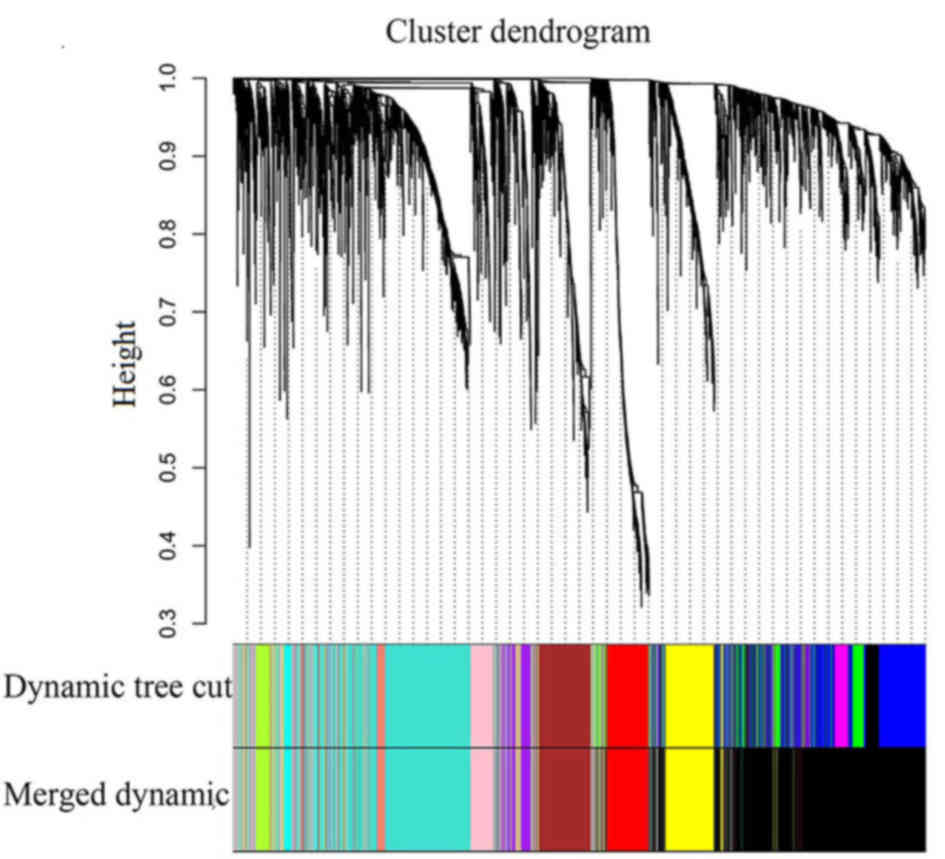
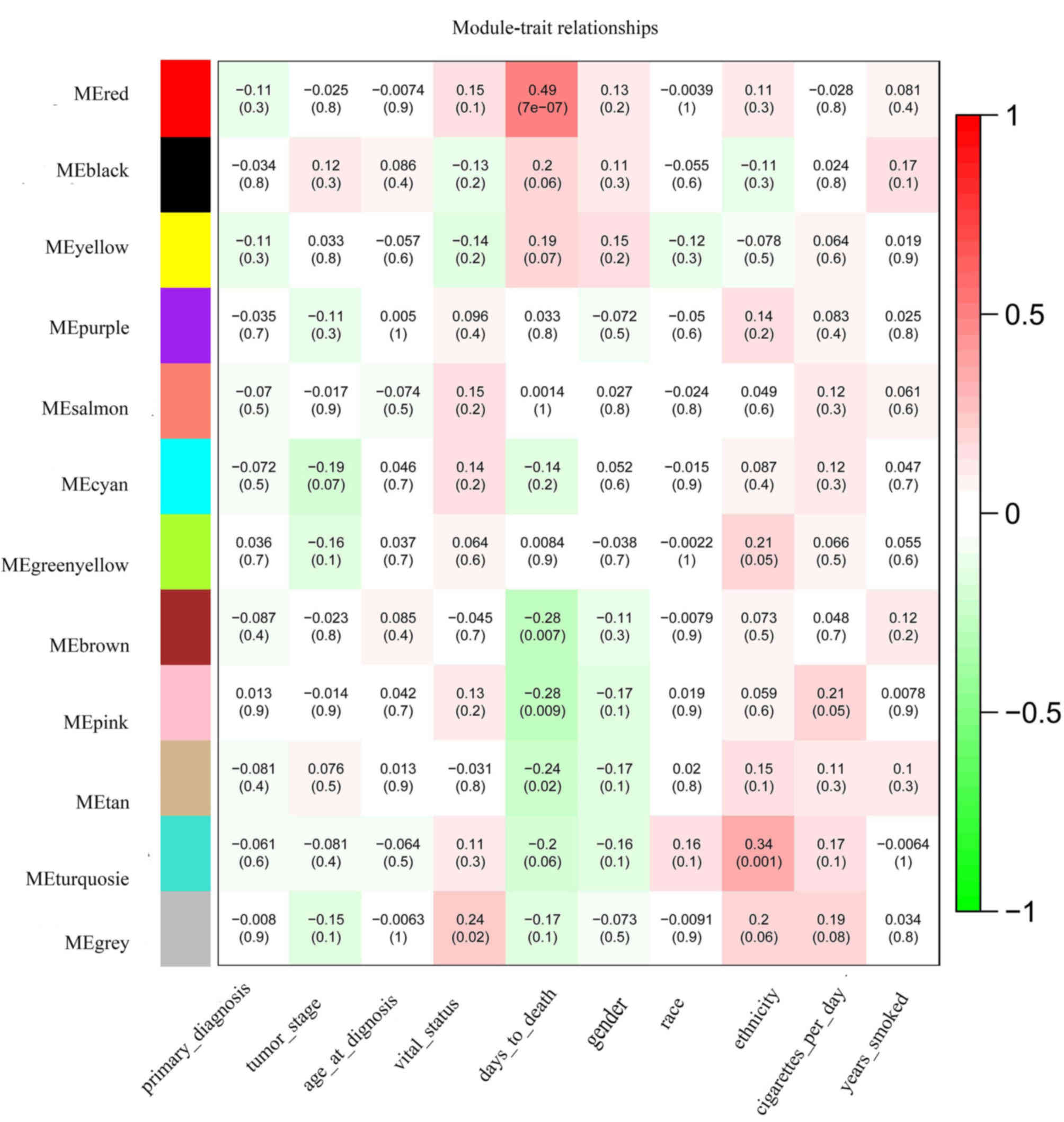
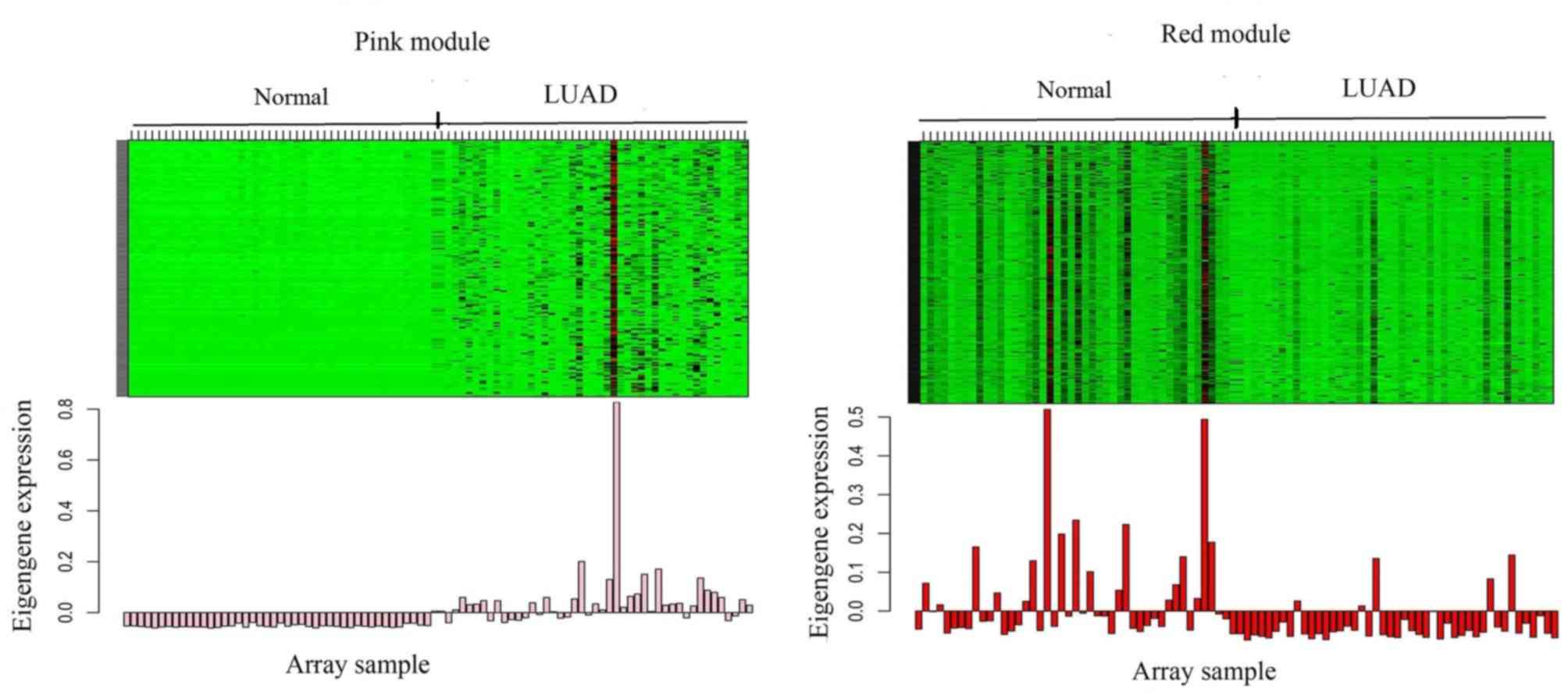
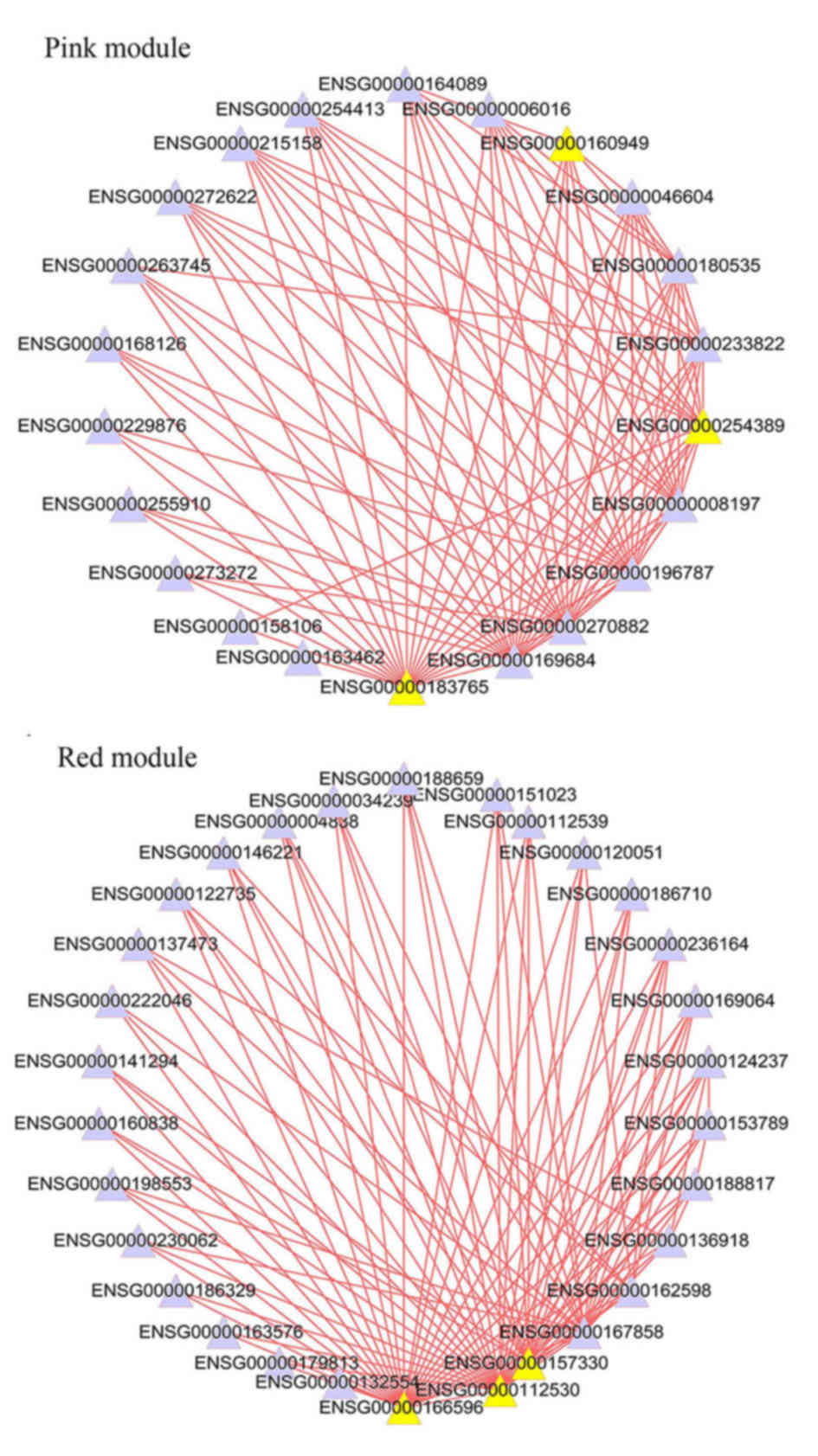
| RNA-seq | Normal |
|
|
|
|
|
TCGA-44-2668-11A-01R-1758-07, |
TCGA-55-6982-11A-01R-1949-07, |
|
|
|
|
TCGA-44-6145-11A-01R-1858-07, |
TCGA-55-6983-11A-01R-1949-07, |
TCGA-50-5930-11A-01R-1755-07, |
|
|
|
TCGA-50-5936-11A-01R-1628-07, |
TCGA-55-6970-11A-01R-1949-07, |
TCGA-44-6146-11A-01R-1858-07, |
|
|
|
TCGA-91-6849-11A-01R-1949-07, |
TCGA-55-6972-11A-01R-1949-07, |
TCGA-49-6744-11A-01R-1858-07, |
|
|
|
TCGA-50-5935-11A-01R-1858-07, |
TCGA-91-6831-11A-02R-1858-07, |
TCGA-50-5931-11A-01R-1858-07, |
TCGA-38-4627-11A-01R-1758-07, |
|
|
TCGA-91-6829-11A-01R-1858-07, |
TCGA-44-2655-11A-01R-1758-07, |
TCGA-49-6743-11A-01R-1858-07, |
TCGA-44-6148-11A-01R-1858-07, |
|
|
TCGA-55-6969-11A-01R-1949-07, |
TCGA-55-6978-11A-01R-1949-07, |
TCGA-91-6836-11A-01R-1858-07, |
TCGA-38-4626-11A-01R-1758-07, |
|
|
TCGA-55-6981-11A-01R-1949-07, |
TCGA-38-4625-11A-01R-1758-07, |
TCGA-44-2665-11A-01R-1758-07, |
TCGA-49-4490-11A-01R-1858-07, |
|
|
TCGA-49-6761-11A-01R-1949-07, |
TCGA-44-5645-11A-01R-1628-07, |
TCGA-49-6745-11A-01R-1858-07, |
TCGA-55-6968-11A-01R-1949-07, |
|
|
TCGA-55-6979-11A-01R-1949-07, |
TCGA-50-5932-11A-01R-1755-07, |
TCGA-44-6147-11A-01R-1858-07, |
TCGA-55-6971-11A-01R-1949-07, |
|
|
TCGA-49-4512-11A-01R-1858-07, |
TCGA-50-5933-11A-01R-1755-07, |
TCGA-91-6847-11A-01R-1949-07, |
TCGA-38-4632-11A-01R-1755-07, |
|
|
TCGA-44-6776-11A-01R-1858-07, |
TCGA-91-6835-11A-01R-1858-07, |
TCGA-44-3396-11A-01R-1758-07, |
TCGA-44-2657-11A-01R-1758-07, |
|
|
TCGA-44-2661-11A-01R-1758-07, |
TCGA-44-6144-11A-01R-1755-07 |
|
|
|
| Cancer |
|
|
|
|
|
TCGA-55-7570-01A-11R-2039-07, |
TCGA-55-7726-01A-11R-2170-07, |
|
|
|
|
TCGA-44-7661-01A-11R-2066-07, |
TCGA-44-7662-01A-11R-2066-07, |
TCGA-55-7910-01A-11R-2170-07, |
TCGA-44-2665-01A-01R-0946-07, |
|
|
TCGA-44-7669-01A-21R-2066-07, |
TCGA-55-7725-01A-11R-2170-07, |
TCGA-44-7671-01A-11R-2066-07, |
TCGA-55-7724-01A-11R-2170-07, |
|
|
TCGA-50-5930-01A-11R-1755-07, |
TCGA-49-6745-01A-11R-1858-07, |
TCGA-50-5933-01A-11R-1755-07, |
TCGA-44-6778-01A-11R-1858-07, |
|
|
TCGA-78-7163-01A-12R-2066-07, |
TCGA-50-7109-01A-11R-2039-07, |
TCGA-55-7283-01A-11R-2039-07, |
TCGA-44-7659-01A-11R-2066-07, |
|
|
TCGA-55-7574-01A-11R-2039-07, |
TCGA-44-7670-01A-11R-2066-07, |
TCGA-44-7667-01A-31R-2066-07, |
TCGA-55-7914-01A-11R-2170-07, |
|
|
TCGA-55-7911-01A-11R-2170-07, |
TCGA-44-7672-01A-11R-2066-07, |
TCGA-44-7660-01A-11R-2066-07, |
TCGA-86-7713-01A-11R-2066-07, |
|
|
TCGA-49-6742-01A-11R-1858-07, |
TCGA-78-7540-01A-11R-2066-07, |
TCGA-49-6743-01A-11R-1858-07, |
TCGA-69-7980-01A-11R-2187-07, |
|
|
TCGA-44-A4SS-01A-11R-A24X-07, |
TCGA-55-8092-01A-11R-2241-07, |
TCGA-44-2659-01A-01R-0946-07, |
TCGA-86-7711-01A-11R-2066-07, |
|
|
TCGA-86-7953-01A-11R-2187-07, |
TCGA-55-6985-01A-11R-1949-07, |
TCGA-67-6217-01A-11R-1755-07, |
TCGA-78-7536-01A-11R-2066-07, |
|
|
TCGA-97-A4M5-01A-11R-A24X-07, |
TCGA-86-8279-01A-11R-2287-07, |
TCGA-44-2655-01A-01R-0946-07, |
TCGA-44-6777-01A-11R-1858-07, |
|
|
TCGA-93-7348-01A-21R-2039-07, |
TCGA-55-7576-01A-11R-2066-07, |
TCGA-55-7903-01A-11R-2170-07 |
|
| miRNA-seq | Normal |
|
|
|
|
|
TCGA-44-2668-11A-01R-1757-13, |
TCGA-93-7348-11A-01H-2038-13, |
|
|
|
|
TCGA-55-7576-11A-01H-2065-13, |
TCGA-55-7903-11A-01H-2169-13, |
TCGA-55-7726-11A-01H-2169-13, |
TCGA-50-5932-11A-01H-2169-1, |
|
|
TCGA-44-7661-11A-01H-2065-13, |
TCGA-44-6776-11A-01H-2169-13, |
TCGA-44-7662-11A-01H-2065-13, |
TCGA-55-7910-11A-01H-2169-13, |
|
|
TCGA-91-6835-11A-01H-2169-13, |
TCGA-55-7570-11A-01H-2038-13, |
TCGA-44-2665-11A-01R-1757-13, |
TCGA-44-3396-11A-01R-1757-13, |
|
|
TCGA-44-7669-11A-01H-2065-13, |
TCGA-55-7725-11A-01H-2169-13, |
TCGA-44-7671-11A-01H-2065-13, |
TCGA-55-7724-11A-01H-2169-13, |
|
|
TCGA-50-5930-11A-01H-2169-13, |
TCGA-50-5933-11A-01H-2169-13, |
TCGA-44-2657-11A-01R-1757-13, |
TCGA-44-6778-11A-01H-2169-13, |
|
|
TCGA-49-6745-11A-01H-2169-13, |
TCGA-78-7163-11A-01H-2065-13, |
TCGA-50-7109-11A-01H-2038-13, |
TCGA-55-7283-11A-01H-2038-13, |
|
|
TCGA-44-7659-11A-01H-2065-13, |
TCGA-44-2661-11A-01R-1757-13, |
TCGA-55-7574-11A-01H-2038-13, |
TCGA-44-7670-11A-01H-2065-13, |
|
|
TCGA-44-7667-11A-01H-2065-13, |
TCGA-55-7914-11A-01H-2169-13, |
TCGA-44-7672-11A-01H-2065-13, |
TCGA-55-7911-11A-01H-2169-13, |
|
|
TCGA-44-7660-11A-01H-2065-13, |
TCGA-86-7713-11A-01H-2065-13, |
TCGA-49-6742-11A-01H-2169-13, |
TCGA-44-6144-11A-01H-2169-13, |
|
|
TCGA-78-7540-11A-01H-2065-13, |
TCGA-49-6743-11A-01H-2169-13, |
TCGA-86-7711-11A-01H-2065-13, |
TCGA-49-6744-11A-01H-2169-13, |
|
|
TCGA-44-2655-11A-01R-1757-13, |
TCGA-91-6836-11A-01H-2169-13, |
TCGA-44-6777-11A-01H-2169-13, |
|
|
| Cancer |
|
|
|
|
|
TCGA-55-6982-01A-11H-1948-13, |
TCGA-50-5935-01A-11H-1754-13, |
|
|
|
|
TCGA-91-6831-01A-11H-1857-13, |
TCGA-50-5931-01A-11H-1754-13, |
TCGA-38-4627-01A-01T-1207-13, |
TCGA-91-6829-01A-21H-1857-13, |
|
|
TCGA-44-2655-01A-01T-0947-13, |
TCGA-49-6743-01A-11H-1857-13, |
TCGA-44-6148-01A-11H-1754-13, |
TCGA-55-6969-01A-11H-1948-13, |
|
|
TCGA-55-6978-01A-11H-1948-13, |
TCGA-44-6145-01A-11H-1754-13, |
TCGA-38-4626-01A-01T-1207-13, |
TCGA-55-6981-01A-11H-1948-13, |
|
|
TCGA-38-4625-01A-01T-1207-13, |
TCGA-44-2665-01A-01T-0947-13, |
TCGA-49-4490-01A-21H-1857-13, |
TCGA-49-6761-01A-31H-1948-13, |
|
|
TCGA-44-5645-01A-01T-1627-13, |
TCGA-49-6745-01A-11H-1857-13, |
TCGA-55-6968-01A-11H-1948-13, |
TCGA-55-6979-01A-11H-1948-13, |
|
|
TCGA-55-6983-01A-11H-1948-13, |
TCGA-44-6147-01A-11H-1754-13, |
TCGA-55-6971-01A-11H-1948-13, |
TCGA-49-4512-01A-21H-1857-13, |
|
|
TCGA-50-5933-01A-11H-1754-13, |
TCGA-91-6847-01A-11H-1948-13, |
TCGA-38-4632-01A-01T-1754-13, |
TCGA-69-7980-01A-11H-2186-13, |
|
|
TCGA-44-A4SS-01A-11H-A24S-13, |
TCGA-55-7727-01A-11H-2169-13, |
TCGA-44-2659-01A-01T-0947-13, |
TCGA-50-5930-01A-11H-1754-13, |
|
|
TCGA-86-7953-01A-11H-2186-13, |
TCGA-55-6985-01A-11H-1948-13, |
TCGA-67-6217-01A-11H-1754-13, |
TCGA-78-7536-01A-11H-2065-13, |
|
|
TCGA-97-A4M5-01A-11H-A24S-13, |
TCGA-86-8279-01A-11H-2286-13, |
TCGA-50-5936-01A-11H-1627-13, |
TCGA-55-6970-01A-11H-1948-13, |
|
|
TCGA-44-6146-01A-11H-A279-13, |
TCGA-91-6849-01A-11H-1948-13, |
TCGA-55-6972-01A-11H-1948-13 |
|