Currently, epidermal growth factor receptor tyrosine
kinase inhibitors (EGFR-TKIs) are efficacious in treating patients
with NSCLC with sensitive EGFR mutations (4). These drugs have become the standard
therapy for patients with advanced NSCLC with sensitive mutations
(4). Compared with platinum-based
chemotherapy, treatment with EGFR-TKI may improve the overall
survival time of patients with NSCLC with EGFR exon 19 deletion and
mutations in exon 21 (L858R), exon 18 (G719X) and exon 20 (S768I)
(5,6). However, following 8–10 months of
treatment, numerous patients who originally responded to EGFR-TKIs
eventually develop drug resistance (7). Strategies designed to reverse primary
or acquired resistance to EGFR-TKIs are required for the
development of more effective treatments. Previous studies
demonstrated that EGFR-TKI-induced tumor microenvironment stresses
and autophagy are important causes of resistance (8,9).
The current review focused on the known molecular
mechanisms of EGFR-mediated regulation of autophagy. The role of
autophagy in EGFR-TKI treatment was also discussed. Furthermore,
co-inhibiting EGFR and autophagy signaling as a novel strategy to
improve the efficacy of EGFR-TKIs was explored.
The EGFR family consists of tyrosine transmembrane
glycoproteins that are encoded by proto-oncogenes, including human
epidermal growth factor receptor (HER)-1, HER-2, HER-3 and HR-4
(10). EGFR is mainly expressed in
epithelial, mesenchymal and neuronal cells (10). Since its discovery in the 1980s, the
EGFR signaling pathway has been implicated in organ development,
including the mammary glands and epiphyseal cartilage (11,12).
EGFR serves roles in tumor cell proliferation, differentiation,
migration, adhesion, treatment resistance, survival and apoptosis
(13–15).
At present, EGFR-targeted drugs consist of two
types: Small molecule TKIs that inhibit the tyrosine kinase
activity of the EGFR intracellular domain and artificially
synthesized EGFR monoclonal antibodies that block the extracellular
domain and inhibit the activation of the EGFR ligand binding domain
(4,5). EGFR-TKI drugs have been used clinically
for a relatively long time and are recommended by the National
Comprehensive Cancer Network and European Society for Medical
Oncology guidelines for the treatment of advanced NSCLC with
sensitive mutations (16,17). EGFR targeting is an emerging strategy
for treating other tumors, including anaplastic thyroid cancer and
colorectal cancer (18,19).
The first-generation EGFR-TKIs, gefitinib, erlotinib
and icotinib, are effective as first-line treatment for patients
with advanced NSCLC harboring activating EGFR mutations (17,20). The
second-generation EGFR-TKIs, afatinib and dacomitinib, irreversibly
bind to the tyrosine kinase of EGFR and other ErbB-family members
(4,21). Afatinib has been approved as a
first-line treatment of patients with advanced NSCLC with sensitive
mutations (21). Dacomitinib reduced
lung cancer progression in patients with NSCLC exhibiting EGFR
activating mutations compared with gefitinib in a phase III trial
(22). Third-generation EGFR-TKIs,
osimertinib (AZD9291), rociletinib (CO-1686), olmutinib (HM61713)
and others (EGF816, ASP8273), suppress EGFR activating and
resistance mutations (23,24). In clinical trials, the
third-generation drugs demonstrated higher response rates among
tumors with acquired EGFR T790M (25,26).
Although treatment efficacy has been reported in
patients treated with EGFR-TKIs, drug resistance eventually
develops following ~10 months (7).
The mechanisms resulting in this resistance are various and not
fully understood (27). The most
common mechanism is the acquisition of secondary EGFR T790M
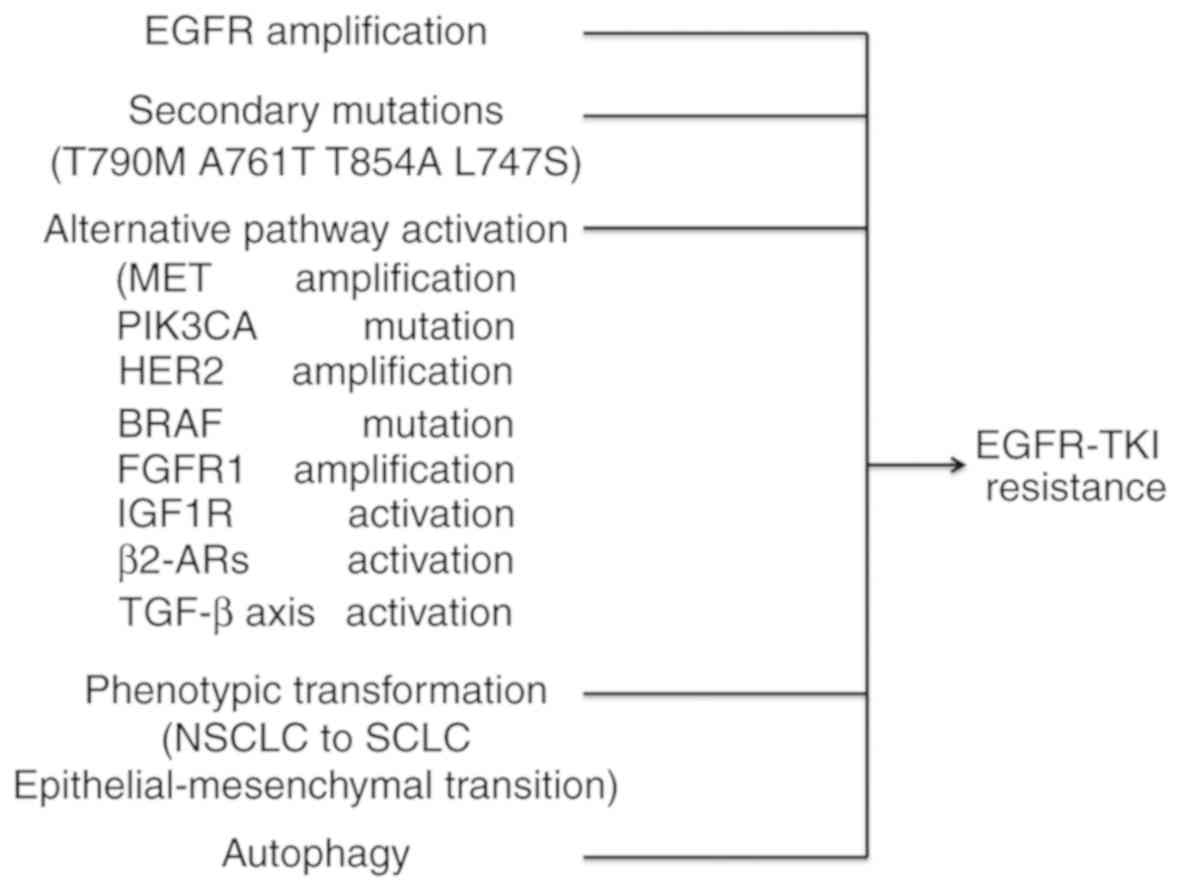
mutations (28,29). Other mechanisms of resistance may
include the following: Secondary mutations or amplification in
EGFR, alternative pathway activation, histologic and phenotypic
transformation, tumor growth factor β-dependent interleukin (IL)-6
secretion (30) or
β2-adrenergic receptors (β2-ARs) activation
(Fig. 1) (31). Previous studies demonstrated that
EGFR-TKI-induced tumor microenvironment stresses and autophagy are
important causes of resistance (8,9).
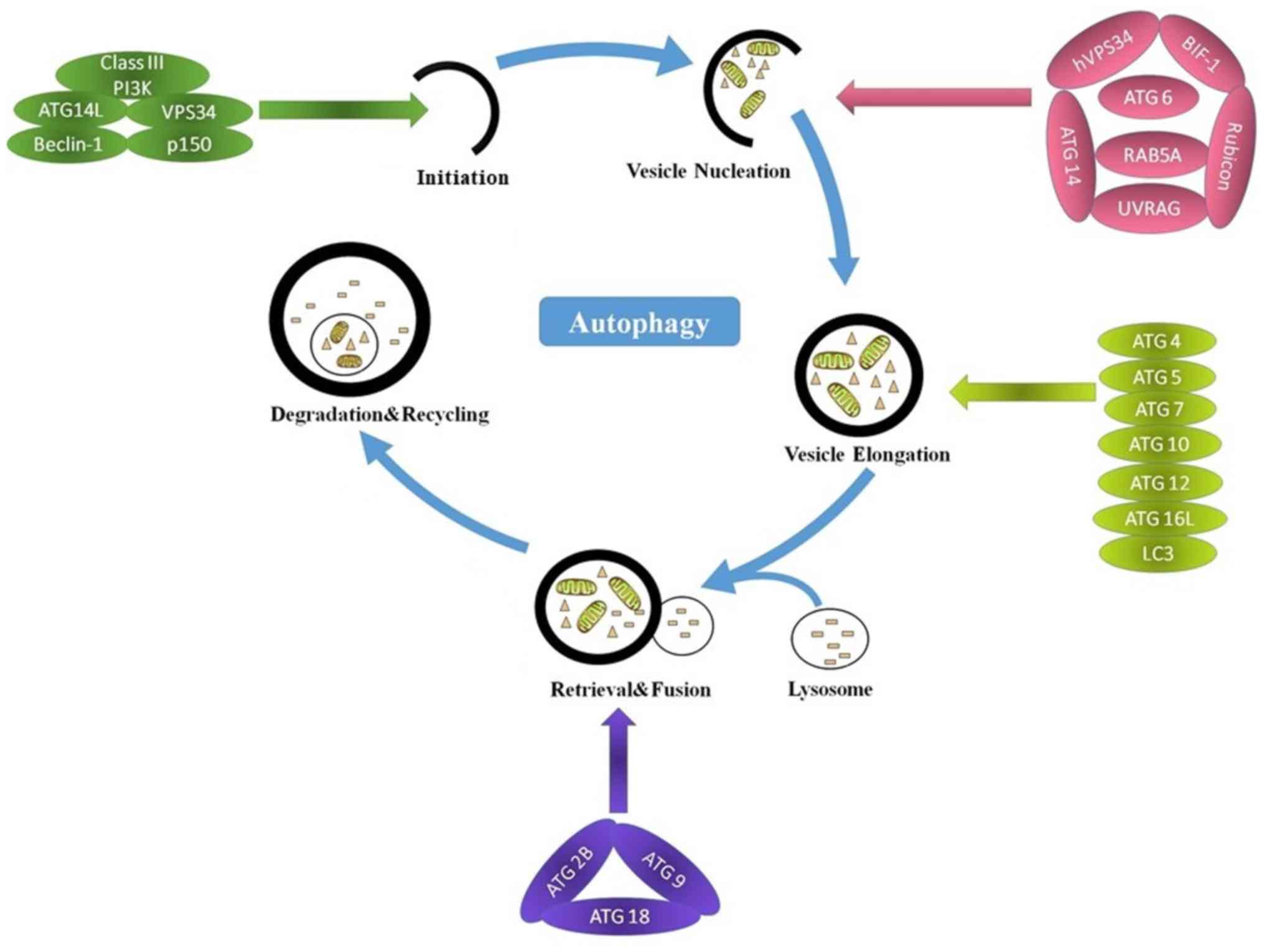
Autophagy can be divided into three main steps:
Initiation, elongation and degradation (32). Autophagy is regulated by complex
signaling pathways and autophagy-related genes (ATGs) (36,37). The
main steps and molecular regulators of autophagy are presented in
Fig. 2. Long non-coding RNA (lncRNA)
has been implicated in the regulation of autophagy (38,39).
microRNAs (miRNAs/miRs) have been reported to regulate certain ATGs
at different stages of autophagy (40). Furthermore, the majority of lncRNAs
serve a regulatory role by acting as sponges to sequester
autophagy-associated miRNAs from their targets (41–43).
Metastasis associated lung adenocarcinoma transcript 1 may
upregulate the expression of the miR-101 targets stathmin 1, RAB5A,
member RAS oncogene family and autophagy related 4D cysteine
peptidase by acting as a sponge of miR-101, thus inducing autophagy
(41). In
anoxia/reoxygenation-induced autophagy of cardiomyocytes, the
lncRNA autophagy promoting factor is upregulated to protect ATG7
from being degraded by miR-188-3p, and this promotes autophagic
death of cardiomyocytes (42). The
lncRNA HOXA transcript antisense RNA, myeloid-specific 1 regulates
autophagy by sequestering miR-20a/106b and miR-125b and their
targets unc-51 like autophagy activating kinase 1 (ULK1), E2F
transcription factor 1 and DNA damage regulated autophagy modulator
2 (43).
The downstream pathways triggered by EGFR activation
include the EGFR-Ras-rapidly accelerated fibrosarcoma (Raf)-c-Jun
N-terminal (JNK), EGFR-PI3K/protein kinase B (AKT)/mechanistic
target of rapamycin (mTOR), and EGFR-janus kinase (JAK)-signal
transducer and activator of transcription 3 (STAT3) signaling
pathways, all of which have regulatory effects on autophagy. The
EGFR-Ras-Raf-JNK signaling pathway exerts a potent stimulatory
effect on autophagy, while the EGFR-phosphoinositide 3-kinase
(PI3K)/AKT/mTOR signaling pathway exerts a potent inhibitory effect
on autophagy. The EGFR-JAK-STAT3 signaling pathway serves
stimulatory and inhibitory roles in autophagy (44).
The RAS oncogene serves important roles in the
regulation of cell survival and growth and is frequently activated
in cancer. Following autophosphorylation, the adaptor protein
growth factor receptor-bound protein 2 binds EGFR at the
phosphorylated sites and activates son of sevenless (SOS), a
guanosine triphosphate (GTP)-exchange factor for RAS. SOS then
converts RAS-guanosine disphosphate into RAS-GTP (45–47).
Previous studies demonstrated that autophagy is required for
oncogenic Ras-induced malignant cell transformation (45). Increased autophagy in these K-ras
mutation tumor cells is required for cell survival and
transformation. Genetic inhibition of autophagy in RAS-transformed
cells leads to decreased cell survival during starvation and
abrogated tumorigenesis in mice (45). Alexandrova et al (46) revealed that oncogenic K-Ras
expression upregulated autophagy through reactive oxygen species,
p38, mitogen-activated protein kinase and JNK activation and
subsequent upregulation of ATG5 and ATG7. Furthermore, JNK
phosphorylates beclin-1 (BECN1) at three different tyrosine
residues, T79, S60 and S87, as well as B-cell lymphoma 2 (Bcl-2),
leading to the separation of BECN1 from the BECN1/Bcl-2 complex in
response to starvation (47). The
release of BECN1 results in autophagy activation (36).
PI3Ks are a family of lipid kinases, and class I are
involved in tumorigenesis. Class I consists of a regulatory subunit
p85 and a catalytic subunit p110. Class I kinases are often
activated by growth factor stimulation through EGFR. The p85
regulatory subunit directly binds to phosphotyrosine residues on
EGFR (48). This binding removes the
intermolecular inhibition of the p110 catalytic subunit, allowing
p110 to phosphorylate phosphatidylinositol-3,4 biphosphate into
phosphatidylinositol-3,4,5 biphosphate (PIP3) (48). AKT is subsequently recruited to the
plasma membrane by PIP3 and phosphorylated by pyruvate
dehydrogenase kinase 1 at Thr308 and Ser473. AKT activates mTOR,
relieving its negative effect on autophagy regulation (48). mTOR is a serine/threonine protein
kinase that phosphorylates and inactivates unc-51-like autophagy
activating kinase (ULK) 1/2 (49).
The inhibited PI3K/AKT1 signaling upregulates the inhibitory
activity of tuberous sclerosis complex 1/2 on Ras homolog,
mammalian target of rapamycin complex-1 (mTORC1) binding, which is
essential for mTOR activity in conditions of starvation or growth
factor receptor inhibition. The decline in mTOR activity
subsequently separates mTORC1 from the ULK1/2 complex [including
ULK1/2, ATG13, ATG101, and RB1 inducible coiled-coil 1 (RB1CC1)]
and thus activates ULK1/2. The activated ULK1/2 phosphorylates
ATG13 and RB1CC1, two components of the ULK1/2 complex, which
subsequently initiates the autophagy cascade (50).
The JAK/STAT signaling pathway is a major pathway
which is activated by EGFR family members (44). The STAT3 gene located on chromosome
17q21 encodes an 89 kDa protein. STAT3 belongs to a family of
transcription factors that mainly exist in the cytoplasm (51). Growth-factor receptor tyrosine
kinases, cytokine-receptor-associated kinases and nonreceptor
tyrosine kinases phosphorylate conserved tyrosine residue 705 on
STAT3, resulting in its activation and translocation from the
cytoplasm to the nucleus (51).
Unphosphorylated STAT3 can form dimers and translocate into the
nucleus; however, tyrosine phosphorylation enhances STAT3
dimerization and translocation into the nucleus (52). Once in the nucleus, STAT3 regulates
genes involved in cell proliferation, differentiation, survival and
angiogenesis (52). STAT3 is
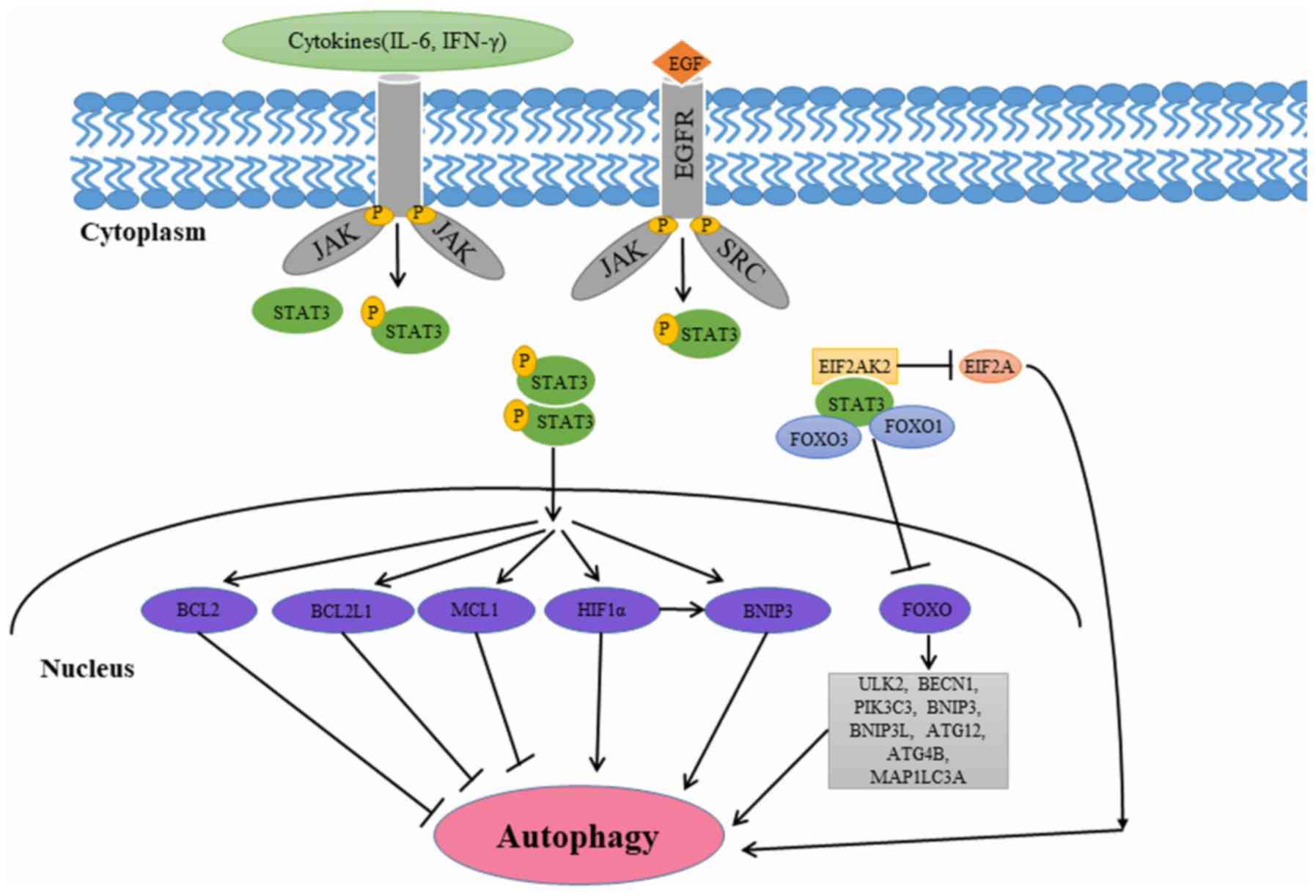
involved in multiple aspects of autophagy (53–58). The
different subcellular localization patterns of STAT3 affect
autophagy in a transcriptional or nontranscriptional manner
(Fig. 3).
Several studies have revealed that cytoplasmic STAT3
suppresses autophagy by interacting with eukaryotic translation
initiation factor 2 a kinase 2 (EIF2AK2), forkhead box protein
(FOX) O1, and FOXO3 (53–56). Niso-Santano et al (53) demonstrated that cytoplasmic STAT3 may
bind to the EIF2AK2 catalytic domain competitively, inhibiting its
function in the process. Thus, STAT3 may function as a competitive
inhibitor of EIF2AK2, inhibiting EIF2AK2 enzymatic activity and
phosphorylation of EIF2A, a known autophagy activator (53). Eukaryotic translation initiation
factor 2A-activating transcription factor 4 signaling
pathway-mediated cyclooxygenase 2 overexpression may contribute to
cadmium-induced autophagy in kidney (54). Furthermore, cytoplasmic STAT3 may
interact with the autophagy-associated proteins FOXO1 and FOXO3
(55). FOXO3 can upregulate multiple
autophagy-associated genes including ULK2, BECN1,
phosphatidylinositol 3-kinase catalytic subunit type 3, Bcl-2
interacting protein 3 (BNIP3), Bcl-2 interacting protein 3 like,
ATG12, ATG4B and microtubule associated protein 1 light chain 3 a
following dephosphorylation and translocation to the nucleus. The
active FOXO1 and FOXO3a exist exclusively in the nucleus of naïve T
cells (56). In the cytoplasm of
activated T cells, the inactive pFOXO1 and pFOXO3a integrate with
unphosphorylated STAT3 (56).
FOXO1/FOXO3a rapidly relocalize to the nucleus following IL-6 or
IL-10 mediated pSTAT3 activation. STAT3 inhibitors completely
inhibit cytokine-induced translocation of FOXO1/FOXO3a into the
nucleus (56).
Nuclear STAT3 increases the expression of negative
regulators of autophagy including Bcl-2, Bcl-2 like 1 and MCL1
apoptosis regulator, Bcl-2 family member (MCL1), thus inhibiting
autophagy (57,58). Following its activation, nuclear
STAT3 increases Bcl-2 expression, leading to autophagy inhibition
(57). In pancreatic ductal
adenocarcinoma cells, miR506 triggers autophagic flux by direct
inhibition of the STAT3-Bcl-2-BECN1 axis (59). Tai et al (60) demonstrated that sorafenib activated
autophagy in a dose- and time-dependent manner through
downregulation of phospho-STAT3 and MCL1 in hepatocellular
carcinoma cell lines. The ectopic expression of MCL1 reversed the
effect of sorafenib on autophagy. Nuclear STAT3 may inhibit
autophagy through the downregulation of phosphatidylinositol
3-kinase catalytic subunit type 3 (60). The reduction of Vps34 protein levels
in fiber type-specific regulation of autophagy and skeletal muscle
atrophy occurs in a STAT3-dependent manner, which decreases
ps34/p150/BECN1/Atg14 complex 1 (61).
Nuclear STAT3 may promote autophagy by modulating
the hypoxic expression of hypoxia-inducible factor 1α (HIF-1α) and
BNIP3 (62–65). STAT3 transcriptionally upregulates
HIF-1α and inhibits its ubiquitination, which is mediated by von
Hippel-Lindau tumor suppressor (62). Previous studies indicated that
autophagy is closely associated with hypoxia (63). HIF-1α serves an essential role in
various cellular responses; in particular, it promotes cell
proliferation and survival (64).
Hypoxia-induced autophagy serves an important role in
HIF-1α-dependent general mechanisms of cell survival (64). BNIP3 and BNIP3L are the downstream
targets of HIF-1α-dependent autophagy (65). BNIP3 is a conserved member of the
BH3-only subfamily of the pro-apoptotic Bcl-2 family, and its
expression is associated with the initiation of autophagy in
different cell models (66).
Furthermore, BNIP3 may dissociate BECN1 from the Bcl-2-BECN1complex
(67). In addition, STAT3 regulates
BNIP3 expression (68). Concanavalin
A (Con A) induced autophagy through a STAT3-macrophage migration
inhibitory factor-BNIP3-dependent signaling pathway (68). Pretreatment with
epigallocatechin-3-gallate (EGCG) abrogated the upregulation of
JAK1, JAK2, p-STAT3 and BNIP3 in a dose-dependent manner (69). The results obtained from the
aforementioned studies indicated that ECGG decreases ConA-induced
autophagy by downregulating the IL-6/JAKs/STAT/BNIP3-mediated
signaling pathway (69).
Numerous studies demonstrated that initiation of
autophagy may increase tumor resistance to anticancer therapies in
different types of cancer cells (70). In lung cancer cells, autophagy may be
activated by EGFR-TKIs, and coinhibiting EGFR signaling and
autophagy demonstrates promising results in vitro (71,72). The
autophagy induced by EGFR-TKIs executed a cytoprotective function
in lung cancer cells. Autophagy inhibition by chloroquine (CQ) and
small interfering RNAs targeting ATG5 and ATG7 increased the
cytotoxic effect of the EGFR-TKIs gefitinib and erlotinib in
vitro (71). Li et al
(72) revealed that erlotinib
induced apoptosis and autophagy in NSCLC cell lines with activating
EGFR mutations (exon 19 deletion). Inhibition of autophagy may
increase the sensitivity of NSCLC cell lines to erlotinib,
suggesting that autophagy functions as a protective mechanism.
Furthermore, the resistance to erlotinib may be reversed through
autophagy inhibition (72). Wang
et al (73) revealed that
erlotinib induced autophagy in TKI-sensitive and TKI-resistant lung
cancer cells. Inhibition of autophagy significantly increased
sensitivity to erlotinib in TKI-resistant cancer cells via
regulation of endoplasmic reticulum stress-induced apoptosis
(73). These results indicated that
cotargeting autophagy and EGFR signaling may present novel clinical
strategies in the treatment of NSCLC. The resistance to erlotinib
in wild-type EGFR-expressing NSCLC cells may be overcome through
autophagy inhibition (74).
Furthermore, a phase I clinical trial investigated the efficacy,
safety and pharmacokinetics of the combination of
hydroxychloroquine (HCQ) with erlotinib in patients with advanced
NSCLC (75). Although this study
revealed no significant improvement in survival time, the safety of
adding HCQ to erlotinib was established (75). The recommended dose of HCQ was 1,000
mg when given in combination with 150 mg of erlotinib in a phase 2
study (75). However, CQ in
combination with chemotherapy and radiation therapy increased the
median survival time of patients with glioblastoma multiforme
compared with controls in a randomized, double-blind,
placebo-controlled trial (76).
There are currently several ongoing clinical trials involving CQ or
HCQ, and preliminary antitumor activity has been demonstrated in
pancreatic adenocarcinoma, melanoma and glioblastoma (77–79).
These observations demonstrated that autophagy may
have context-dependent and even opposing effects on the behavior of
cancer cells. The efficacy of autophagy inhibition in different
types of human cancer, including pancreatic adenocarcinoma,
melanoma, and glioblastoma have also varied widely (77–79).
Autophagy may be stimulated or inhibited during cancer treatment,
depending on the type of cell, the stress signals, such as
chemotherapy, radiotherapy or target therapy, and other
circumstances, such as hypoxia or starvation. The identification of
novel biomarkers for evaluating dynamic changes in autophagy and
new methods to evaluate autophagy in clinical samples may improve
patient outcomes.
EGFR-TKIs are effective for the treatment of
patients with NSCLC with EGFR-sensitive mutations, although
acquired resistance inevitably emerges (7). EGFR and its downstream signaling
pathways serve an essential role in autophagy regulation (44). Autophagy exhibits complex,
context-dependent roles in cancer, and interventions to enhance or
inhibit autophagy have been proposed as an addition to
EGFR-TKI-based therapies (71,80).
Co-targeting autophagy signaling may be a novel therapeutic
strategy for cancer treatment. However, elucidation of the
mechanisms involved in autophagy is required prior to the use of
such strategies. Autophagy inhibition may reduce the antitumor
immune response (83). Rao et
al (83) revealed that autophagy
deficiency favored oncogenesis via changes in the tumor
microenvironment that involved the regulatory T-cell-mediated
inhibition of anticancer immunosurveillance. Autophagy also serves
important roles in the survival of dormant cancer cells (84). A recent study using a Drosophila
melanogaster tumor model demonstrated that dormant tumors from
autophagy-deficient animals reactivated tumor growth when
transplanted into autophagy-proficient animals (84). This suggested that autonomous
autophagy in the surrounding nontumor cells of the microenvironment
may be involved in the regrowth of dormant tumors.
The combination of EGFR-TKIs and autophagy
inhibitors or inducers is attracting increased attention in NSCLC
therapy. Understanding the mechanisms underlying the
context-dependent effects of autophagy on cancer may provide a
basis for making rational decisions on strategies to manipulate
autophagy during cancer therapy.
Not applicable.
The present review was supported by a grant from the
General Research Program of Zhejiang Provincial Health Department
of China (grant no. 2017KY127).
Not applicable.
XW, WL, NZ and XZ were responsible for the
literature search and manuscript preparation. ZJ was responsible
for manuscript co-writing and correction. All authors revised the
article and approved the final version for publication.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mao Y, Yang D, He J and Krasna MJ:
Epidemiology of lung cancer. Surg Oncol Clin N Am. 25:439–445.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al-Farsi A and Ellis PM: Treatment
paradigms for patients with metastatic non-small cell lung cancer,
squamous lung cancer: First, second, and third-line. Front Oncol.
4:1572014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Inal C, Yilmaz E, Piperdi B, Perez-Soler R
and Cheng H: Emerging treatment for advanced lung cancer with egfr
mutation. Expert Opin Emerg Drugs. 20:597–612. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pao W and Chmielecki J: Rational,
biologically based treatment of EGFR-mutant non-small-cell lung
cancer. Nat Rev Cancer. 10:760–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carrera S, Buque A, Azkona E, Aresti U,
Calvo B, Sancho A, Arruti M, Nuño M, Rubio I, de Lobera AR, et al:
Epidermal growth factor receptor tyrosine-kinase inhibitor
treatment resistance in non-small cell lung cancer: Biological
basis and therapeutic strategies. Clin Transl Oncol. 16:339–350.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu D, Yang Y and Zhao S: Autophagy
facilitates the EGFR-TKI acquired resistance of non-small-cell lung
cancer cells. J Formos Med Assoc. 113:141–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sakuma Y, Matsukuma S, Nakamura Y,
Yoshihara M, Koizume S, Sekiguchi H, Saito H, Nakayama H, Kameda Y,
Yokose T, et al: Enhanced autophagy is required for survival in
EGFR-independent EGFR-mutant lung adenocarcinoma cells. Lab Invest.
93:1137–1146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lemmon MA, Schlessinger J and Ferguson KM:
The egfr family: Not so prototypical receptor tyrosine kinases.
Cold Spring Harb Perspect Biol. 6:a0207682014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang X, Zhu J, Li Y, Lin T, Siclari VA,
Chandra A, Candela EM, Koyama E, Enomoto-Iwamoto M and Qin L:
Epidermal growth factor receptor (EGFR) signaling regulates
epiphyseal cartilage development through β-catenin-dependent and
-independent pathways. J Biol Chem. 288:32229–32240. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brisken C and O'Malley B: Hormone action
in the mammary gland. Cold Spring Harb Perspect Biol.
2:a0031782010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee HC, Su MY, Lo HC, Wu CC, Hu JR, Lo DM,
Chao TY, Tsai HJ and Dai MS: Cancer metastasis and EGFR signaling
is suppressed by amiodarone-induced versican V2. Oncotarget.
6:42976–42987. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Clapéron A, Mergey M, Nguyen Ho-Bouldoires
TH, Vignjevic D, Wendum D, Chrétien Y, Merabtene F, Frazao A,
Paradis V, Housset C, et al: EGF/EGFR axis contributes to the
progression of cholangiocarcinoma through the induction of an
epithelial-mesenchymal transition. J Hepatol. 61:325–332. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Masuda H, Zhang D, Bartholomeusz C,
Doihara H, Hortobagyi GN and Ueno NT: Role of epidermal growth
factor receptor in breast cancer. Breast Cancer Res Treat.
136:331–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi YK, Wang L, Han BH, Li W, Yu P, Liu
YP, Ding CM, Song X, Ma ZY, Ren XL, et al: First-line icotinib
versus cisplatin/pemetrexed plus pemetrexed maintenance therapy for
patients with advanced EGFR mutation-positive lung adenocarcinoma
(CONVINCE): A phase 3, open-label, randomized study. Ann Oncol.
28:2443–2450. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi Y, Zhang L, Liu X, Zhou C, Zhang L,
Zhang S, Wang D, Li Q, Qin S, Hu C, et al: Icotinib versus
gefitinib in previously treated advanced non-small-cell lung cancer
(ICOGEN): A randomised, double-blind phase 3 non-inferiority trial.
Lancet Oncol. 14:953–961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Antonelli A, Fallahi P, Ulisse S, Ferrari
SM, Minuto M, Saraceno G, Santini F, Mazzi V, D'Armiento M and
Miccoli P: New targeted therapies for anaplastic thyroid cancer.
Anticancer Agents Med Chem. 12:87–93. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Koustas E, Karamouzis MV, Mihailidou C,
Schizas D and Papavassiliou AG: Co-targeting of EGFR and autophagy
signaling is an emerging treatment strategy in metastatic
colorectal cancer. Cancer Lett. 396:94–102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Costa DB, Nguyen KS, Cho BC, Sequist LV,
Jackman DM, Riely GJ, Yeap BY, Halmos B, Kim JH, Jänne PA, et al:
Effects of erlotinib in EGFR mutated non-small cell lung cancers
with resistance to gefitinib. Clin Cancer Res. 14:7060–7067. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu HA and Pao W: Targeted therapies:
Afatinib-new therapy option for EGFR-mutant lung cancer. Nat Rev
Clin Oncol. 10:551–552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa
K, Niho S, Tsuji F, Linke R, Rosell R, Corral J, et al: Dacomitinib
versus gefitinib as first-line treatment for patients with
EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A
randomised, open-label, phase 3 trial. Lancet Oncol. 18:1454–1466.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang S, Cang S and Liu D: Third-generation
inhibitors targeting EGFR T790M mutation in advanced non-small cell
lung cancer. J Hematol Oncol. 9:342016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Russo A, Franchina T, Ricciardi GRR,
Smiroldo V, Picciotto M, Zanghi M, Rolfo C and Adamo V: Third
generation EGFR TKIs in EGFR-mutated NSCLC: Where are we now and
where are we going. Crit Rev Oncol Hematol. 117:38–47. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Soria JC, Ohe Y, Vansteenkiste J,
Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura
F, Nogami N, Kurata T, et al: Osimertinib in untreated EGFR-mutated
advanced non-small-cell lung cancer. N Engl J Med. 378:113–125.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Santarpia M, Liguori A, Karachaliou N,
Gonzalez-Cao M, Daffinà MG, D'Aveni A, Marabello G, Altavilla G and
Rosell R: Osimertinib in the treatment of non-small-cell lung
cancer: Design, development and place in therapy. Lung Cancer
(Auckl). 8:109–125. 2017.PubMed/NCBI
|
|
27
|
Piotrowska Z and Sequist LV: Epidermal
growth factor receptor-mutant lung cancer: New drugs, new
resistance mechanisms, and future treatment options. Cancer J.
21:371–377. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu HA, Riely GJ and Lovly CM: Therapeutic
strategies utilized in the setting of acquired resistance to EGFR
tyrosine kinase inhibitors. Clin Cancer Res. 20:5898–5907. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rotow J and Bivona TG: Understanding and
targeting resistance mechanisms in NSCLC. Nat Rev Cancer.
17:637–658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yao Z, Fenoglio S, Gao DC, Camiolo M,
Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V,
et al: TGF-beta IL-6 axis mediates selective and adaptive
mechanisms of resistance to molecular targeted therapy in lung
cancer. Proc Natl Acad Sci USA. 107:15535–15540. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nilsson MB, Sun H, Diao L, Tong P, Liu D,
Li L, Fan Y, Poteete A, Lim SO, Howells K, et al: Stress hormones
promote EGFR inhibitor resistance in NSCLC: Implications for
combinations with β-blockers. Sci Transl Med. 9(pii): eaao43072017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Levine B and Kroemer G: Autophagy in
aging, disease and death: The true identity of a cell death
impostor. Cell Death Differ. 16:1–2. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cuervo AM and Wong E: Chaperone-mediated
autophagy: Roles in disease and aging. Cell Res. 24:92–104. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang Z, Guo M, Zhao S, Xu W, Shao J,
Zhang F, Wu L, Lu Y and Zheng S: The update on transcriptional
regulation of autophagy in normal and pathologic cells: A novel
therapeutic target. Biomed Pharmacother. 74:17–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jing Z, Han W, Sui X, Xie J and Pan H:
Interaction of autophagy with microRNAs and their potential
therapeutic implications in human cancers. Cancer Lett.
356:332–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li L, Chen H, Gao Y, Wang YW, Zhang GQ,
Pan SH, Ji L, Kong R, Wang G, Jia YH, et al: Long noncoding RNA
MALAT1 promotes aggressive pancreatic cancer proliferation and
metastasis via the stimulation of autophagy. Mol Cancer Ther.
15:2232–2243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget.
6:8474–8490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fu Z, Luo W, Wang J, Peng T, Sun G, Shi J,
Li Z and Zhang B: Malat1 activates autophagy and promotes cell
proliferation by sponging miR-101 and upregulating STMN1, RAB5A and
ATG4D expression in glioma. Biochem Biophys Res Commun.
492:480–486. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang K, Liu CY, Zhou LY, Wang JX, Wang M,
Zhao B, Zhao WK, Xu SJ, Fan LH, Zhang XJ, et al: APF lncRNA
regulates autophagy and myocardial infarction by targeting
miR-188-3p. Nat Commun. 6:67792015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen ZH, Wang WT, Huang W, Fang K, Sun YM,
Liu SR, Luo XQ and Chen YQ: The lncRNA HOTAIRM1 regulates the
degradation of PML-RARA oncoprotein and myeloid cell
differentiation by enhancing the autophagy pathway. Cell Death
Differ. 24:212–224. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Henson E, Chen Y and Gibson S: EGFR family
members' regulation of autophagy is at a crossroads of cell
survival and death in cancer. Cancers (Basel). 9(pii): E272017.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nyfeler B and Eng CH: Revisiting autophagy
addiction of tumor cells. Autophagy. 12:1206–1207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Alexandrova AY, Kopnin PB, Vasiliev JM and
Kopnin BP: ROS up-regulation mediates Ras-induced changes of cell
morphology and motility. Exp Cell Res. 312:2066–2073. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhou YY, Li Y, Jiang WQ and Zhou LF:
MAPK/JNK signalling: A potential autophagy regulation pathway.
Biosci Rep. 35(pii): e001992015.PubMed/NCBI
|
|
48
|
Yip PY: Phosphatidylinositol
3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR)
signaling pathway in non-small cell lung cancer. Transl Lung Cancer
Res. 4:165–176. 2015.PubMed/NCBI
|
|
49
|
Jung CH, Seo M, Otto NM and Kim DH: ULK1
inhibits the kinase activity of mTORC1 and cell proliferation.
Autophagy. 7:1212–1221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ernst M and Putoczki TL: Stat3: Linking
inflammation to (gastrointestinal) tumourigenesis. Clin Exp
Pharmacol Physiol. 39:711–718. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Demaria M, Camporeale A and Poli V: Stat3
and metabolism: How many ways to use a single molecule? Int J
Cancer. 135:1997–2003. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Niso-Santano M, Shen S, Adjemian S, Malik
SA, Mariño G, Lachkar S, Senovilla L, Kepp O, Galluzzi L, Maiuri MC
and Kroemer G: Direct interaction between STAT3 and EIF2AK2
controls fatty acid-induced autophagy. Autophagy. 9:415–417. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Luo B, Lin Y, Jiang S, Huang L, Yao H,
Zhuang Q, Zhao R, Liu H, He C and Lin Z: Endoplasmic reticulum
stress eIF2α-ATF4 pathway-mediated cyclooxygenase-2 induction
regulates cadmium-induced autophagy in kidney. Cell Death Dis.
7:e22512016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Oh HM, Yu CR, Golestaneh N, Amadi-Obi A,
Lee YS, Eseonu A, Mahdi RM and Egwuagu CE: STAT3 protein promotes
T-cell survival and inhibits interleukin-2 production through
up-regulation of Class O Forkhead transcription factors. J Biol
Chem. 286:30888–30897. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Oh HM, Yu CR, Dambuza I, Marrero B and
Egwuagu CE: STAT3 protein interacts with Class O Forkhead
transcription factors in the cytoplasm and regulates
nuclear/cytoplasmic localization of FoxO1 and FoxO3a proteins in
CD4(+) T cells. J Biol Chem. 287:30436–30443. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ray S, Zhao Y, Jamaluddin M, Edeh CB, Lee
C and Brasier AR: Inducible STAT3 NH2 terminal mono-ubiquitination
promotes BRD4 complex formation to regulate apoptosis. Cell Signal.
26:1445–1455. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kiprianova I, Remy J, Milosch N, Mohrenz
IV, Seifert V, Aigner A and Kögel D: Sorafenib sensitizes glioma
cells to the BH3 mimetic ABT-737 by targeting MCL1 in a
STAT3-dependent manner. Neoplasia. 17:564–573. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sun L, Hu L, Cogdell D, Lu L, Gao C, Tian
W, Zhang Z, Kang Y, Fleming JB and Zhang W: MIR506 induces
autophagy-related cell death in pancreatic cancer cells by
targeting the STAT3 pathway. Autophagy. 13:703–714. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS,
Cheng AL, Chen PJ and Chen KF: Mcl-1-dependent activation of Beclin
1 mediates autophagic cell death induced by sorafenib and SC-59 in
hepatocellular carcinoma cells. Cell Death Dis. 4:e4852013.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yamada E, Bastie CC, Koga H, Wang Y,
Cuervo AM and Pessin JE: Mouse skeletal muscle fiber-type-specific
macroautophagy and muscle wasting are regulated by a
Fyn/STAT3/Vps34 signaling pathway. Cell Rep. 1:557–569. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Nechemia-Arbely Y, Khamaisi M, Rosenberger
C, Koesters R, Shina A, Geva C, Shriki A, Klaus S, Rosen S,
Rose-John S, et al: In vivo evidence suggesting reciprocal renal
hypoxia-inducible factor-1 upregulation and signal transducer and
activator of transcription 3 activation in response to hypoxic and
non-hypoxic stimuli. Clin Exp Pharmacol Physiol. 40:262–272. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li M, Tan J, Miao Y, Lei P and Zhang Q:
The dual role of autophagy under hypoxia-involvement of interaction
between autophagy and apoptosis. Apoptosis. 20:769–777. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Abdul Rahim SA, Dirkse A, Oudin A,
Schuster A, Bohler J, Barthelemy V, Muller A, Vallar L, Janji B,
Golebiewska A and Niclou SP: Regulation of hypoxia-induced
autophagy in glioblastoma involves ATG9A. Br J Cancer. 117:813–825.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hsieh DJ, Kuo WW, Lai YP, Shibu MA, Shen
CY, Pai P, Yeh YL, Lin JY, Viswanadha VP and Huang CY:
17β-estradiol and/or estrogen receptor β attenuate the autophagic
and apoptotic effects induced by prolonged hypoxia through
HIF-1α-mediated BNIP3 and IGFBP-3 signaling blockage. Cell Physiol
Biochem. 36:274–284. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wilkinson S and Ryan KM: Growth factor
signaling permits hypoxia-induced autophagy by a
HIF1alpha-dependent, BNIP3/3L-independent transcriptional program
in human cancer cells. Autophagy. 5:1068–1069. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Karpathiou G, Sivridis E, Koukourakis M,
Mikroulis D, Bouros D, Froudarakis M, Bougioukas G, Maltezos E and
Giatromanolaki A: Autophagy and Bcl-2/BNIP3 death regulatory
pathway in non-small cell lung carcinomas. APMIS. 121:592–604.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lai YC, Chuang YC, Chang CP and Yeh TM:
Macrophage migration inhibitory factor has a permissive role in
concanavalin A-induced cell death of human hepatoma cells through
autophagy. Cell Death Dis. 6:e20082015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li S, Xia Y, Chen K, Li J, Liu T, Wang F,
Lu J, Zhou Y and Guo C: Epigallocatechin-3-gallate attenuates
apoptosis and autophagy in concanavalin A-induced hepatitis by
inhibiting BNIP3. Drug Des Devel Ther. 10:631–647. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Huang Z, Zhou L, Chen Z, Nice EC and Huang
C: Stress management by autophagy: Implications for
chemoresistance. Int J Cancer. 139:23–32. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J,
Ge W, Feng L, Lin X, Wang X, et al: EGFR tyrosine kinase inhibitors
activate autophagy as a cytoprotective response in human lung
cancer cells. PLoS One. 6:e186912011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li YY, Lam SK, Mak JC, Zheng CY and Ho JC:
Erlotinib-induced autophagy in epidermal growth factor receptor
mutated non-small cell lung cancer. Lung Cancer. 81:354–361. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang Z, Du T, Dong X, Li Z, Wu G and Zhang
R: Autophagy inhibition facilitates erlotinib cytotoxicity in lung
cancer cells through modulation of endoplasmic reticulum stress.
Int J Oncol. 48:2558–2566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zou Y, Ling YH, Sironi J, Schwartz EL,
Perez-Soler R and Piperdi B: The autophagy inhibitor chloroquine
overcomes the innate resistance of wild-type EGFR non-small-cell
lung cancer cells to erlotinib. J Thorac Oncol. 8:693–702. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Goldberg SB, Supko JG, Neal JW, Muzikansky
A, Digumarthy S, Fidias P, Temel JS, Heist RS, Shaw AT, McCarthy
PO, et al: A phase I study of erlotinib and hydroxychloroquine in
advanced non-small-cell lung cancer. J Thorac Oncol. 7:1602–1608.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sotelo J, Briceño E and López-González MA:
Adding chloroquine to conventional treatment for glioblastoma
multiforme: A randomized, double-blind, placebo-controlled trial.
Ann Intern Med. 144:337–343. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Boone BA, Bahary N, Zureikat AH, Moser AJ,
Normolle DP, Wu WC, Singhi AD, Bao P, Bartlett DL, Liotta LA, et
al: Safety and biologic response of pre-operative autophagy
inhibition in combination with gemcitabine in patients with
pancreatic adenocarcinoma. Ann Surg Oncol. 22:4402–4410. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Rangwala R, Leone R, Chang YC, Fecher LA,
Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M,
et al: Phase I trial of hydroxychloroquine with dose-intense
temozolomide in patients with advanced solid tumors and melanoma.
Autophagy. 10:1369–1379. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Rosenfeld MR, Ye X, Supko JG, Desideri S,
Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, et al: A
phase I/II trial of hydroxychloroquine in conjunction with
radiation therapy and concurrent and adjuvant temozolomide in
patients with newly diagnosed glioblastoma multiforme. Autophagy.
10:1359–1368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
La Monica S, Galetti M, Alfieri RR,
Cavazzoni A, Ardizzoni A, Tiseo M, Capelletti M, Goldoni M,
Tagliaferri S, Mutti A, et al: Everolimus restores gefitinib
sensitivity in resistant non-small cell lung cancer cell lines.
Biochem Pharmacol. 78:460–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
So KS, Kim CH, Rho JK, Kim SY, Choi YJ,
Song JS, Kim WS, Choi CM, Chun YJ and Lee JC:
Autophagosome-mediated EGFR down-regulation induced by the CK2
inhibitor enhances the efficacy of EGFR-TKI on EGFR-mutant lung
cancer cells with resistance by T790M. PLoS One. 9:e1140002014.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Sano T, Takeuchi S, Nakagawa T, Ishikawa
D, Nanjo S, Yamada T, Nakamura T, Matsumoto K and Yano S: The novel
phosphoinositide 3-kinase-mammalian target of rapamycin inhibitor,
BEZ235, circumvents erlotinib resistance of epidermal growth factor
receptor mutant lung cancer cells triggered by hepatocyte growth
factor. Int J Cancer. 133:505–513. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Rao S, Yang H, Penninger JM and Kroemer G:
Autophagy in non-small cell lung carcinogenesis: A positive
regulator of antitumor immunosurveillance. Autophagy. 10:529–531.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Katheder NS, Khezri R, O'Farrell F,
Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen
T, Juhász G, et al: Microenvironmental autophagy promotes tumour
growth. Nature. 541:417–420. 2017. View Article : Google Scholar : PubMed/NCBI
|