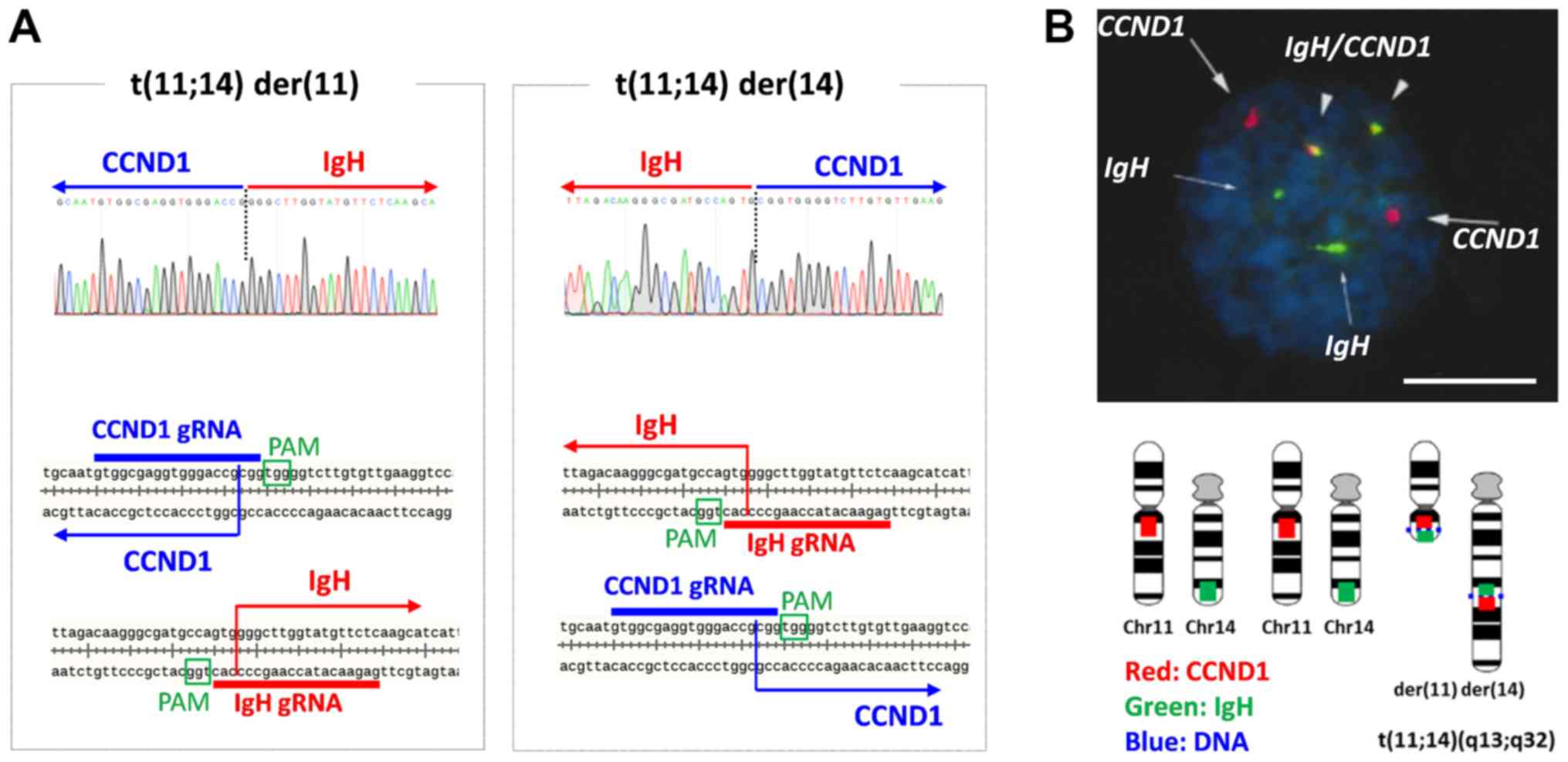
shown). Direct sequencing of translocation-specific PCR products
showed that the junction points of t(11;14) der(11) with the Chr11
centromere and der(14) with the Chr14 centromere were 3 bp from the
PAM sequences shown in Fig. 3A. As
reported, the Cas9 editing site is 3 bp from the PAM; direct
rejoining occurred after editing in this cell clone. Reciprocal
chromosomal translocation was also confirmed by fluorescence in
situ hybridization (FISH) probing (IgH [green] and
CCND1 [red] in Fig. 4B). In
triploid karyotype of 293T cells, only one pair of chromosome 11
and 14 was engaged with t(11;14), whereas other two were not.
Characteristics of t(11;14)-positive
cells
IgH enhancer-driven CCND1 overexpression by
t(11;14) is one of the key factors in oncogenic cell proliferation
in MM cells, because the immunoglobulin genes are highly active to
produce antibodies only in the B lymphocyte lineage. Therefore, it
is difficult to observe IgH enhancer-mediated activation of
CCND1 gene unless the cells with t(11;14) differentiate to B
lymphocytes. However, silent IgH gene in other cell species
including 293, which is embryonal kidney origin, may negatively
affect neighboring gene expression (33). And it is possible that such
macroscopic changes induced by chromosomal translocation could
alter the cellular characteristics through gene expression.
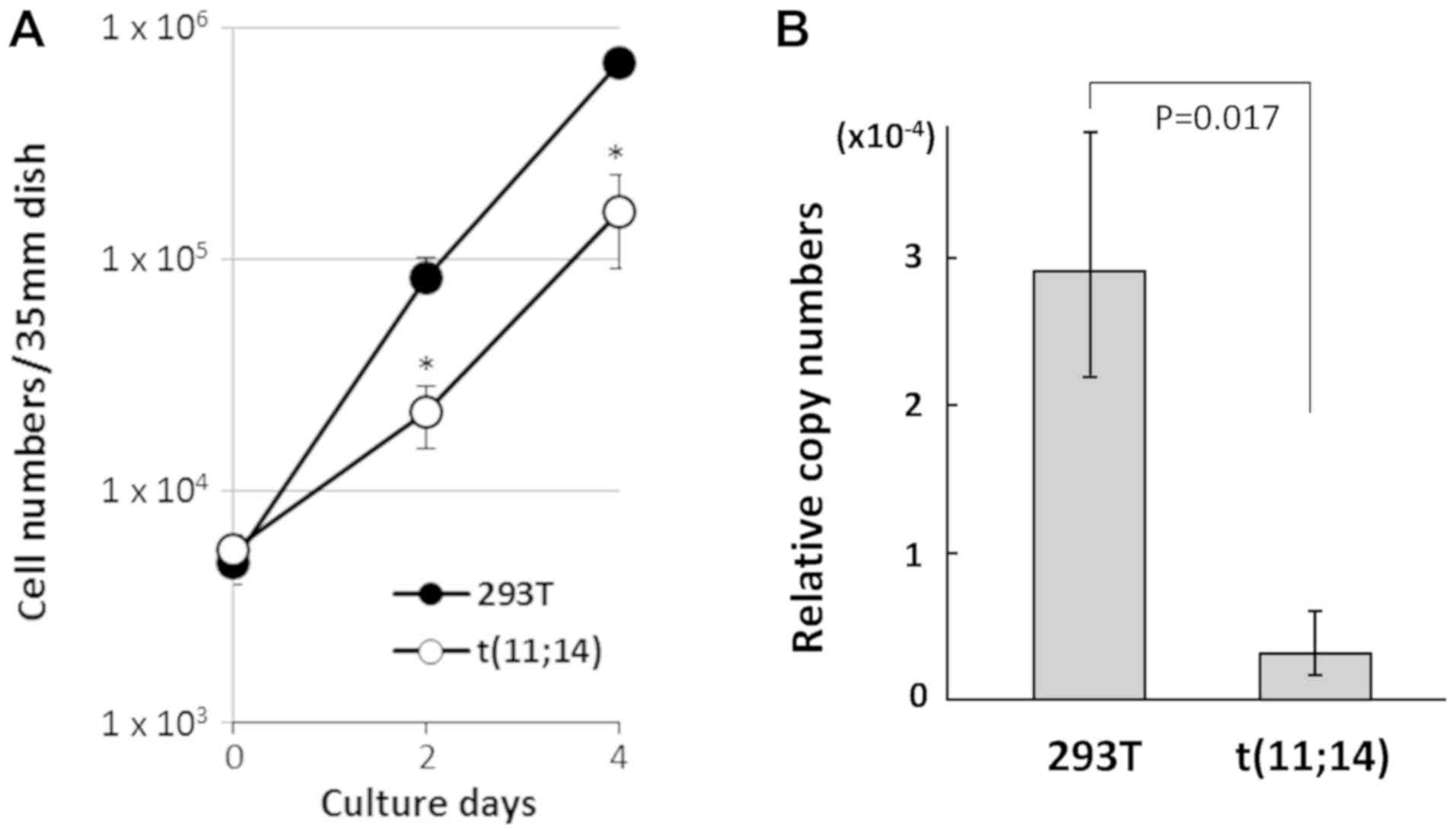
Accordingly, we compared the cell proliferation rate and
CCND1 gene expression in t(11;14)-positive cells to that of
parental 293T cells. As shown in Fig.
5A, t(11;14)-positive cells exhibited slower proliferation than
293T cells. A comparison of gene expression of the CCND1
using GAPDH as control by RT-Q-PCR revealed lower
CCND1 expression in the t(11;14)-positive cells compared
with parental 293T cells (Fig. 5B).
As a previous report suggested that the level of CCND1
expression is positively correlated with cell proliferation
(34), it is possible that decreased
CCND1 expression caused by the silent IgH enhancer
repositioned in the upstream of CCND1 gene in
t(11;14)-positive cells could result in slower growth compared with
parental 293T cells in strong contrast to MM cells. In addition,
MYEOV gene which locates in the upstream of CCND1
gene and is often overexpressed in MM cells by the translocated
IgH variable region in t(11;14) der(11) (35) was also analyzed. But we couldn't
detect MYEOV gene expression by RT-PCR both in
t(11;14)-positive cells and parental 293T cells because of the
restricted expression of this gene (gtexportal.org/home/gene/ENSG00000172927.3, The
Genotype-Tissue Expression database).
Discussion
In this study, we successfully induced artificial
chromosomal translocation between the IgH constant region
promoter/enhancer and the CCND1 protein-coding region using
an IgH and CCND1-specific CRISPR/Cas9 genome editing
system. In contrast to previous reports describing induction of
growth-enhancing fusion genes using CRISPR/Cas9 (22–27), we
encountered growth repression in the non-B lymphocyte target cells,
which could have been associated with the transcriptionally silent
IgH gene. These data suggest that selection pressure
specific to translocation-positive cells is important to isolate
the cells with such growth-suppressing translocations.
Recent reports have described efficient induction of
targeted chromosomal translocation by introduction of a HDR
template DNA which mimics translocation junction sequences
(27,36). The template DNA carries a
5′-translocation donor gene fragment and 3′-recipient gene fragment
separated by a selection marker, and is introduced into cells with
routine translocation site-specific CRISPR/Cas9 vectors. After the
two target sequences are specifically cut by Cas9s, breakpoints are
repaired by HDR using the template DNA to form chromosomal
translocation. Furthermore, the use of HSVtk for negative selection
of off-target recombination of template DNA (37), knockout of p53 to suppress apoptotic
cell death (38,39), and addition of NHEJ inhibitors to
shift DNA double strand break repair system to HDR (40–42) also
enhance HDR-template mediated genome editing. As application of
these techniques can accelerate selective induction of difficult
chromosomal translocations, we are planning to explore use of these
techniques in combinations for future studies.
Although CCND1 expression in cells with
translocations is thought to be associated with the transcriptional
silence of IgH in cells other than B lymphocytes, we were
able to obtain cells with t(11;14), suggesting that we should be
able to induce this type of translocation in other cells. In
addition, different types of immunoglobulin-associated
translocations observed in B lymphocytic malignancies could be
induced using a similar CRISPR/Cas9 system. For studying the
process of myelomagenesis in the viewpoint of IgH
enhancer-driven CCND1 overexpression, we plan to induce
t(11;14) in B lymphocyte-derived iPS cells (BiPSCs) in which
IgH genes are silent (43)
using this technique. As we have already confirmed that our BiPSCs
can be differentiated into hematopoietic stem cells (HSCs), we will
check if t(11;14)-carrying BiPSCs are also capable of
differentiating into HSCs and B lymphocyte lineage in vitro.
Moreover, we will analyze the possible function of t(11;14) and
CCND1 overexpression on B-cell differentiation and tumor
development after transplantation into mouse bone marrow by
comparing t(11;14)-BiPSC and parental BiPSC.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by
grants-in-aid for Scientific Research (grant nos. 15K00543 and
16K18424) from the Japanese Ministry of Education, Culture, Sports,
Science, and Technology.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
NT designed and performed the study. YA conducted
the growth analysis and RT-qPCR. AY, YY and MS contributed to the
cell culture and gene delivery experiments. AK and FK were involved
in the design and construction of the CRISPR vectors. KK was
involved in the experimental design. AS conceived and designed the
study. All authors read and approved the manuscript, and agreed to
be accountable for all aspects of the work in ensuring that
questions related to the accuracy or integrity of any part of the
work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rabbitts TH: Chromosomal translocations in
human cancer. Nature. 372:143–149. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nambiar M, Kari V and Raghavan SC:
Chromosomal translocations in cancer. Biochim Biophys Acta.
1786:139–152. 2008.PubMed/NCBI
|
|
3
|
Lieber MR: Mechanisms of human lymphoid
chromosomal translocations. Nat Rev Cancer. 16:387–398. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chesi M, Bergsagel PL, Brents LA, Smith
CM, Gerhard DS and Kuehl WM: Dysregulation of cyclin D1 by
translocation into an IgH gamma switch region in two multiple
myeloma cell lines. Blood. 88:674–681. 1996.PubMed/NCBI
|
|
5
|
Chesi M, Nardini E, Brents LA, Schrock E,
Ried T, Kuehl WM and Bergsagel PL: Frequent translocation
t(4;14)(p16.3;q32.3) in multiple myeloma is associated with
increased expression and activating mutations of fibroblast growth
factor receptor 3. Nat Genet. 16:260–264. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chesi M, Bergsagel PL, Shonukan OO,
Martelli ML, Brents LA, Chen T, Schrock E, Ried T and Kuehl WM:
Frequent dysregulation of the c-maf proto-oncogene at 16q23 by
translocation to an Ig locus in multiple myeloma. Blood.
91:4457–4463. 1998.PubMed/NCBI
|
|
7
|
Inaba T, Matsushime H, Valentine M,
Roussel MF, Sherr CJ and Look AT: Genomic organization, chromosomal
localization, and independent expression of human cyclin D genes.
Genomics. 13:565–574. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Szepetowski P, Perucca-Lostanlen D and
Gaudray P: Mapping genes according to their amplification status in
tumor cells: Contribution to the map of 11q13. Genomics.
16:745–750. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sherr CJ: Mammalian G1 cyclins. Cell.
73:1059–1065. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fonseca R, Blood EA, Oken MM, Kyle RA,
Dewald GW, Bailey RJ, Van Wier SA, Henderson KJ, Hoyer JD,
Harrington D, et al: Myeloma and the t(11;14)(q13;q32); evidence
for a biologically defined unique subset of patients. Blood.
99:3735–3741. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fenton JA, Pratt G, Rothwell DG, Rawstron
AC and Morgan GJ: Translocation t(11;14) in multiple myeloma:
Analysis of translocation breakpoints on der(11) and der(14)
chromosomes suggests complex molecular mechanisms of recombination.
Genes Chromosomes Cancer. 39:151–155. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Walker BA, Wardell CP, Johnson DC, Kaiser
MF, Begum DB, Dahir NB, Ross FM, Davies FE, Gonzalez D and Morgan
GJ: Characterization of IGH locus breakpoints in multiple myeloma
indicates a subset of translocations appear to occur in pregerminal
center B cells. Blood. 121:3413–3419. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu Z, Zan H, Pone EJ, Mai T and Casali P:
Immunoglobulin class-switch DNA recombination: Induction, targeting
and beyond. Nat Rev Immunol. 12:517–531. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Casellas R, Basu U, Yewdell WT, Chaudhuri
J, Robbiani DF and Di Noia JM: Mutations, kataegis and
translocations in B cells: Understanding AID promiscuous activity.
Nat Rev Immunol. 16:164–176. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nambiar M and Raghavan SC: How does DNA
break during chromosomal translocations? Nucleic Acids Res.
39:5813–5825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Van der Oost J: Molecular biology. New
tool for genome surgery. Science. 339:768–770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gaj T, Gersbach CA and Barbas CF III: ZFN,
TALEN, and CRISPR/Cas-based methods for genome engineering. Trends
Biotechnol. 31:397–405. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jinek M, Chylinski K, Fonfara I, Hauer M,
Doudna JA and Charpentier E: A programmable dual-RNA-guided DNA
endonuclease in adaptive bacterial immunity. Science. 337:816–821.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cong L, Ran FA, Cox D, Lin S, Barretto R,
Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA and Zhang F:
Multiplex genome engineering using CRISPR/Cas systems. Science.
339:819–823. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mali P, Yang L, Esvelt KM, Aach J, Guell
M, DiCarlo JE, Norville JE and Church GM: RNA-guided human genome
engineering via Cas9. Science. 339:823–826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moreno-Mateos MA, Vejnar CE, Beaudoin JD,
Fernandez JP, Mis EK, Khokha MK and Giraldez AJ: CRISPRscan:
Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in
vivo. Nat Methods. 12:982–988. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choi PS and Meyerson M: Targeted genomic
rearrangements using CRISPR/Cas technology. Nat Commun. 5:37282014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Breese EH, Buechele C, Dawson C, Cleary ML
and Porteus MH: Use of genome engineering to create patient
specific MLL translocations in primary human hematopoietic stem and
progenitor cells. PLoS One. 10:e01366442015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang J, Zhang L, Zhou X, Chen X, Huang G,
Li F, Wang R, Wu N, Yan Y, Tong C, et al: Induction of
site-specific chromosomal translocations in embryonic stem cells by
CRISPR/Cas9. Sci Rep. 6:219182016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lekomtsev S, Aligianni S, Lapao A and
Bürckstummer T: Efficient generation and reversion of chromosomal
translocations using CRISPR/Cas technology. BMC Genomics.
17:7392016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reimer J, Knoss S, Labuhn M, Charpentier
EM, Gohring G, Schlegelberger B, Klusmann JH and Heckl D:
CRISPR-Cas9-induced t(11;19)/MLL-ENL translocations initiate
leukemia in human hematopoietic progenitor cells in vivo.
Haematologica. 102:1558–1566. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vanoli F, Tomishima M, Feng W, Lamribet K,
Babin L, Brunet E and Jasin M: CRISPR-Cas9-guided oncogenic
chromosomal translocations with conditional fusion protein
expression in human mesenchymal cells. Proc Natl Acad Sci USA.
114:3696–3701. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mashiko D, Fujihara Y, Satouh Y, Miyata H,
Isotani A and Ikawa M: Generation of mutant mice by pronuclear
injection of circular plasmid expressing Cas9 and single guided
RNA. Sci Rep. 3:33552013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cepko C: Large-scale preparation and
concentration of retrovirus stocks. Curr Protoc Mol Biol. Chapter
9: Unit 0.12. 1997.doi: 10.1002/0471142727.mb0912s37.
|
|
30
|
Doench JG, Fusi N, Sullender M, Hegde M,
Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et
al: Optimized sgRNA design to maximize activity and minimize
off-target effects of CRISPR-Cas9. Nat Biotechnol. 34:184–191.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ronchetti D, Finelli P, Richelda R,
Baldini L, Rocchi M, Viggiano L, Cuneo A, Bogni S, Fabris S,
Lombardi L, et al: Molecular analysis of 11q13 breakpoints in
multiple myeloma. Blood. 93:1330–1337. 1999.PubMed/NCBI
|
|
33
|
Jhunjhunwala S, van Zelm MC, Peak MM,
Cutchin S, Riblet R, van Dongen JJ, Grosveld FG, Knoch TA and Murre
C: The 3D structure of the immunoglobulin heavy-chain locus:
Implications for long-range genomic interactions. Cell.
133:265–279. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zwijsen RM, Klompmaker R, Wientjens EB,
Kristel PM, van der Burg B and Michalides RJ: cyclin D1 triggers
autonomous growth of breast cancer cells by governing cell cycle
exit. Mol Cell Biol. 16:2554–2560. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Janssen JW, Vaandrager JW, Heuser T, Jauch
A, Kluin PM, Geelen E, Bergsagel PL, Kuehl WM, Drexler HG, Otsuki
T, et al: Concurrent activation of a novel putative transforming
gene, myeov, and cyclin D1 in a subset of multiple myeloma cell
lines with t(11;14)(q13;q32). Blood. 95:2691–2698. 2000.PubMed/NCBI
|
|
36
|
Spraggon L, Martelotto LG, Hmeljak J,
Hitchman TD, Wang J, Wang L, Slotkin EK, Fan PD, Reis-Filho JS and
Ladanyi M: Generation of conditional oncogenic chromosomal
translocations using CRISPR-Cas9 genomic editing and
homology-directed repair. J Pathol. 242:102–112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gondo Y, Nakamura K, Nakao K, Sasaoka T,
Ito K, Kimura M and Katsuki M: Gene replacement of the p53 gene
with the lacZ gene in mouse embryonic stem cells and mice by using
two steps of homologous recombination. Biochem Biophys Res Commun.
202:830–837. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Haapaniemi E, Botla S, Persson J,
Schmierer B and Taipale J: CRISPR-Cas9 genome editing induces a
p53-mediated DNA damage response. Nat Med. 24:927–930. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ihry RJ, Worringer KA, Salick MR, Frias E,
Ho D, Theriault K, Kommineni S, Chen J, Sondey M, Ye C, et al: p53
inhibits CRISPR-Cas9 engineering in human pluripotent stem cells.
Nat Med. 24:939–946. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu C, Liu Y, Ma T, Liu K, Xu S, Zhang Y,
Liu H, La Russa M, Xie M, Ding S and Qi LS: Small molecules enhance
CRISPR genome editing in pluripotent stem cells. Cell Stem Cell.
16:142–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song J, Yang D, Xu J, Zhu T, Chen YE and
Zhang J: RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in
efficiency. Nat Commun. 7:105482016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li G, Zhang X, Zhong C, Mo J, Quan R, Yang
J, Liu D, Li Z, Yang H and Wu Z: Small molecules enhance
CRISPR/Cas9-mediated homology-directed genome editing in primary
cells. Sci Rep. 7:89432017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kawamura F, Inaki M, Katafuchi A, Abe Y,
Tsuyama N, Kurosu Y, Yanagi A, Higuchi M, Muto S, Yamaura T, et al:
Establishment of induced pluripotent stem cells from normal B cells
and inducing AID expression in their differentiation into
hematopoietic progenitor cells. Sci Rep. 7:16592017. View Article : Google Scholar : PubMed/NCBI
|