Introduction
Renal cell carcinoma (RCC) is globally the most
prevalent cancer affecting the kidney in adults (1). It was reported that ~64,000 new cases
were diagnosed in 2017 in the USA (1), and this value has risen by 2–4% each
year (2). Chromophobe RCC (chRCC) is
the third most common histological subtype of RCC (3), comprising 5–7% of all RCC cases
(4). Due to advances in technology
for the diagnosis and treatment of chRCC, the 5-year survival rate
of chRCC is >75% (5) and the
outcome is typically favorable when compared with that of other
subtypes (6). However, patients with
this disease still have a 5–10% probability of eventually
developing progression and metastasis (7). Therefore, it is essential to identify
tumor-specific biomarkers and the underlying molecular mechanisms
of chRCC, which may be conducive to improved risk assessment of the
disease, guiding clinical decision-making, and developing novel
diagnostic and therapeutic strategies for chRCC.
The molecular pathogenesis of cancer is complex,
involving the inactivation and mutation of tumor suppressor genes
and the activation of oncogenes (8).
Recently, bioinformatics analysis using high-throughput platforms
has emerged as an efficacious approach to identifying new targets
and comprehending the underlying molecular mechanisms of carcinoma
(9). For instance, Cao et al
(10) reported that five genes,
COL1A2, COL1A1, COL4A1, THBS2 and ITGA5, which they
determined to be significantly overexpressed in gastric cancer
(GC), were associated with the prognosis of GC and were potential
biomarkers and therapeutic targets for GC. In addition, Wang et
al (11) identified 227
differentially expressed genes (DEGs) between breast cancer and
normal breast tissues, and found that the hub gene NDC80 may
be a key prognostic factor and potential target.
In the present study, three raw gene chips [GSE6280
(12), GSE11151 (13) and GSE15641 (14)] were downloaded from the NCBI-Gene
Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) in order to detect
the DEGs between chRCC tissues and normal renal tissues. Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analysis (15) and Gene Ontology
(GO) functional annotation analysis (16) was applied. A protein-protein
interaction (PPI) network was subsequently generated to identify
hub genes associated with chRCC. To further confirm the association
between the hub genes and chRCC, Oncomine dataset (https://www.oncomine.org) and UALCAN (http://ualcan.path.uab.edu) analyses were performed to
examine the expression of the hub genes and associated patient
survival rates.
Materials and methods
Microarray data
A total of 3 profile datasets (GSE6280, GSE11151 and
GSE15641) were downloaded from the GEO database, a public
functional genomics dataset. The platform for GSE6280 and GSE15641
was GPL96, (HG-U133A) Affymetrix Human Genome U133A Array, and the
platform for GSE11151 was GPL570, (HG-U133_Plus_2) Affymetrix Human
Genome U133 Plus 2.0 Array. The raw data consisted of 11 chRCC
tissues (1 in GSE6280, 4 in GSE11151 and 6 in GSE15641) and 32
matched normal tissues (6 in GSE6280, 3 in GSE11151 and 23 in
GSE15641).
Expression analysis of DEGs
All raw data were processed with the R version 3.5.1
software package (https://www.r-project.org/). The ‘limma’ package
(http://www.bioconductor.org/pack-ages/release/bioc/html/limma.html)
in R was utilized for data normalization. The Affy package
(http://www.
bioconductor.org/packages/release/bioc/html/affy.html) was
utilized for gene differential expression analysis. Genes with |log
fold-change (FC)|>1 and P<0.05 were considered to be
DEGs.
GO enrichment analysis
The Database for Annotation, Visualization and
Integrated Discovery (DAVID) (15)
(https://david-d.ncifcrf.gov; version
6.8) provides a comprehensive set of functional annotation tools
for investigators to better understand the biological significance
of certain genes. Based on DAVID, GO analysis, including analysis
of cellular component (CC), molecular function (MF) and biological
process (BP) terms, was performed. P-values of <0.01 and gene
counts of >10 were considered significant thresholds.
KEGG analysis
KOBAS (16)
(http://kobas.cbi.pku.edu.cn; ver. 3.0),
a web server for gene or protein functional annotation and
functional gene set enrichment, was used for pathway enrichment
analysis. Pathways with P-values of <0.01 were screened as
statistically significant.
PPI network
With the confidence level >0.7 and ‘Homo
sapiens’ as a limit, a PPI of DEGs was gathered from the Search
Tool for the Retrieval of Interacting Genes/Proteins (17) (https://string-db.org; ver.10.5). The network
visualization software CytoScape version 3.6 (https://cytoscape.org) was utilized to generate PPI
networks. The top 10 genes were subsequently selected and
considered to be hub genes using the plug-in unit CytoHubba.
Expression and survival analysis of
hub genes
The Oncomine platform featuring scalability, high
quality, consistency and standardized analysis was utilized to
investigate hub gene expression within chRCC across multiple
datasets. Patients were divided into low- and high-expression
groups according to the median gene expression. UALCAN (18), a user-friendly, interactive web
resource for analyzing cancer transcriptome data based on The
Cancer Genome Atlas dataset, was utilized to construct an overall
survival analysis for the hub genes.
Results
Identification of DEGs in chRCC
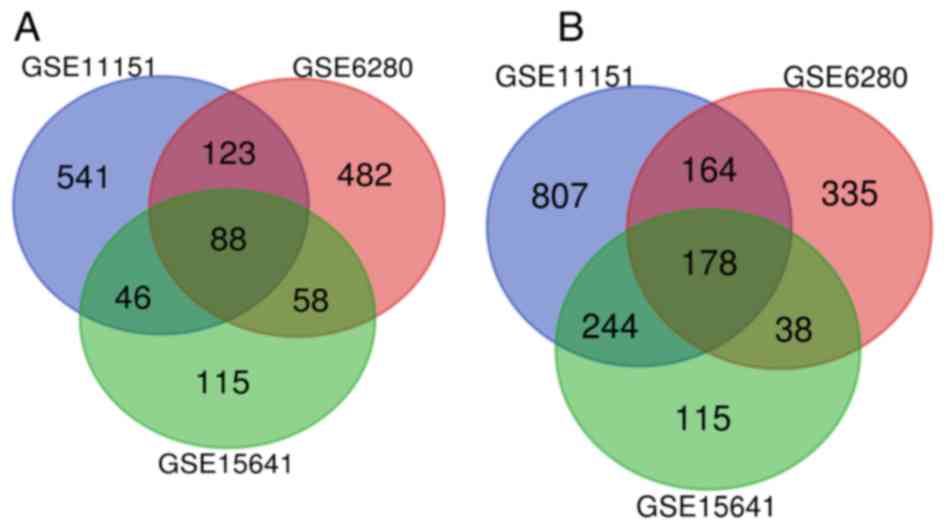
After normalization, a total of 266 overlapping DEGs
(Fig. 1 and Table SI) were identified from 3 profile
datasets (GSE6280, GSE11151 and GSE15641), including 88 upregulated
genes and 178 downregulated genes (|logFC|>1 and P<0.05). The
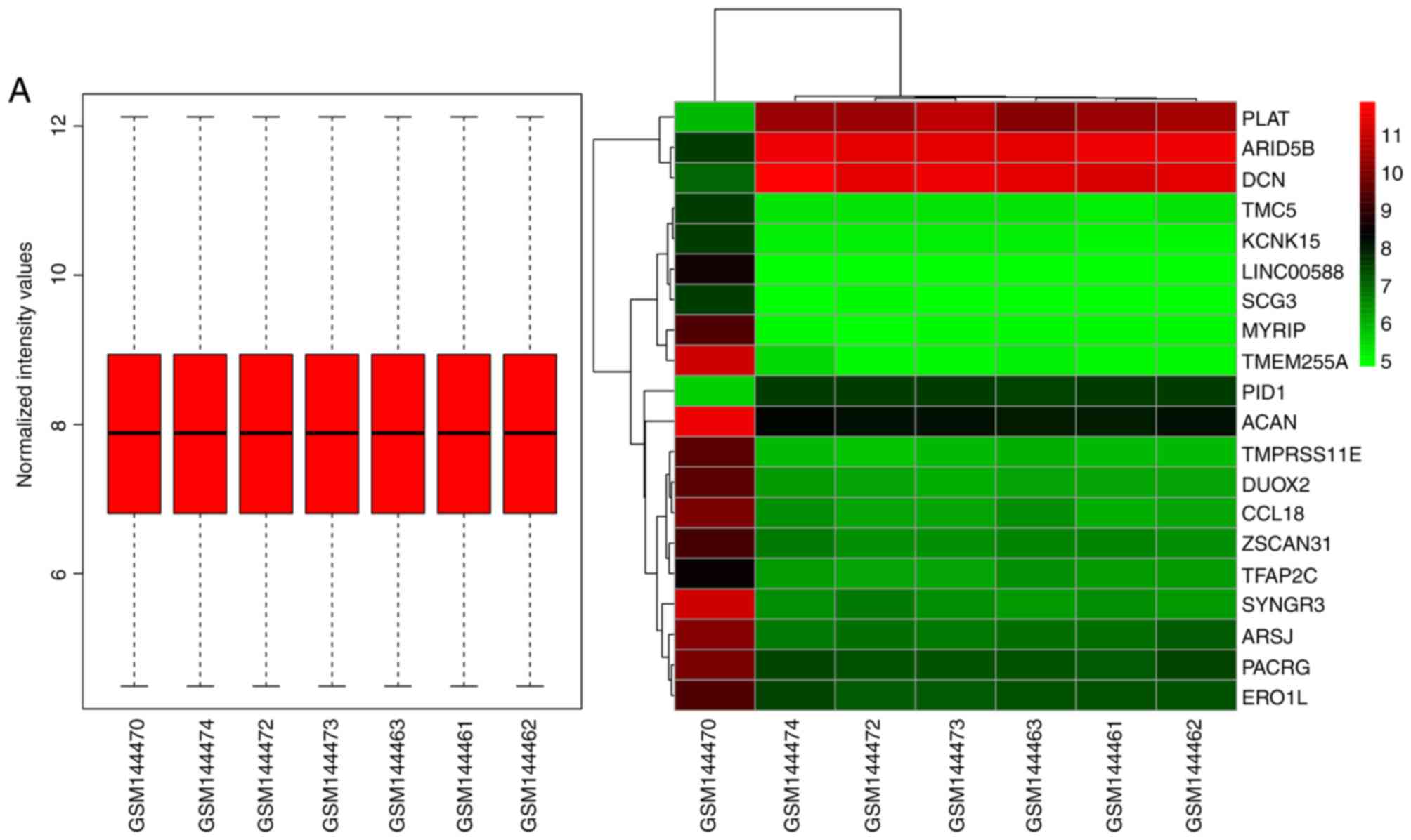
heatmaps of the top 20 DEGs and the results of the normalization of
each dataset are presented in Fig.
2.
GO enrichment analysis
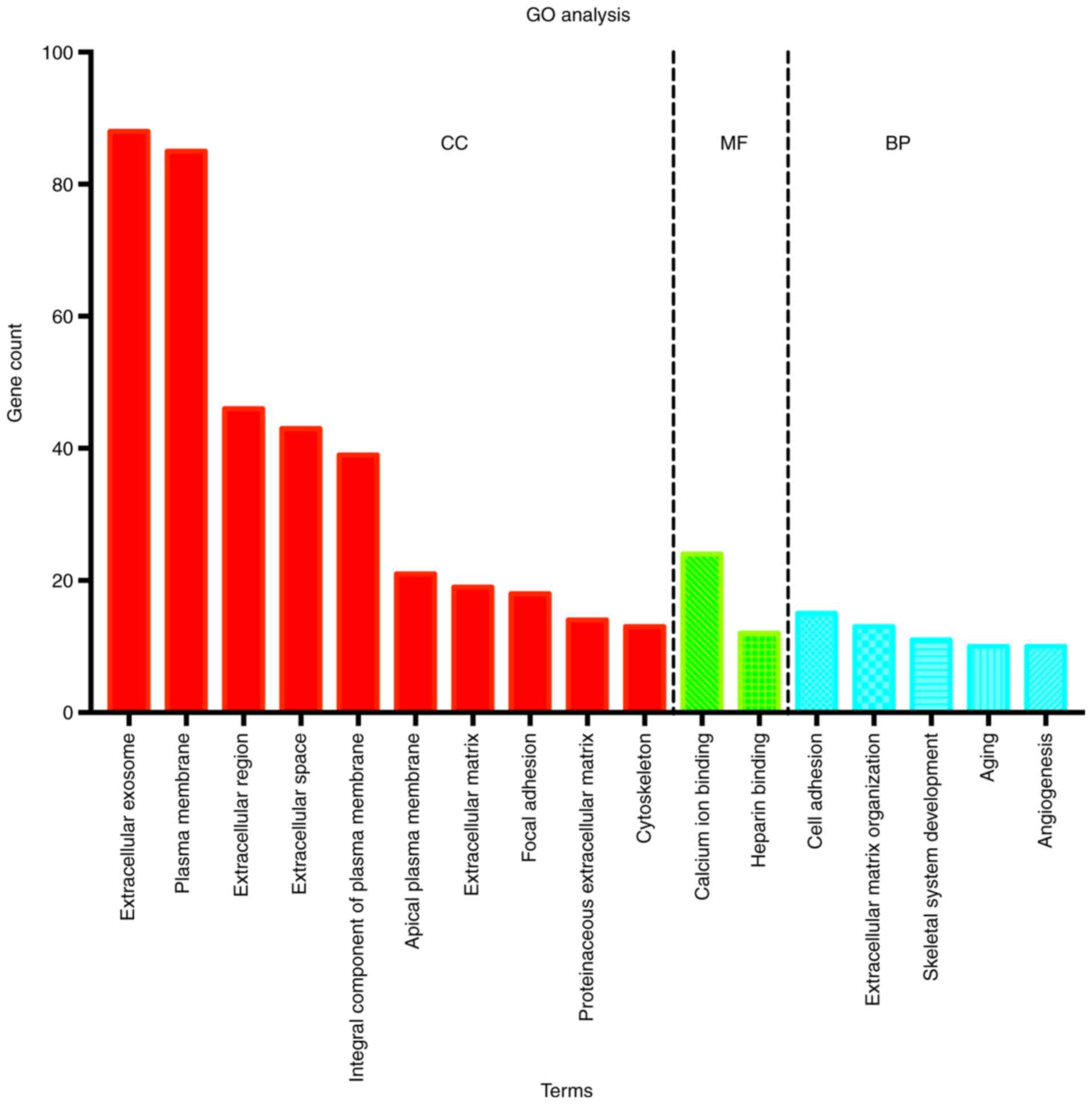
All DEGs were input into the online tool DAVID to
perform GO analysis. The results demonstrated that, for CC, DEGs of
chRCC were mainly enriched in 10 terms, including ‘extracellular
exosome’, ‘plasma membrane’, ‘extracellular region’ and
‘extracellular matrix’. For MF, DEGs were mainly enriched in 2
terms, namely ‘calcium ion binding’ and ‘heparin binding’, while
for BP, DEGs were mainly enriched in 5 terms, namely ‘cell
adhesion’, ‘extracellular matrix organization’, ‘skeletal system
development’, ‘aging’ and ‘angiogenesis’ (Fig. 3).
KEGG analysis
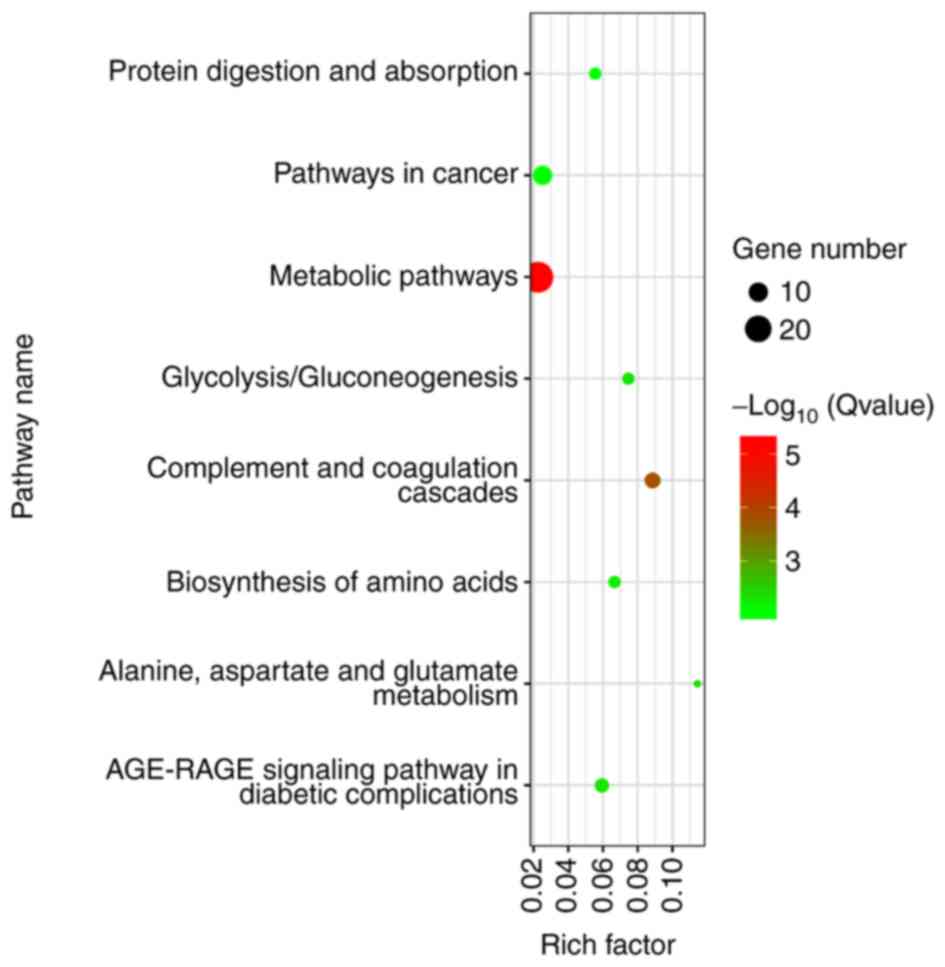
After gene ID conversion, all DEGs were uploaded to
KOBAS to analyze the pathways at the functional level. There were 9
KEGG pathways associated with enriched DEGs, comprising ‘pathways
in cancer’ and ‘metabolic pathways’, among others (Fig. 4).
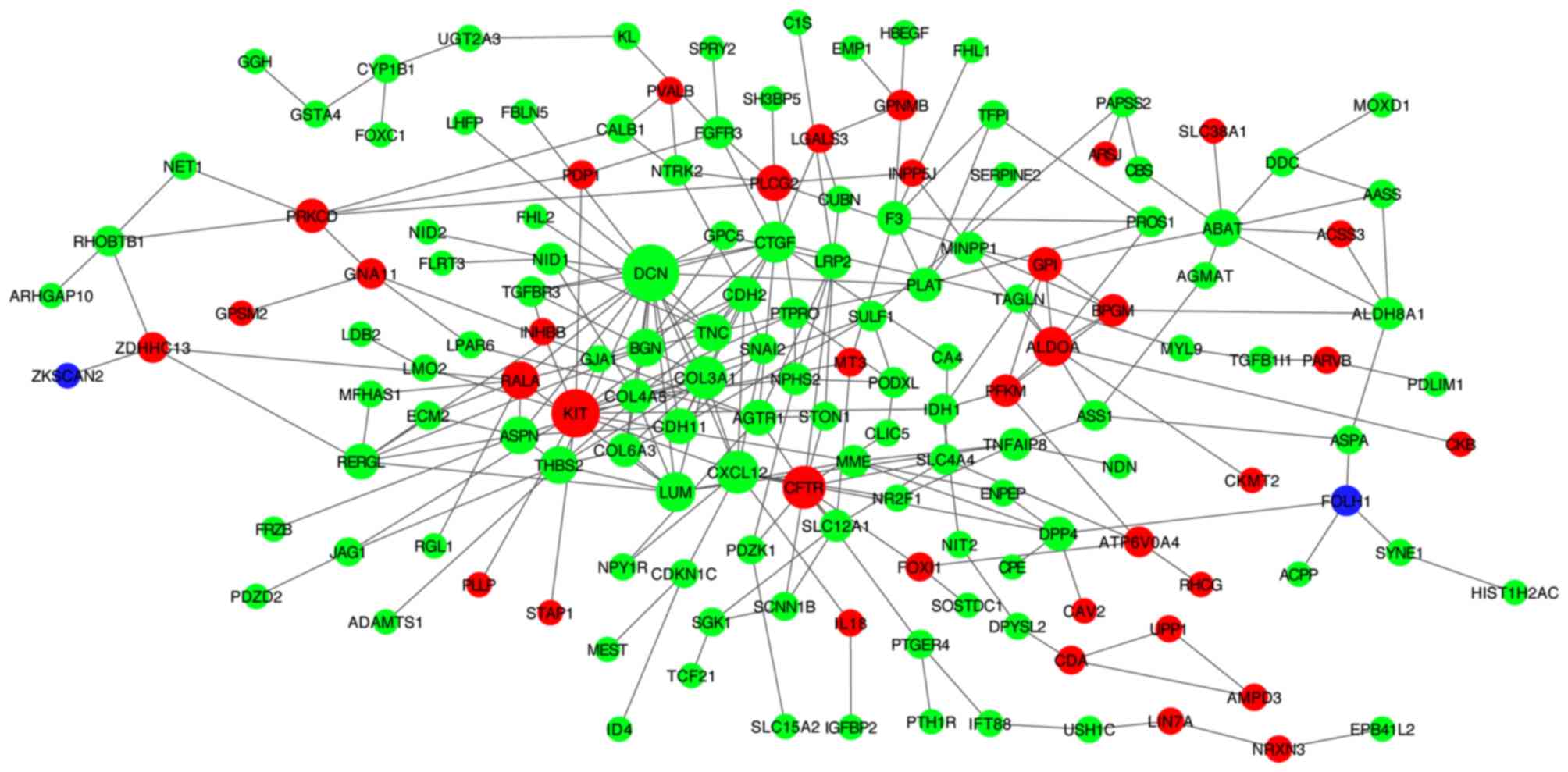
PPI network
In the PPI network (Fig.
5), red, green and violet nodes represent upregulated genes,
downregulated genes and other human proteins interacting with DEGs,
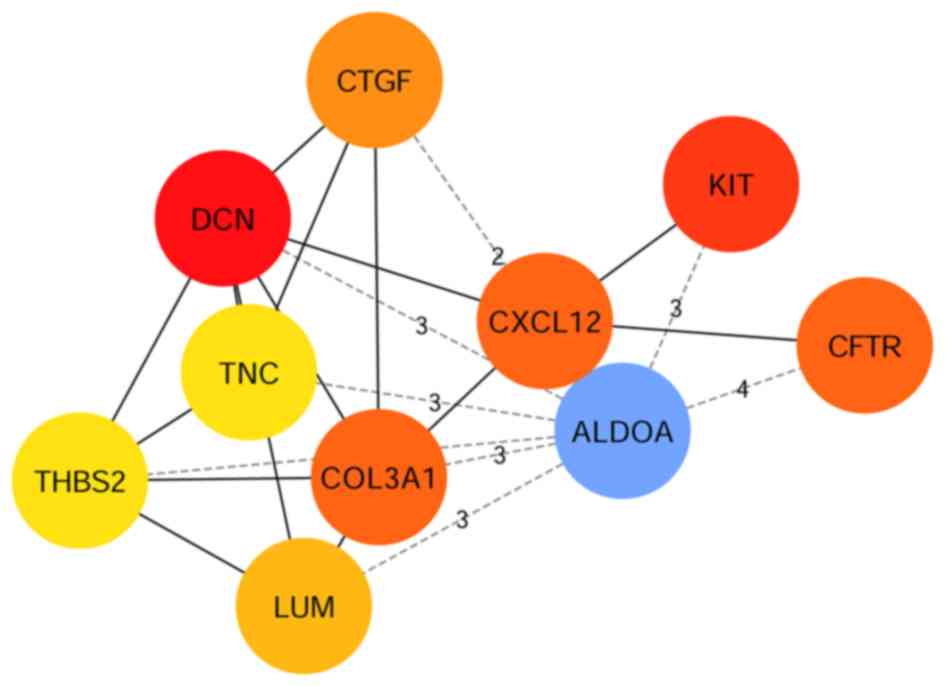
respectively. Using the plug-in unit cytoHubba, 10 hub genes with
the highest degree of interaction were screened (Fig. 6), including 3 upregulated genes
(KIT, CFTR and ALDOA) and 7 downregulated genes
(DCN, COL3A1, CXCL12, CTGF, LUM, TNC and THBS2). The
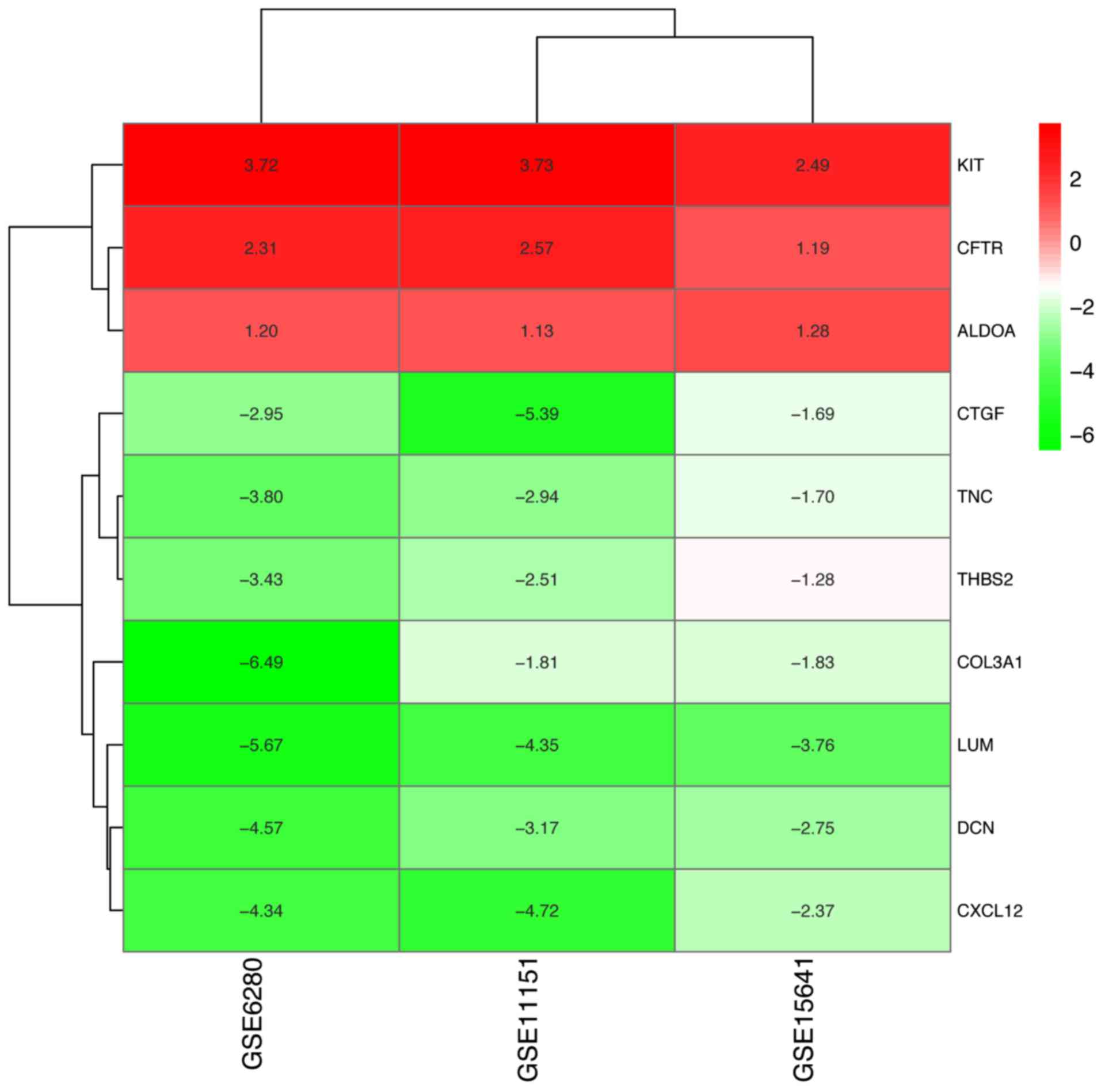
heatmap of the 10 hub genes is presented in Fig. 7.
Comparison of hub genes across
multiple analyses
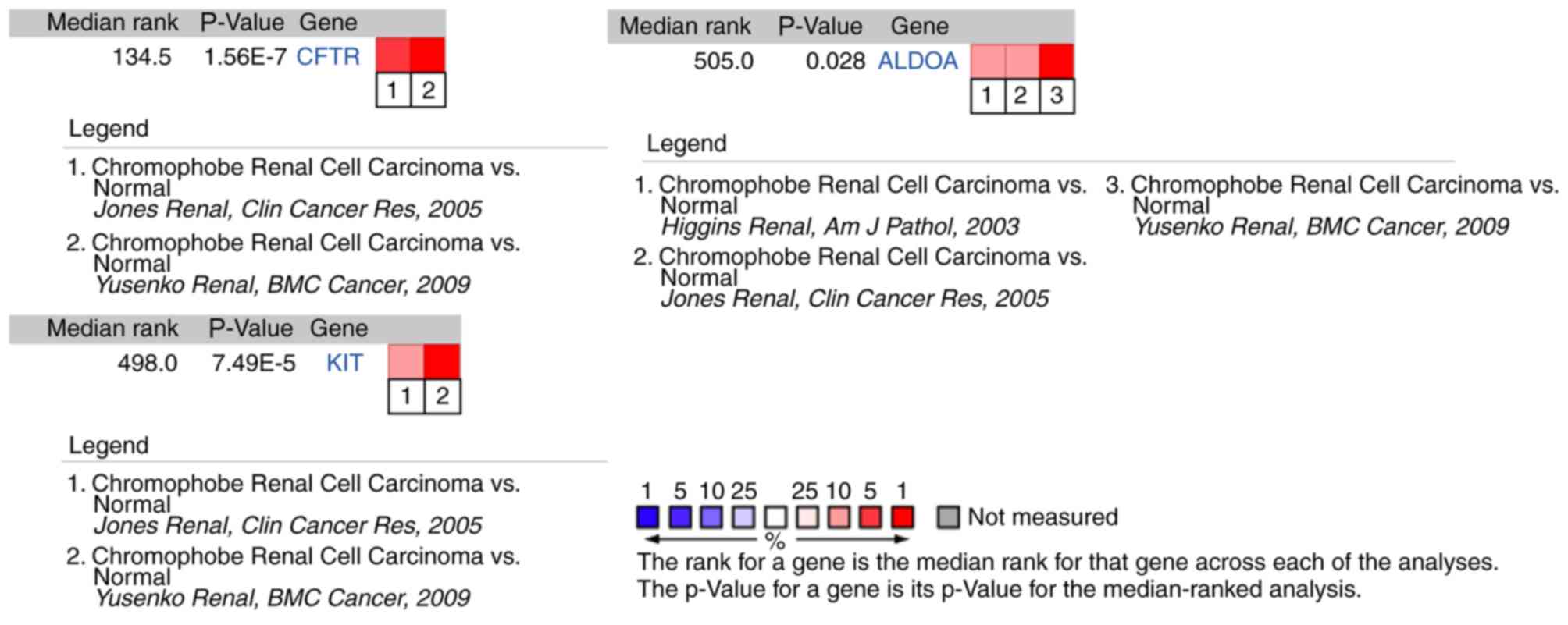
The results of hub gene expression level analysis in
chRCC revealed that the expression of KIT, CFTR and
ALDOA had differences among different analysis datasets
(Fig. 8; Fig. S1).
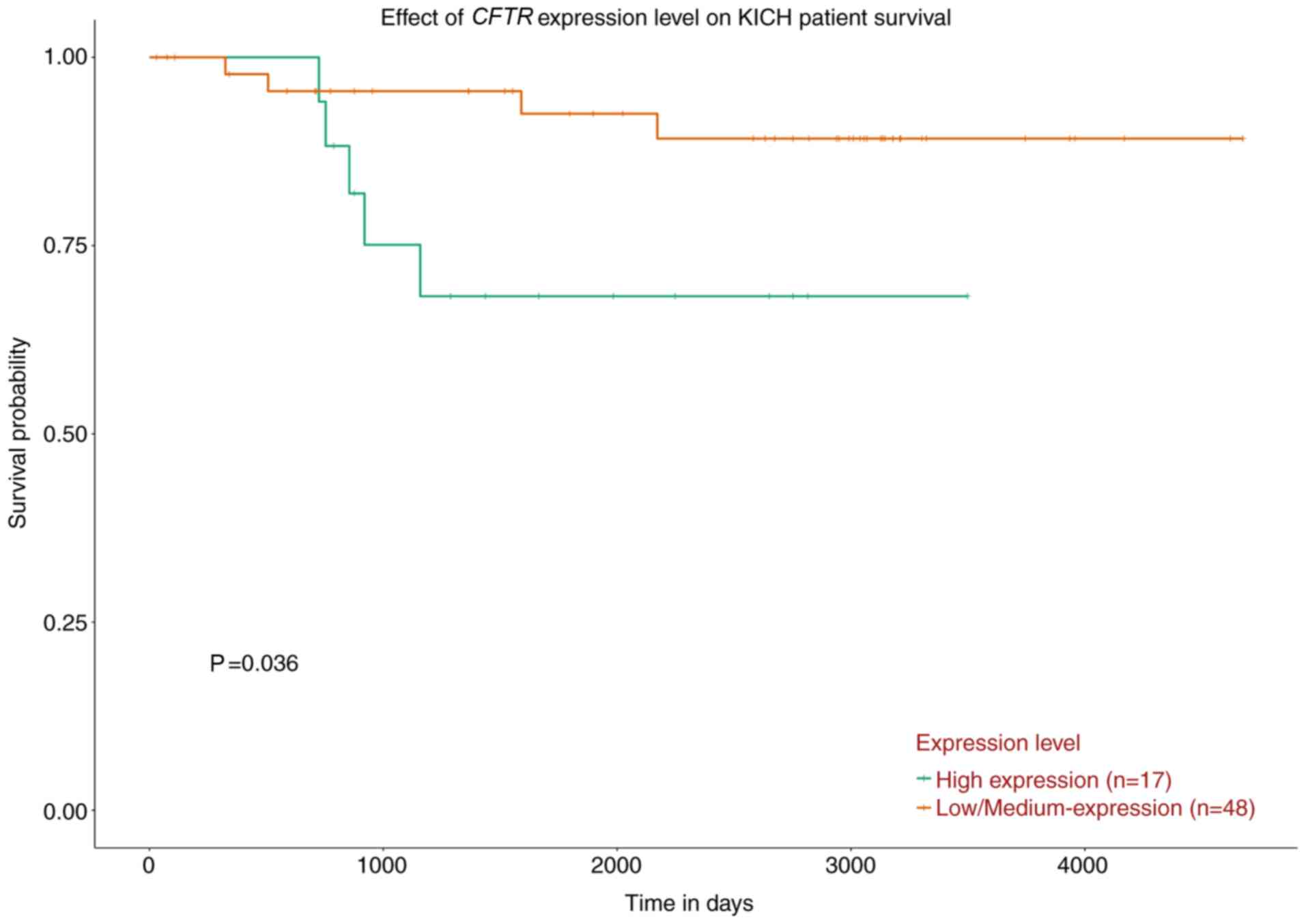
Survival analysis
The overall survival analysis of the 10 hub genes
demonstrated that only high expression levels of CFTR were
associated with a worse survival rate in patients with chRCC
(Fig. 9; Fig. S2).
Discussion
chRCC is the third most common histological subtype
of RCC, behind clear cell RCC and papillary RCC (3); it accounts for 5–7% of all RCC cases
(4). Although patients with chRCC
have a better prognosis compared with other subtypes, the long-term
outcomes are highly variable and there is a 5–10% probability of
eventually developing metastasis (7). Therefore, it is essential to identify
the tumor-specific biomarkers and the underlying molecular
mechanisms of chRCC, which may be conducive to developing novel
diagnostic and therapeutic strategies for chRCC. Microarray
analyses with high-throughput sequencing technologies have been
widely used to determine potential diagnostic and therapeutic
targets in the progression of cancer (19,20).
In the present study, a total of 266 overlapping
DEGs, including 88 upregulated genes and 178 downregulated genes,
were identified from 3 profile datasets. GO analysis revealed that
266 DEGs were mainly enriched in 17 terms, including ‘extracellular
exosome’, ‘plasma membrane’, ‘extracellular region’, ‘extracellular
matrix’, ‘cell adhesion’ and ‘extracellular matrix organization’.
In addition, 266 DEGs underwent KEGG analysis and were shown to be
enriched mainly in 9 pathways. In the PPI network, 10 genes with a
high degree of interaction were chosen as hub genes, including 3
upregulated genes (KIT, CFTR and ALDOA) and 7
downregulated genes (DCN, COL3A1, CXCL12, CTGF, LUM, TNC and
THBS2).
KIT, a receptor tyrosine kinase, can activate
several signaling pathways, including the PI3K-Akt signaling
pathway (21). Mutations of
KIT are associated with gastrointestinal stromal tumors,
lung cancer and other tumor types (22). ALDOA, a member of the class I
fructose- bisphosphate aldolase protein family, may contribute to
tumorigenesis and the progression of pancreatic and lung cancer
(23). DCN plays a vital role
in tumor suppression, including a stimulatory effect on autophagy
and inflammation, and an inhibitory effect on angiogenesis and
tumorigenesis, after binding to multiple cell surface receptors
(24). CXCL12 is associated
with diverse cellular functions, including immune surveillance,
tumor growth and metastasis, and the inflammatory response
(25). Tang et al (26) reported that high expression of
tenascin C, an extracellular matrix protein, was significantly
associated with poor disease-free survival in patients with lung
cancer. Thrombospondin 2, as a potent inhibitor of tumor growth and
angiogenesis, may be involved in cell adhesion and migration
(27).
In order to further verify the association between
the 10 hub genes and chRCC, the present study compared the
expression of 10 hub genes across multiple datasets using the
Oncomine platform; 3 genes (KIT, CFTR and ALDOA) were
indicated to have differences among the datasets. Furthermore, the
overall survival analysis based on UALCAN revealed that high
expression levels of CFTR were associated with a worse survival
rate in patients with chRCC. In summary, CFTR may be a potential
prognostic biomarker and novel therapeutic target for chRCC.
CFTR, a cAMP-activated chloride channel
widely distributed in the epithelial cells of various tissues
(28), plays an important role in
maintaining cell homeostasis and is associated with metabolism
(29). Mutations in CFTR are
responsible for regulation of epithelial ion and water transport
and fluid homeostasis, which affects the epithelial tissue of
various organ systems, including the urogenital, respiratory and
gastrointestinal systems (30). In
addition, CFTR mutation increases the risk of various types
of cancer, including lung, breast and colon cancer (31). Xu et al (32) reported that CFTR could promote
the aggression of ovarian cancer and that knockdown of CFTR
suppressed the aggressive behavior of ovarian cancer. In addition,
Peng et al (33) found that
higher expression of CFTR was associated with aggressive behaviors,
progression and a poor prognosis in cervical cancer, suggesting
that CFTR may be a novel therapeutic target and prognostic
indicator for cervical cancer. A number of studies have reported
that the downregulation of CFTR promotes invasion and proliferation
and is associated with poor prognosis in several types of cancer,
including lung (34), intestinal
(35) and esophageal cancer
(36). To date, to the best of our
knowledge, there have been no studies on the association between
CFTR and RCC. Therefore, further experimental investigation
is required to examine the influence of CFTR mutations on
RCC, both in vivo and in vitro.
In conclusion, based on integrated bioinformatics
analysis, the present study identified 266 DEGs. It was indicated
that CFTR may be involved in the progression and poor
prognosis of chRCC, and that it may function as a novel therapeutic
target and prognostic biomarker for chRCC. These results improve
our understanding of chRCC at the molecular level. However, further
investigation of CFTR both in vivo and in vitro are
required to confirm the findings of this study, in order to verify
the functions and elucidate the underlying mechanisms of chRCC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by Ke-qun Chai Inheritance
Studio of National Prominent Chinese Medicine Doctor, State
Administration of Traditional Chinese Medicine (Hangzhou, China;
grant no. 2A21533).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SW and KC conceived and designed the study. ZY and
SW performed the experiment. SW wrote the paper. KC and ZY reviewed
and edited the manuscript. All authors approved the manuscript and
agree to be accountable for all aspects of the research with regard
to ensuring that the accuracy or integrity of any part of the work
are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sanchez DJ and Simon MC: Genetic and
metabolic hallmarks of clear cell renal cell carcinoma. Biochim
Biophys Acta Rev Cancer. 1870:23–31. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guo K, Chen Q, He X, Yao K, Li Z, Liu Z,
Chen J, Liu Z, Guo C, Lu J, et al: Expression and significance of
Cystatin-C in clear cell renal cell carcinoma. Biomed Pharmacother.
107:1237–1245. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weng WH, Chen YT, Yu KJ, Chang YH, Chuang
CK and Pang ST: Genetic alterations of her genes in chromophobe
renal cell carcinoma. Oncol Lett. 11:2111–2116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Noguchi G, Tsutsumi S, Yasui M, Ohtake S,
Umemoto S, Nakaigawa N, Yao M and Kishida T: Significant response
to nivolumab for metastatic chromophobe renal cell carcinoma with
sarcomatoid differentiation: A case report. BMC Urol. 18:262018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Drendel V, Heckelmann B, Schell C, Kook L,
Biniossek ML, Werner M, Jilg CA and Schilling O: Proteomic
distinction of renal oncocytomas and chromophobe renal cell
carcinomas. Clin Proteomics. 15:252018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He HT, Xu M, Kuang Y, Han XY, Wang MQ and
Yang Q: Biomarker and competing endogenous RNA potential of
tumor-specific long noncoding RNA in chromophobe renal cell
carcinoma. Onco Targets Ther. 9:6399–6406. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Casuscelli J, Weinhold N, Gundem G, Wang
L, Zabor EC, Drill E, Wang PI, Nanjangud GJ, Redzematovic A,
Nargund AM, et al: Genomic landscape and evolution of metastatic
chromophobe renal cell carcinoma. JCI Insight. 2:926882017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vastrad C and Vastrad B: Bioinformatics
analysis of gene expression profiles to diagnose crucial and novel
genes in glioblastoma multiform. Pathol Res Pract. 214:1395–1461.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li L, Cai S, Liu S, Feng H and Zhang J:
Bioinformatics analysis to screen the key prognostic genes in
ovarian cancer. J Ovarian Res. 10:272017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cao L, Chen Y, Zhang M, Xu D, Liu Y, Liu
T, Liu SX and Wang P: Identification of hub genes and potential
molecular mechanisms in gastric cancer by integrated bioinformatics
analysis. PeerJ. 6:e51802018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Zhang Y, Huang Q and Li C:
Integrated bioinformatics analysis reveals key candidate genes and
pathways in breast cancer. Mol Med Rep. 17:8091–8100.
2018.PubMed/NCBI
|
|
12
|
Yusenko MV, Zubakov D and Kovacs G: Gene
expression profiling of chromophobe renal cell carcinomas and renal
oncocytomas by Affymetrix GeneChip using pooled and individual
tumours. Int J Biol Sci. 29:517–527. 2009. View Article : Google Scholar
|
|
13
|
Yusenko MV, Ruppert T and Kovacs G:
Analysis of differentially expressed mitochondrial proteins in
chromophobe renal cell carcinomas and renal oncocytomas by 2-D gel
electrophoresis. Int J Biol Sci. 6:213–224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jones J, Out H, Spentzos D, Kolia S, Inan
M, Beecken WD, Fellbaum C, Gu X, Joseph M, Pantuck AJ, et al: Gene
signatures of progression and metastasis in renal cell cancer. Clin
Cancer Res. 11:5730–5739. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong
S, Kong L, Gao G, Li CY and Wei L: Kobas 2.0: A web server for
annotation and identification of enriched pathways and diseases.
Nucleic Acids Res 39 (Web Server Issue). W316–W322. 2011.
View Article : Google Scholar
|
|
17
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The string database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45:D362–D368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zang Y, Gu L, Zhang Y, Wang Y and Xue F:
Identification of key genes and pathways in uterine leiomyosarcoma
through bioinformatics analysis. Oncol Lett. 15:9361–9368.
2018.PubMed/NCBI
|
|
20
|
Zhu N, Hou J, Wu Y, Li G, Liu J, Ma G,
Chen B and Song Y: Identification of key genes in rheumatoid
arthritis and osteoarthritis based on bioinformatics analysis.
Medicine (Baltimore). 97:e109972018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marech I, Ammendola M, Leporini C, Patruno
R, Luposella M, Zizzo N, Passantino G, Sacco R, Farooqi AA, Zuccalà
V, et al: C-kit receptor and tryptase expressing mast cells
correlate with angiogenesis in breast cancer patients. Oncotarget.
9:7918–7927. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pai T, Bal M, Shetty O, Gurav M, Ostwal V,
Ramaswamy A, Ramadwar M and Desai S: Unraveling the spectrum of kit
mutations in gastrointestinal stromal tumors: An Indian tertiary
cancer center experience. South Asian J Cancer. 6:113–117. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ji S, Zhang B, Liu J, Qin Y, Liang C, Shi
S, Jin K, Liang D, Xu W, Xu H, et al: ALDOA functions as an
oncogene in the highly metastatic pancreatic cancer. Cancer Lett.
374:127–135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mlakar V, Berginc G, Volavsek M, Stor Z,
Rems M and Glavac D: Presence of activating KRAS mutations
correlates significantly with expression of tumour suppressor genes
DCN and TPM1 in colorectal cancer. BMC Cancer. 9:2822009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Katsura M, Shoji F, Okamoto T, Shimamatsu
S, Hirai F, Toyokawa G, Morodomi Y, Tagawa T, Oda Y and Maehara Y:
Correlation between CXCR4/CXCR7/CXCL12 chemokine axis expression
and prognosis in lymph-node-positive lung cancer patients. Cancer
Sci. 109:154–165. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang YA, Chen CH, Sun HS, Cheng CP, Tseng
VS, Hsu HS, Su WC, Lai WW and Wang YC: Global Oct4 target gene
analysis reveals novel downstream PTEN and TNC genes required for
drug-resistance and metastasis in lung cancer. Nucleic Acids Res.
43:1593–1608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang X, Zhang L, Li H, Sun W, Zhang H and
Lai M: THBS2 is a potential prognostic biomarker in colorectal
cancer. Sci Rep. 6:333662016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tu Z, Chen Q, Zhang JT, Jiang X, Xia Y and
Chan HC: CFTR is a potential marker for nasopharyngeal carcinoma
prognosis and metastasis. Oncotarget. 7:76955–76965. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xia X, Wang J, Liu Y and Yue M: Lower
cystic fibrosis transmembrane conductance regulator (CFTR) promotes
the proliferation and migration of endometrial carcinoma. Med Sci
Monit. 23:966–974. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oh IH, Oh C, Yoon TY, Choi JM, Kim SK,
Park HJ, Eun YG, Chung DH, Kwon KH and Choe BK: Association of CFTR
gene polymorphisms with papillary thyroid cancer. Oncol Lett.
3:455–461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qiao D, Yi L, Hua L, Xu Z, Ding Y, Shi D,
Ni L, Song N, Wang Y and Wu H: Cystic fibrosis transmembrane
conductance regulator (CFTR) gene 5t allele may protect against
prostate cancer: A case-control study in chinese han population. J
Cyst Fibros. 7:210–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu J, Yong M, Li J, Dong X, Yu T, Fu X and
Hu L: High level of CFTR expression is associated with tumor
aggression and knockdown of CFTR suppresses proliferation of
ovarian cancer in vitro and in vivo. Oncol Rep.
33:2227–2234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Peng X, Wu Z, Yu L, Li J, Xu W, Chan HC,
Zhang Y and Hu L: Overexpression of cystic fibrosis transmembrane
conductance regulator (CFTR) is associated with human cervical
cancer malignancy, progression and prognosis. Gynecol Oncol.
125:470–476. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li W, Wang C, Peng X, Zhang H, Huang H and
Liu H: CFTR inhibits the invasion and growth of esophageal cancer
cells by inhibiting the expression of NF-κB. Cell Biol Int.
42:1680–1687. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Than BL, Linnekamp JF, Starr TK,
Largaespada DA, Rod A, Zhang Y, Bruner V, Abrahante J, Schumann A,
Luczak T, et al: CFTR is a tumor suppressor gene in murine and
human intestinal cancer. Oncogene. 35:4179–4187. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Son JW, Kim YJ, Cho HM, Lee SY, Lee SM,
Kang JK, Lee JU, Lee YM, Kwon SJ, Choi E, et al: Promoter
hypermethylation of the CFTR, gene and clinical/pathological
features associated with non-small cell lung cancer. Respirology.
16:1203–1209. 2011. View Article : Google Scholar : PubMed/NCBI
|