Introduction
Prostate cancer (PC) is the most frequently
diagnosed type of cancer among men throughout the developed world
(1). It originates from high-grade
prostatic intra-epithelial neoplasia and often progresses to
metastatic PC. Commonly, androgen deprivation therapy is the
first-line treatment for patients with PC (2). However, most patients will relapse due
to the development of castration-resistant PC (CRPC) after 2–3
years (3).
Previous evidence has suggested that the acquisition
of androgen independence may be due to the upregulation of growth
factor/receptor signaling pathways, principally that of the
epidermal growth factor receptor (EGFR) (4,5), which
is an attractive target for therapeutic intervention. EGFR is a
multifunctional membrane glycoprotein expressed in numerous types
of tissue (6). Furthermore, EGFR is
overexpressed in many tumors, and leads to excessive cell
proliferation and the development of malignancy (7). EGFR interacts with its ligand, EGF, to
regulate cell growth, differentiation, motility, adhesion and
tumorigenesis. EGF engagement activates the intrinsic kinase
activity of EGFR, leading to the activation of several downstream
intracellular signaling pathways, including those of MEK, ERK and
PI3K/Akt, and STAT3 (8). STAT3 is
activated by EGF and further regulates the expression of numerous
genes involved in the cell cycle and proliferation, including
cyclin D1, BCL-2 and surviving (9,10).
Previous studies have demonstrated that constitutively activated
STAT proteins are present in numerous types of tumor cells and
cancer tissues, and that STAT3 is the most active transcription
factor (11,12). Inhibition of EGFR inhibits these
signaling cascades and further promotes apoptosis of cancer cells
(13).
Epigenetic mechanisms, such as DNA methylation and
histone modification, have important roles in cancer development.
Histone deacetylases (HDACs), enzymes involved in removing acetyl
groups from histones, remodel chromatin and regulate gene
transcription, thereby serving crucial roles in cell proliferation,
cell cycle regulation and cell differentiation (14,15).
HDACs are overexpressed in a number of types of tumor (16,17),
which indicates that HDACs may be potential targets for epigenetic
intervention. Trichostatin A (TSA), a class I and II HDAC
inhibitor, exerts anti-tumor effects in colon carcinoma, breast
adenocarcinoma and esophageal squamous cells (18–20).
Zhang et al (21) revealed
that higher dosages of TSA (5–40 µM) exert potent activity against
prostate cancer cells through the induction of cell cycle arrest
and apoptosis.
In the present study, the anti-proliferative effect
of a lower concentration of TSA (<1 µM) on PC3 cells was
investigated as well as the potential of combination treatment with
doxorubicin (DOX) in CRPC.
Materials and methods
Reagents
TSA and MG132 were purchased from Sigma-Aldrich
(Merck KGaA). Stock solutions of TSA (2 mM) and MG132 (10 mM) were
prepared using dimethyl sulfoxide (DMSO). The final concentration
of DMSO in the medium was maintained at 0.1%. MG132, proteasome
inhibitor was used to determine if decreased EGFR expression by TSA
treatment was via proteasomal degradation.
Cell lines and culture
PC3 human prostate cancer cells from American Type
Culture Collection were cultured in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(HyClone; GE Healthcare Life Sciences) and penicillin (100
U/ml)/streptomycin (100 µg/ml), and were maintained at 37°C in a
humidified atmosphere containing 5% CO2.
MTT assay
A total of 3,000 cells in the logarithmic growth
phase were seeded into 96-well plates. After 24 h of incubation,
the cells were exposed to a range of concentrations (0–1 µM) of TSA
and incubated for 72 h. After this, 10 µl MTT (5 mg/ml) solution
was added to each well and incubated for 4 h at 37°C and 5%
CO2. Following incubation, the supernatant was removed,
and the cells were suspended in 100 µl of 100% DMSO. The absorbance
values were measured at 570 nm and were normalized to the DMSO
control by using a Thermo Multiskan spectrum plate reader (Thermo
Fisher Scientific, Inc.). The experiments were conducted in
triplicate. The status of the cells in each group was evaluated at
×100 magnification using a light microscope (CKX41; Olympus
Corporation).
Cell cycle assay
Cells were treated with 0.5 µM TSA or the DMSO
control for 24 h. Cells were then harvested and fixed in 70% cold
ethanol at −20°C overnight. Cells were washed with 1X PBS and
incubated with RNase at 37°C for 30 min, followed by propidium
iodide (PI; 50 µg/ml) staining for another 30 min at 4°C. Cell
suspensions were analyzed with a BD FACSCalibur™ flow cytometer (BD
Biosciences). Data were analyzed with ModFit LT software (version
3.3; Verity Software House, Inc.).
Immunoblotting
PC3 cells were treated with TSA or combined with
MG132 or doxorubicin for 24 h. Since EGF usually induces
phosphorylation of EGFR within 30 min, phosphorylation of EGFR was
determined at 10 min after EGF induction in PC3 cells treated with
TSA for 30 min prior to EGF induction. After treatment, cells were
washed with cold PBS and disrupted with 1X cell lysis buffer (Cell
Signaling Technology, Inc.), followed by brief sonication
(amplitude 20%, duration 3 sec, twice, on ice). The protein
concentration was analyzed using a Bio-Rad protein assay kit II
with bovine serum albumin standards, according to the
manufacturer's protocol (cat. no. 5000002; Bio-Rad Laboratories,
Inc.). Cell lysates were separated by SDS-PAGE (30 µg protein each
lane; 10% gel for high or middle molecular weight and 15% gel for
low molecular weight protein) and then transferred onto
nitrocellulose membranes (Hybond-C; Amersham Pharmacia Biotech,
Inc.). Following blocking with PBS-Tween-20 containing 5% non-fat
dried milk for 1 h at room temperature, the membranes were
incubated with primary antibodies against EGFR (1:1,000; cat. no.
2646), phospho-EGFR (1:1,000; cat. no. 4407), CDK6 (1:1,000; cat.
no. 13331), CDK4 (1:1,000; cat. no. 12790), phospho-retinoblastoma
(RB) protein (1:1,000; cat. no. 3590), RB (1:1,000; cat. no. 9309),
BAX (1:1,000; cat. no. 5023), BCL-2 (1:1,000; cat. no. 2876),
cleaved caspase-3 (1:1,000; cat. no. 9664) (all Cell Signaling
Technology, Inc.), STAT3 (1:2,000; cat. no. ab68153), Cyclin D1
(1:1,000; cat. no. ab134175) and BCL-XL (1:1,000; cat. no. ab32370)
(all from Abcam; overnight at 4°C. A horseradish
peroxidase-conjugated secondary antibody (1:1,000; cat. no. 7074 or
7076; Cell Signaling Technology, Inc.) was then incubated with the
membranes for 1 h at room temperature. Immunoreactivity bands were
detected using an enhanced chemiluminescence kit (cat. no. NCI4106;
Pierce; Thermo Fisher Scientific, Inc.). β-actin (1:1,000; cat. no.
3700; Cell Signaling Technology, Inc.) was used as the loading
control.
Reverse transcription-quantitative
(RT-qPCR)
Total RNA was extracted from the PC3 cells treated
with TSA with TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. cDNA
was synthesized from 4 µg total RNA with Oligo (dT)18
primer using the EasyScript first-strand cDNA synthesis superMix
(TransGen Biotech Co., Ltd.) and ARKTIK Thermal Cycler (Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
The temperature protocol was as follows: Denaturation, 65°C for 5
min; annealing, on ice for 2 min; elongation, 42°C for 30 min; and
termination, 85°C for 5 sec. PCR amplification, using 4 µl cDNA
with TransStart® Green qPCR SuperMix (TransGen Biotech
Co., Ltd.), for CDK6 and cyclin D1 genes was performed with the
Mx3005p Real-Time PCR system (Agilent Technologies, Inc.) at 94°C
for initial denaturation for 30 sec, followed by 40 cycles at 94°C
for 6 sec and 60°C for 30 sec. The primer sequences were: CDK6
forward, 5′-CATTCAAAATCTGCCCAACC-3′ and reverse,
5′-GGTCCTGGAAGTATGGGTGA-3′; cyclin D1 forward,
5′-GCTTCCTCTCCAGAGTGATC-3′ and reverse, 5′-GTCCATGTTCTGCTGGGCCT-3′.
GAPDH forward, 5′-GTGAAGGTCGGAGTCAACGG-3′ and reverse,
5′-CCTGGAAGATGGTGATGGGA-3′. GAPDH was used as the reference gene.
Gene expression was calculated using the 2−ΔΔCq method
(22).
Statistical analysis
SPSS (version 21; IBM Corp.) software was used to
perform the statistical analysis. Data are presented as the mean ±
SD, with a minimum of 3 repeats. A Student's t-test (two-tailed)
was used to compare between two groups. One-way analysis of
variance was performed to compare multiple groups followed by
Tukey's test to determine multiple comparisons between the groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
TSA inhibits PC3 cells by inactivating
the EGFR
To evaluate the cytotoxic effect of TSA, the
inhibitory effect of TSA on the proliferation of PC3 cells was
investigated using an MTT assay. TSA inhibited PC3 cell
proliferation in a dose- and time-dependent manner (Fig. 1A and B). Following treatment with 0.5
and 1.0 µM TSA for 72 h, the inhibitory rate was ~50 and 72%,
respectively. The suppressive effect of TSA on cell proliferation
was further confirmed by microscopic observation. Treatment with
TSA decreased cell density (Fig.
1C). EGFR was evaluated as it is overexpressed in prostate
cancer and has an important role in prostate cancer progression
(23). Treatment with TSA for 24 h
decreased the expression of EGFR and its downstream molecule STAT3
(Fig. 1D). Activity of EGFR was
further determined by analyzing the phosphorylation level of EGFR.
Since EGF usually induces phosphorylation of EGFR within 30 min,
phosphorylation of EGFR was determined at 10 min after EGF
induction in PC3 cells treated with TSA for 30 min prior to EGF
induction. The results of the present study indicated that TSA
treatment for 30 min decreased EGFR phosphorylation, but not
expression, indicating that TSA treatment decreased both the
expression and activity of EGFR (Fig.
1E). Treatment with HDAC inhibitors can induce ubiquitination
and proteasomal degradation of the erbB family in head and neck
squamous tumors (24). To test
whether TSA suppressed EGFR expression in a proteasomal pathway,
PC3 cells were treated with the proteasome inhibitor MG132.
However, EGFR protein levels remained suppressed by TSA in the
presence of MG132 (Fig. 1F),
indicating that proteasomal degradation is not involved in the
TSA-induced EGFR turnover.
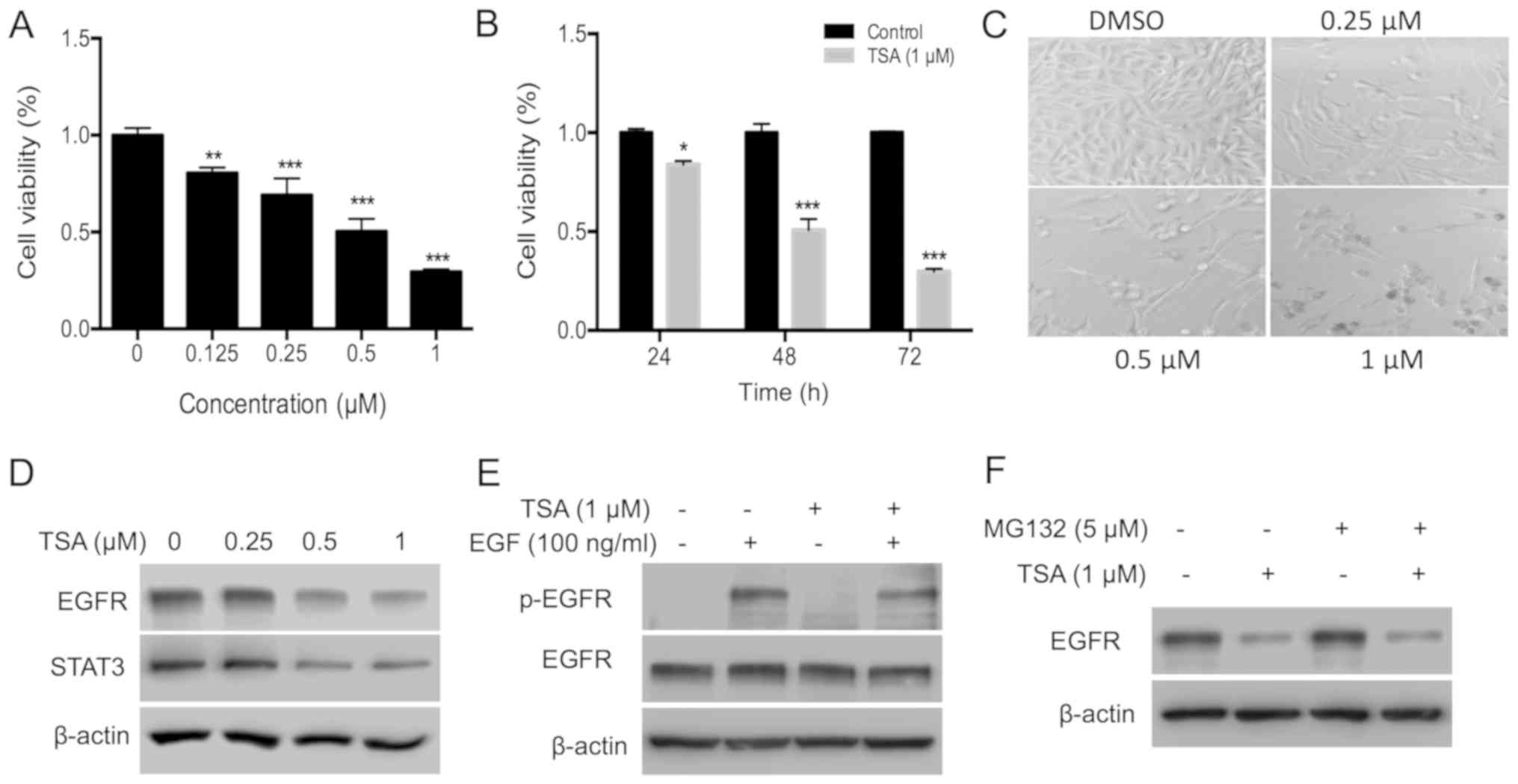 | Figure 1.TSA inhibits PC3 cell proliferation by
disrupting the EGFR-STAT3 pathway. (A) PC3 cells (3×103)
were plated in a 96-well plate and treated with different doses of
TSA (0, 0.25, 0.5 and 1 µM) for 72 h; an MTT assay was used to
measure the effects of TSA on PC3 cell proliferation. (B) PC3 cells
were subjected to the aforementioned treatments, and the
time-course effect of TSA (1.0 µM) was measured using an MTT assay.
(C) Effects of TSA on the density and morphology of PC3 cells were
observed via microscopy. The magnification is ×100. (D) Expression
of EGFR and STAT3 in PC3 cells treated with TSA. PC3 cells
(4×105) were plated in 6-cm dishes, and 0.25, 0.5 or 1
µM TSA was added for 24 h. DMSO was added into the control group
wells. Cells were collected and lysed with lysis buffer and the
protein expression analyzed via western blotting. (E) TSA inhibits
the phosphorylation of EGFR induced by EGF. Cells were starved in
serum-free medium for 12 h and then treated with 1 µM TSA for 30
min, followed by EGF (100 ng/ml) for 10 min. Cells were collected,
and western blotting was used to evaluate the phosphorylation of
EGFR. (F) Expression of EGFR in PC3 cells treated with TSA and
MG132. Cells were treated with 1 µM TSA and 5 µM MG132 for 24 h.
Cells were collected, and western blotting was used to evaluate the
expression of EGFR. *P<0.05; **P<0.01; ***P<0.001 vs.
respective control. TSA, trichostatin A; EGFR, epidermal growth
factor; p, phosphorylated; DMSO, dimethyl sulfoxide. |
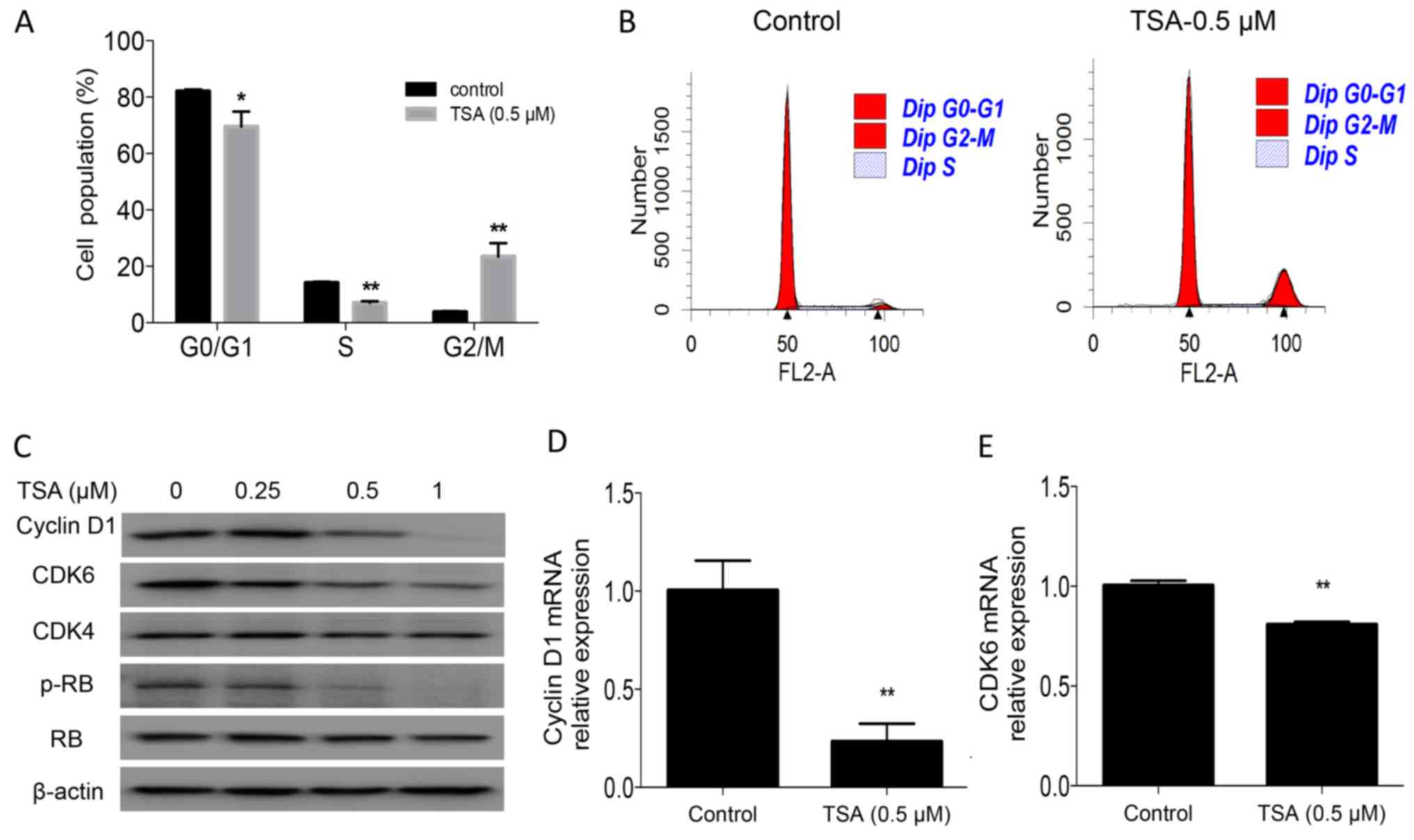
TSA induces cell cycle arrest by
downregulating the expression of CDK6 and cyclin D1, and the
phosphorylation of RB
To identify the underlying mechanism associated with
the suppression of cell proliferation by TSA, cell cycle analysis
was performed in the present study. The results revealed that
treatment with TSA for 24 h led to an accumulation of PC3 cells in
the G2/M phase (3.8 vs. 23.4%; P<0.01) and a concomitant
decrease in the S-phase cell fraction (14.1 vs. 7%; P<0.01;
Fig. 2A and B). TSA-induced cell
cycle arrest was further confirmed by examining the expression of
the cell cycle regulatory protein cyclin D1, CDK6, and the
phosphorylation of RB in PC3 cells. Western blot analysis and
RT-qPCR indicated that TSA treatment decreased cyclin D1 and CDK6
levels, and the phosphorylation of RB (Fig. 2C-E).
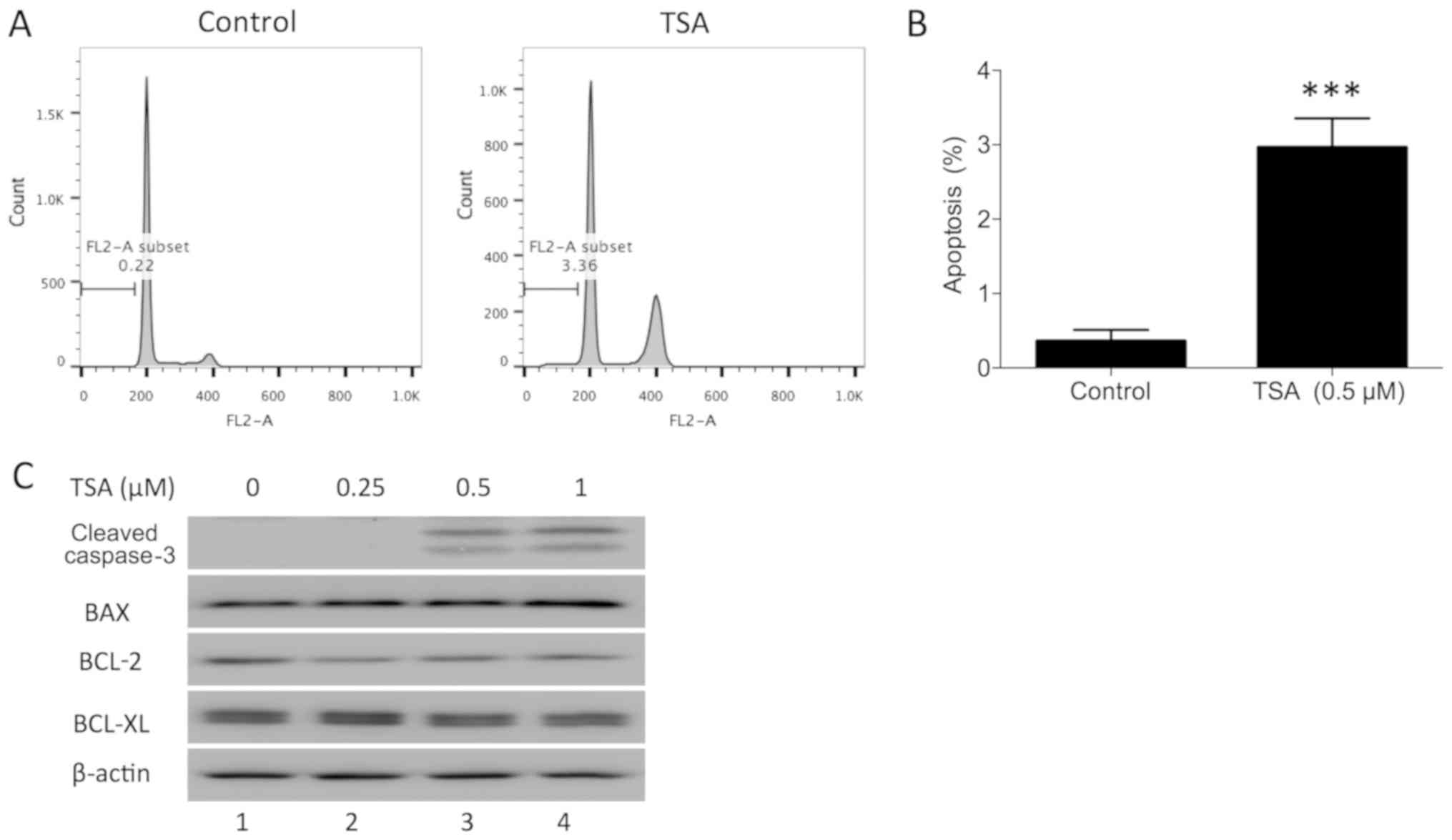
TSA induces apoptosis in PC3
cells
In addition to blocking the cell cycle phase
transition, it has been demonstrated that TSA induces cell
apoptosis in human breast cancer (18). To determine whether TSA induces
apoptosis in prostate cancer cells, PC3 cells were treated with a
TSA dose series for 24 h in the present study. The apoptotic
percentage of PC3 cells following treatment was evaluated using
flow cytometry. The sub-G1 fraction was markedly increased compared
with the control (Fig. 3A and B).
TSA-induced apoptosis was further confirmed by examining the
expression of cleaved caspase-3 in PC3 cells. Western blot analysis
indicated that TSA treatment increased cleaved caspase-3 in PC3
cells (Fig. 3C). Furthermore, BAX,
BCL-2 and BCL-XL protein expression was measured in PC3 cells
following TSA treatment. BAX protein was upregulated and BCL-2
protein was downregulated following treatment, while BCL-XL was not
notably affected. These results indicated that TSA may induce PC3
apoptosis via the mitochondrial pathway.
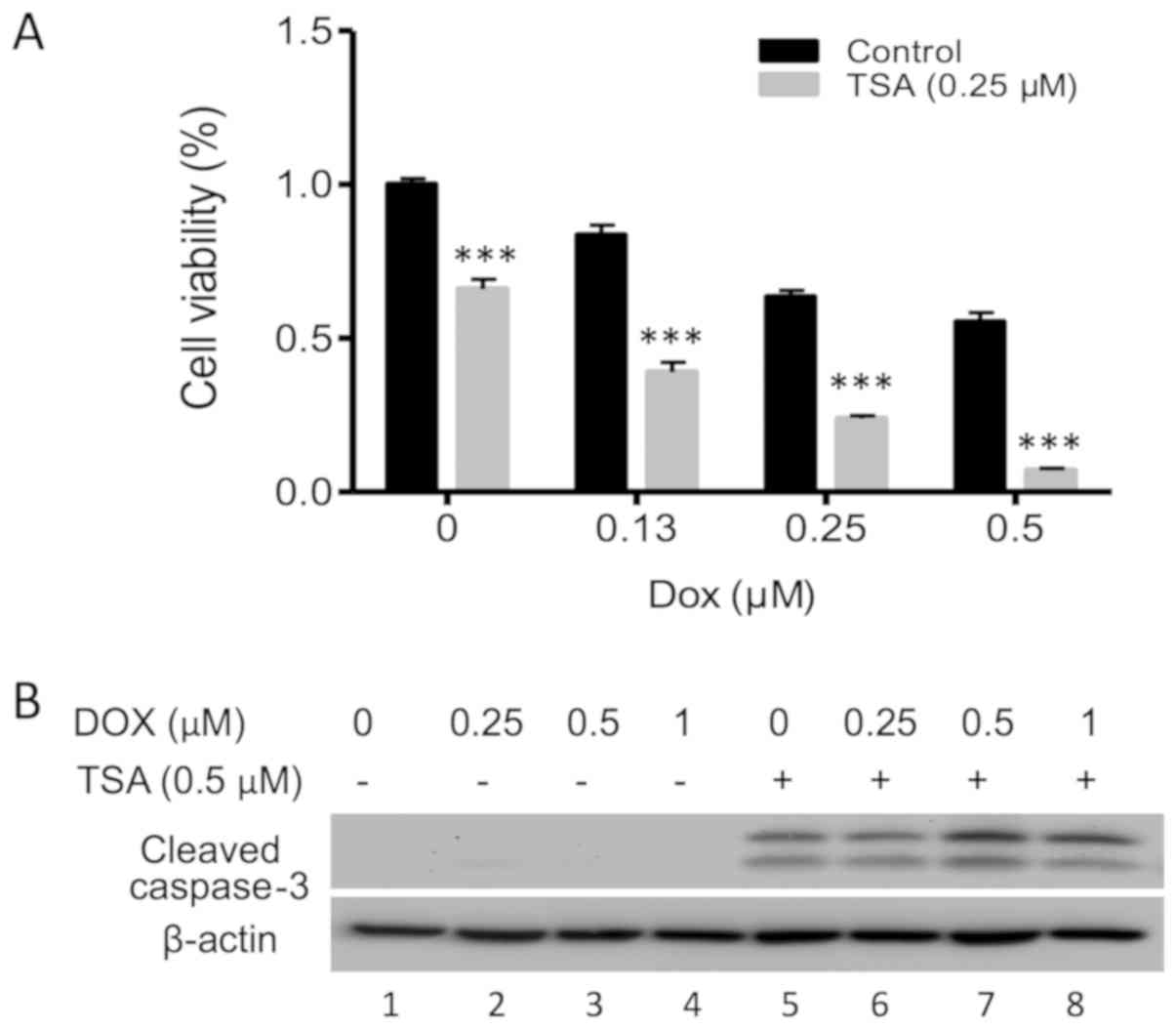
TSA enhances apoptosis induced by
DOX
The majority of cases of prostate cancer exhibit
radio- and chemoresistance. In recent years, combinations of HDAC
inhibitors with chemotherapy drugs have been used to treat various
malignancies. In the present study the synergistic effect of TSA
and DOX on the inhibition of PC3 cell proliferation was
investigated. Notably, DOX and TSA in combination demonstrated
consistent inhibitory effects on PC3 cells compared with either
compound alone (Fig. 4A), as well as
a clear elevation of cleaved caspase-3 (Fig. 4B; lanes 7 and 8), indicating a
synergistic enhancement of PC3 cell apoptosis.
Discussion
The epigenetic regulation of gene expression may
represent a potential new therapeutic strategy for cancer
treatment. Histone acetylation is determined using histone
acetyltransferases and HDACs. A variety of HDAC inhibitors have
been demonstrated to induce apoptosis in solid tumors (18,21,25–27). In
the present study, the effects of TSA, a potent HDAC inhibitor, on
disrupting EGFR signaling were characterized, demonstrating the
induction of apoptosis in prostate cancer cells. Molecular evidence
for cell cycle arrest was provided in the decreased expression of
cyclin D1 and CDK6, and the decreased phosphorylation of RB, in
addition to the increased levels of BAX and decreased levels of
BCL-2. Notably, blocking the EGFR signaling pathway sensitized PC3
cells to DOX.
HDAC inhibitors exert transcriptional repression of
certain oncogenes, such as MYC, MYCN and EGFR (28–30). In
the present study, it was demonstrated that TSA treatment led to
downregulated EGFR and STAT3 expression, resulting in decreased
cyclin D1 and CDK6 expression, and thus G2/M cell cycle arrest,
which is in accordance with the results of the aforementioned
studies. These results are, however, different to previous
conclusions (21) that higher
concentrations of TSA induce both G0/G1 and G2/M arrest in PC3
cells, which indicates that the biological function of TSA is
dose-dependent. In addition to blocking cell cycle progression,
HDAC inhibitors induce apoptosis via the mitochondria-mediated
apoptosis pathway (25), which
disrupts the ratio of BAX to BCL-2 or BCL-XL at the mitochondrial
membrane, causing the release of cytochrome c and other
pro-apoptotic molecules into the cytoplasm, which in turn leads to
the activation of caspase-3 to cleave poly (ADP-ribose) polymerase
and execute cell death. In the present study, the sub-G1 fraction
in the cell cycle analysis was determined to represent apoptosis
induction by TSA. This method is an older technique used for the
detection of apoptosis, which could be replaced by Annexin V and PI
double staining to distinguish apoptotic and necrotic cells. In
order to demonstrate the inducible role of TSA in apoptosis, the
level of cleaved caspase-3 was determined using western blot
analysis, which is a common character of apoptosis. It was revealed
that TSA increased cleaved caspase-3 expression, confirming the
induction of apoptosis. Consistent with a previous study, the
present study demonstrated that TSA decreased BCL-2 expression
(21). Furthermore, it was revealed
that TSA treatment increased BAX expression, thereby activating the
mitochondria-mediated apoptosis pathway (31). Blocking cell cycle progression and
inducing apoptosis is a common mechanism of chemotherapeutic agents
to exert anti-proliferative effects. Consistent with a previous
study, TSA treatment led to simultaneous cell cycle arrest and
apoptosis (21).
DOX is an anthracycline antibiotic that is effective
in treating a wide range of different types of cancer (32). However, the toxic side effects of
DOX, particularly those involving cardiac damage, limit its
clinical application for long-term therapy (33). Drug combinations are a widely used
strategy to increase the efficacy and decrease the side effects of
chemotherapy. Herein, the combination of TSA and low-dose DOX
exerted synergistic effects in reducing PC3 cell viability. Higher
levels of cleaved caspase-3 were induced by this combination, and
indicated that TSA could promote cell apoptosis induced by DOX
(34).
In conclusion, the HDAC inhibitor TSA induced cell
cycle arrest and apoptosis in PC3 cells by inactivating the
EGFR-STAT3 signaling pathway. When combined, TSA and DOX exert
synergistic effects in reducing PC3 cell viability by promoting
apoptosis. Although TSA is an epigenetic regulator, the present
study did not establish a mechanistic association between TSA and
the epigenetic regulation of gene expression in PC3 cells. Another
limitation of the present study is that only the single cell line
PC3 was used to evaluate TSA effect. Further studies are required
to determine how TSA blocks the EGFR pathway in an epigenetic
manner using multiple cell lines, including DU145 and LNcap. The
present study supports the rationale for TSA alone or in
combination with DOX as a therapeutic approach to CRPC cases, which
are largely refractory to current therapeutic approaches, and may
promote the rapid clinical evaluation of this strategy.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HZ, XZ, HL, HJ and YJ conceived, designed and
performed the experiments and analyzed the data. HZ, XZ, HL and HJ
contributed to the reagents and/or materials used in the present
study. HZ and YJ wrote the manuscript. All authors confirm the
accuracy of the study.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TSA
|
trichostatin A
|
|
EGFR
|
epidermal growth factor receptor
|
|
HDAC
|
histone deacetylase
|
References
|
1
|
Kgatle MM, Kalla AA, Islam MM, Sathekge M
and Moorad R: Prostate cancer: Epigenetic alterations, risk
factors, and therapy. Prostate Cancer. 2016:56538622016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu C, Zhu Y, Lou W, Nadiminty N, Chen X,
Zhou Q, Shi XB, deVere White RW and Gao AC: Functional p53
determines docetaxel sensitivity in prostate cancer cells.
Prostate. 73:418–427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grasso CS, Wu YM, Robinson DR, Cao X,
Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC,
et al: The mutational landscape of lethal castration-resistant
prostate cancer. Nature. 487:239–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Traish AM and Morgentaler A: Epidermal
growth factor receptor expression escapes androgen regulation in
prostate cancer: A potential molecular switch for tumour growth. Br
J Cancer. 101:1949–1956. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kambhampati S, Ray G, Sengupta K, Reddy
VP, Banerjee SK and Van Veldhuizen PJ: Growth factors involved in
prostate carcinogenesis. Front Biosci. 10:1355–1367. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tzouvelekis A, Ntolios P, Karameris A,
Vilaras G, Boglou P, Koulelidis A, Archontogeorgis K, Kaltsas K,
Zacharis G, Sarikloglou E, et al: Increased expression of epidermal
growth factor receptor (EGF-R) in patients with different forms of
lung fibrosis. Biomed Res Int. 2013:6543542013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu YH, Wei XL, Hu GQ and Wang TX:
Quinolone-indolone conjugate induces apoptosis by inhibiting the
EGFR-STAT3-HK2 pathway in human cancer cells. Mol Med Rep.
12:2749–2756. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wee P and Wang Z: Epidermal growth factor
receptor cell proliferation signaling pathways. Cancers (Basel).
9:522017. View Article : Google Scholar
|
|
9
|
Weerasinghe P, Garcia GE, Zhu Q, Yuan P,
Feng L, Mao L and Jing N: Inhibition of Stat3 activation and tumor
growth suppression of non-small cell lung cancer by G-quartet
oligonucleotides. Int J Oncol. 31:129–136. 2007.PubMed/NCBI
|
|
10
|
Zhang X, Yue P, Page BD, Li T, Zhao W,
Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT and Turkson J:
Orally bioavailable small-molecule inhibitor of transcription
factor Stat3 regresses human breast and lung cancer xenografts.
Proc Natl Acad Sci USA. 109:9623–9628. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tai WT, Cheng AL, Shiau CW, Huang HP,
Huang JW, Chen PJ and Chen KF: Signal transducer and activator of
transcription 3 is a major kinase-independent target of sorafenib
in hepatocellular carcinoma. J Hepatol. 55:1041–1048. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kortylewski M and Yu H: Stat3 as a
potential target for cancer immunotherapy. J Immunother.
30:131–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Su JC, Lin KL, Chien CM, Chuang PW, Chang
LS and Lin SR: Concomitant inactivation of the epidermal growth
factor receptor, phosphatidylinositol 3-kinase/Akt and Janus
tyrosine kinase 2/signal transducer and activator of transcription
3 signalling pathways in cardiotoxin III-treated A549 cells. Clin
Exp Pharmacol Physiol. 37:833–840. 2010.PubMed/NCBI
|
|
14
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reichert N, Choukrallah MA and Matthias P:
Multiple roles of class I HDACs in proliferation, differentiation,
and development. Cell Mol Life Sci. 69:2173–2187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kwiecińska P, Wróbel A, Taubøll E and
Gregoraszczuk EŁ: Valproic acid, but not levetiracetam, selectively
decreases HDAC7 and HDAC2 expression in human ovarian cancer cells.
Toxicol Lett. 224:225–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weichert W, Röske A, Gekeler V, Beckers T,
Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M
and Kristiansen G: Histone deacetylases 1, 2 and 3 are highly
expressed in prostate cancer and HDAC2 expression is associated
with shorter PSA relapse time after radical prostatectomy. Br J
Cancer. 98:604–610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun S, Han Y, Liu J, Fang Y, Tian Y, Zhou
J, Ma D and Wu P: Trichostatin A targets the mitochondrial
respiratory chain, increasing mitochondrial reactive oxygen species
production to trigger apoptosis in human breast cancer cells. PLoS
One. 9:e916102014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Watson JA, McKenna DJ, Maxwell P, Diamond
J, Arthur K, McKelvey-Martin VJ and Hamilton PW: Hyperacetylation
in prostate cancer induces cell cycle aberrations, chromatin
reorganization and altered gene expression profiles. J Cell Mol
Med. 14:1668–1682. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma J, Guo X, Zhang S, Liu H, Lu J, Dong Z,
Liu K and Ming L: Trichostatin A, a histone deacetylase inhibitor,
suppresses proliferation and promotes apoptosis of esophageal
squamous cell lines. Mol Med Rep. 11:4525–4531. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Xu Q, Liu G, Huang H, Lin W,
Huang Y, Chi Z, Chen S, Lan K, Lin J and Zhang Y: Effect of histone
deacetylase on prostate carcinoma. Int J Clin Exp Pathol.
8:15030–15034. 2015.PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gan Y, Shi C, Inge L, Hibner M, Balducci J
and Huang Y: Differential roles of ERK and Akt pathways in
regulation of EGFR-mediated signaling and motility in prostate
cancer cells. Oncogene. 29:4947–4958. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bruzzese F, Leone A, Rocco M, Carbone C,
Piro G, Caraglia M, Di Gennaro E and Budillon A: HDAC inhibitor
vorinostat enhances the antitumor effect of gefitinib in squamous
cell carcinoma of head and neck by modulating ErbB receptor
expression and reverting EMT. J Cell Physiol. 226:2378–2390. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
You BR and Park WH: Trichostatin A induces
apoptotic cell death of HeLa cells in a Bcl-2 and oxidative
stress-dependent manner. Int J Oncol. 42:359–366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Frew AJ, Johnstone RW and Bolden JE:
Enhancing the apoptotic and therapeutic effects of HDAC inhibitors.
Cancer Lett. 280:125–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Emanuele S, Lauricella M, Carlisi D,
Vassallo B, D'Anneo A, Di Fazio P, Vento R and Tesoriere G: SAHA
induces apoptosis in hepatoma cells and synergistically interacts
with the proteasome inhibitor Bortezomib. Apoptosis. 12:1327–1338.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chou CW, Wu MS, Huang WC and Chen CC: HDAC
inhibition decreases the expression of EGFR in colorectal cancer
cells. PLoS One. 6:e180872011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
LaBonte MJ, Wilson PM, Fazzone W, Russell
J, Louie SG, El-Khoueiry A, Lenz HJ and Ladner RD: The dual
EGFR/HER2 inhibitor lapatinib synergistically enhances the
antitumor activity of the histone deacetylase inhibitor
panobinostat in colorectal cancer models. Cancer Res. 71:3635–3648.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yokoyama S, Feige E, Poling LL, Levy C,
Widlund HR, Khaled M, Kung AL and Fisher DE: Pharmacologic
suppression of MITF expression via HDAC inhibitors in the
melanocyte lineage. Pigment Cell Melanoma Res. 21:457–463. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kiliccioglu I, Konac E, Varol N, Gurocak S
and Yucel Bilen C: Apoptotic effects of proteasome and histone
deacetylase inhibitors in prostate cancer cell lines. Genet Mol
Res. 13:3721–3731. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lou Y, Wang Z, Xu Y, Zhou P, Cao J, Li Y,
Chen Y, Sun J and Fu L: Resveratrol prevents doxorubicin-induced
cardiotoxicity in H9c2 cells through the inhibition of endoplasmic
reticulum stress and the activation of the Sirt1 pathway. Int J Mol
Med. 36:873–880. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Menna P, Paz OG, Chello M, Covino E,
Salvatorelli E and Minotti G: Anthracycline cardiotoxicity. Expert
Opin Drug Saf. 11 (Suppl 1):S21–S36. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X, Xu J, Wang H, Wu L, Yuan W, Du J
and Cai S: Trichostatin A, a histone deacetylase inhibitor,
reverses epithelial-mesenchymal transition in colorectal cancer
SW480 and prostate cancer PC3 cells. Biochem Biophys Res Commun.
456:320–326. 2015. View Article : Google Scholar : PubMed/NCBI
|