Introduction
Glioblastoma is the most common type of malignant
brain cancer in adults around the world (1). Despite the rapid development of
surgical techniques and radiotherapy, and the widespread use of
chemotherapy drugs, such as temozolomide (TMZ), glioblastoma
continues to be associated with poor prognosis, with a 5-year
survival rate of 5–13%, along with a high recurrence rate (2). Therefore, it is important to explore
the molecular mechanisms underlying the occurrence and progression
of glioblastoma, and to identify novel therapeutic targets to
improve glioblastoma treatment and prognosis.
TP53 induced glycolysis regulatory phosphatase
(TIGAR) is located on chromosome 12p13.3 and includes six coding
exons and two p53 binding sites (3).
A previous study has demonstrated that TIGAR is similar to
phosphoglucose mutagenesis enzyme and has biphosphatase activity to
decompose fructose 2,6-bisphosphate (FB) (4). FB is the allosteric activator of
phosphofructose kinase (PFK). Therefore, TIGAR inhibits PFK
activity by decreasing FB and directs metabolic flow from
glycolysis to the pentose phosphate pathway (PPP) (4). The PPP provides phosphate ribose for
nucleic acid synthesis. Reduced nicotinamide adenine dinucleotide
phosphate (NADPH), as a byproduct of the PPP, is the main reactive
oxygen species (ROS) scavenger in cells. Additionally, TIGAR is
overexpressed in several types of cancer, including leukemia
(5), lung cancer (6), breast cancer (7), liver cancer (8) and colon cancer (9). TIGAR is considered to protect cancer
cells against ROS-induced apoptosis and to induce DNA damage repair
(10).
To the best of our knowledge, a role for TIGAR in
glioblastoma has not been reported. The present study revealed that
TIGAR was overexpressed in glioblastoma and is a potential novel
prognostic and migration marker in patients with glioblastoma.
Additionally, TIGAR decreased oxidative stress in glioblastoma
cells through PPP-mediated NADPH generation. The present study
demonstrated that TIGAR knockdown led to significant inhibition of
proliferation, migration and invasion in U-87MG cells in an
oxidative stress-independent manner. In addition, TIGAR interacted
with protein kinase B (AKT) and promoted AKT activation. The
results of the present study suggested that TIGAR, as an important
mediator of glioma progression, may be a potential therapeutic
target in glioblastoma.
Materials and methods
Reagents
Bovine serum albumin (BSA), NADPH (cat. no. ST360),
dichloro-dihydro-fluorescein diacetate (DCFH-DA; cat. no. S0033-1)
and rabbit immunoglobulin G (IgG; cat. no. A7016) were purchased
from Beyotime Institute of Biotechnology. Fetal bovine serum (FBS)
was purchased from Thermo Fisher Scientific, Inc.
Lipofectamine® 2000 reagent was purchased from
Invitrogen (Thermo Fisher Scientific, Inc.). The GTVisin™
anti-mouse/anti-rabbit immunohistochemical analysis kit was
purchased from Gene Company, Ltd. Anti-TIGAR (cat. no. sc-166290;
1:1,000 dilution), anti-B cell lymphoma 2 (Bcl2; cat. no. sc-509;
1:1,000 dilution), anti-Bcl2-associated X protein (BAX; cat. no.
sc-20067; 1:1,000 dilution), anti-α-smooth muscle actin (α-SMA;
cat. no. sc-53142; 1:1,000 dilution), anti-E-cadherin (cat. no.
sc-71009; 1:1,000 dilution), anti-N-cadherin (cat. no. sc-59987;
1:1,000 dilution), anti-Snail (cat. no. sc-271977; 1:1,000
dilution), anti-Vimentin (cat. no. sc-80975; 1:1,000 dilution) and
HRP-conjugated goat anti-mouse IgG (m-IgGκ BP-HRP; cat. no.
sc-516102; 1:4,000 dilution) were purchased from Santa Cruz
Biotechnology, Inc. Anti-phosphoinositide 3-kinase (PI3K; cat. no.
ab191606; 1:1,000 dilution) and anti-phosphorylated (p)-PI3K p85 α
(cat. no. ab182651; 1:1,000 dilution) were obtained from Abcam.
Anti-AKT (cat. no. 4685; 1:1,000 dilution), anti-p-AKT (Ser473;
cat. no. 4060; 1:1,000 dilution), anti-β-Actin (cat. no. 3700;
1:3,000 dilution) and HRP-conjugated goat anti-rabbit IgG (cat. no.
7074; 1:4,000 dilution) antibodies, as well as a mouse IgG control
(cat. no. 5415), rabbit IgG control (cat. no. 3900) and
radioimmunoprecipitation assay (RIPA) buffer (cat. no. 9806), were
purchased from Cell Signaling Technology, Inc. Dimethyl sulfoxide
(DMSO), isopropanol, ethanol and chloroform were purchased from
Sinopharm Chemical Reagent Co., Ltd.
Cell culture
The human glioblastoma cell lines LN-18, LN-229,
U-87MG, U-251MG and SNB-19 were purchased from the Shanghai
Institute of Biochemistry and Cell Biology and were authenticated
by short tandem repeat profiling. The U-87MG American Type Culture
Collection cell line used in the present study is most likely a
glioblastoma cell line, but of unknown origin. The cells were
cultured in Dulbecco's Modified Eagle's medium (DMEM, Gibco; Thermo
Fisher Scientific, Inc.) without any antibiotics, supplemented with
10% FBS (Hangzhou Sijiqing Bio-engineering Material Co.) at 37°C in
a humidified atmosphere with 5% CO2.
RNA interference and transfection
A total of 2×104 U-87MG cells were
infected with 4 µl of 1×109 viral particles of
LV-TIGAR-short hairpin RNA (shRNA; 5′-GATTAGCAGCCAGTGTCTTAG-3′) or
negative control (nc)-shRNA (5′-TTACCGAGACCGTACGTAT-3′), which were
synthesized by Shanghai GenePharma Co., Ltd. After 2 days, the
transfected cells were selected for using DMEM containing 2 µg/ml
puromycin in subsequent experiments.
pcDNA3.1 and pcDNA3.1-TIGAR (human) were purchased
from Cyagen Biosciences, Inc. A total of 1.8×105 U-87MG
cells in 6-well plates were transfected with 2 µg of either plasmid
using 4 µl of Lipofectamine® 2000, and complete medium
was added to the cells 6 h after transfection. After further
culturing for 36 h, subsequent experiments were performed. For
TIGAR dependent AKT activation, 1.8×105 U-87MG cells in
6-well plates were transfected with 1, 2 or 3 µg pcDNA3.1-TIGAR
using 4 µl of Lipofectamine® 2000, and complete medium
was added to the cells 6 h after transfection. After a further 36
h, cells were lysed, and AKT and p-AKT were detected using western
blot assay.
Cell viability
U-87MG/NC cells, U-87MG/sh-TIGAR cells and U-87MG
cells transfected with pcDNA3.1 or pcDNA3.1-TIGAR were seeded into
a 96-well culture plate at a density of 3×104
cells/well. Cell proliferation was measured at 24, 48, 72 and 96 h.
MTT (1 mg/ml dissolved in PBS; 100 µl/well; Sigma-Aldrich; Merck
KGaA) was added, and the cells were cultured for 4 h at 37°C prior
to adding 100 µl DMSO. The absorbance was determined using a
multiplate reader (SpectraMax 190; Molecular Devices, LLC) at a
wavelength of 570 nm. For temozolomide (TMZ)-induced cellular
toxicity, cells were treated with 100 µM of TMZ (Selleck, cat. no.
S1237) for 24 h and apoptosis was detected using western
blotting.
Colony formation assay
U-87MG/NC, U-87MG/sh-TIGAR cells, U-87MG/pcDNA3.1
and U-87MG/pcDNA3.1-TIGAR cells (200 cells/well) were plated into a
6-well plate and cultured for 14 days at 37°C. The media were then
removed, and cells were fixed with 4% paraformaldehyde at room
temperature for 10 min and stained with 1% crystal violet in 2%
ethanol at room temperature for 20 min. Visible colonies were
counted by eye.
Western blotting assay
Total protein was extracted from whole cell lysates
using RIPA buffer, and the protein concentration was measured using
a bicinchoninic acid assay. The protein samples (30 µg/lane) were
separated by 12.5% SDS-PAGE. Proteins were transferred to
polyvinylidene difluoride membranes (EMD Millipore) at 300 mA for
90 min. Subsequently, the membranes were blocked at room
temperature for 1 h with Tris-buffered saline containing 0.1%
Tween-20 (TBST) and 5% dry milk and incubated overnight with
primary antibodies at 4°C. Membranes were washed three times with
TBST and incubated for 2 h with secondary antibodies at room
temperature. After washing for three times with TBST, blots were
detected using enhanced chemiluminescence (ECL) reagents (Thermo
Fisher Scientific, Inc) and captured by a Luminescent Image
Analyzer LAS-3000 (Fujifilm Holdings Corporation), and the optical
densities of antibody-specific bands were analyzed using ImageJ
version 1.37 (National Institutes of Health).
Co-immunoprecipitation (Co-IP)
Cells were harvested by centrifugation at 500 × g
for 5 min at 4°C and lysed in RIPA lysis buffer for 30 min.
Following centrifugation at 13,000 × g for 15 min 4°C, the 1/10
volume of supernatant was collected as input, the 1/2 volume of
remaining supernatant was incubated with 1 µg anti-TIGAR (1:200
dilution) or anti-AKT (1:100 dilution) antibody and 40 µl 50%
protein A/G agarose slurry at 4°C overnight. The other remaining
supernatant was incubated with 1 µg control IgG (1:200 dilution)
and 40 µl 50% protein A/G agarose slurry at 4°C overnight. The
protein A/G agarose was recovered by centrifugation at 3,000 × g
for 5 min at 4°C and washed four times with ice-cold lysis buffer.
Proteins were eluted with 2× loading buffer by boiling for 10 min
and subjected to immunoblot analysis according to the
aforementioned western blotting assay.
ROS, NADPH, glutathione (GSH) and
malondialdehyde (MDA) detection
Intracellular ROS were determined using DCFH-DA. A
total of 4×105 cells were washed with 0.01 M PBS and
incubated with 20 µM DCFH-DA at 37°C for 30 min. The cells were
visualized using an inverted fluorescence microscope
(magnification, ×20; Olympus Corporation), and their fluorescence
intensity was measured via fluorescence spectrometry (Spectra
MaxGemini; Molecular Devices LLC). An NADPH detection kit (cat. no.
ECNP-100) was obtained from BioAssay Systems, and GSH (cat. no.
S0053) and MDA (cat. no. S0131) were purchased from Beyotime
Institute of Biotechnology. The NADPH level, GSH content and MDA
level were measured according to the manufacturers' protocols.
Migration and invasion
A Transwell plate (Costar; Corning Inc.) with an
8-µm pore insert was utilized to measure migration and invasion.
DMEM containing 10% FBS (0.6 ml) was added to the lower chamber. A
total of 5×104 U-87MG/nc or U-87MG/sh-TIGAR cells in
serum-free DMEM were directly added to the upper chamber and
incubated at 37°C for 12 h for the migration assay. To measure
invasion, 100 µl diluted Matrigel (1 mg/ml; BD Biosciences) in
serum-free cold DMEM was placed in the upper chamber and in the
lower chamber, DMEM containing 15% FBS was added, and the cells
were incubated at 37°C for 4 h to allow it to set. A total of
5×104 U-87MG/nc or U-87MG/sh-TIGAR cells in serum-free
DMEM were added directly to the upper chamber and incubated at 37°C
for 24 h. Cells in the lower chamber were fixed with 4%
paraformaldehyde at room temperature for 10 min and stained with 1%
crystal violet in 2% ethanol at room temperature for 20 min. The
numbers of cells in the lower chamber were counted under a light
microscope (magnification, ×20). At least five fields were analyzed
per section.
Immunohistochemistry
Immunohistochemistry was used to detect TIGAR
expression levels in surgical resections of glioblastoma tissues
collected from 15 male patients (45–55 years old) first diagnosed
and admitted to Hunan Cancer Hospital and The Affiliated Cancer
Hospital of Xiangya School of Medicine (Changsha, China) between
January 2018 and May 2018. The use of glioblastoma samples was
approved by the Ethics Committee of Hunan Cancer Hospital and
Affiliated Cancer Hospital of Xiangya School of Medicine (no.
20180104). Written informed consent was obtained from all patients
prior to enrollment.
The glioblastoma sections were fixed in 4%
paraformaldehyde for 2 days at room temperature followed by
paraffin-embedding. From the embedded tissue, 5-µm thick sections
were cut and deparaffinized in dimethylbenzene, rehydrated in
ethanol solutions of 100, 95, and 70% ethanol, and subsequently
washed in PBS for 10 min each. Antigen retrieval was performed at
95°C for 20 min, followed by washing with PBS for three times and
blocking with 3% BSA for 1 h at room temperature. Sections were
incubated with an anti-TIGAR antibody (1:100 dilution) at room
temperature for 2 h, and the slides were washed three times with
PBS and incubated with components of the GTVisin™
anti-mouse/anti-rabbit immunohistochemical analysis kit, according
to the manufacturer's protocols. Glioblastoma tissues and paired
normal-appearing tissues in sections were confirmed by Pathology
department. Images were captured under a light microscope
(magnification, ×20). At least five fields were analyzed per
section and quantify staining was analyzed using ImageJ version
1.37 (National Institutes of Health)
Bioinformatics
Kaplan-Meier survival analysis between TIGAR high
expression and low expression in glioma was carried out. Overall
survival time based on GSE4412-GPL96 dataset in PrognoScan was
performed on PrognoScan (http://dna00.bio.kyutech.ac.jp/PrognoScan/) (11). A total of 74 patients were divided
into TIGAR high expression group (n=36) and TIGAR low expression
group (n=38) and Cox P-value (P=0.007189) was
generated automatically.
Statistical analysis
Experimental data are expressed as the mean ±
standard deviation of at least three independent experiments.
Statistical analyses were performed using SPSS v18.0 statistical
software (SPSS, Inc.). Paired or unpaired Student's t-test were
used to compare the significance between two paired groups and two
independent groups respectively, and a one-way ANOVA followed by a
post-hos Tukey's test was used to determine the significance
between three or more independent groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
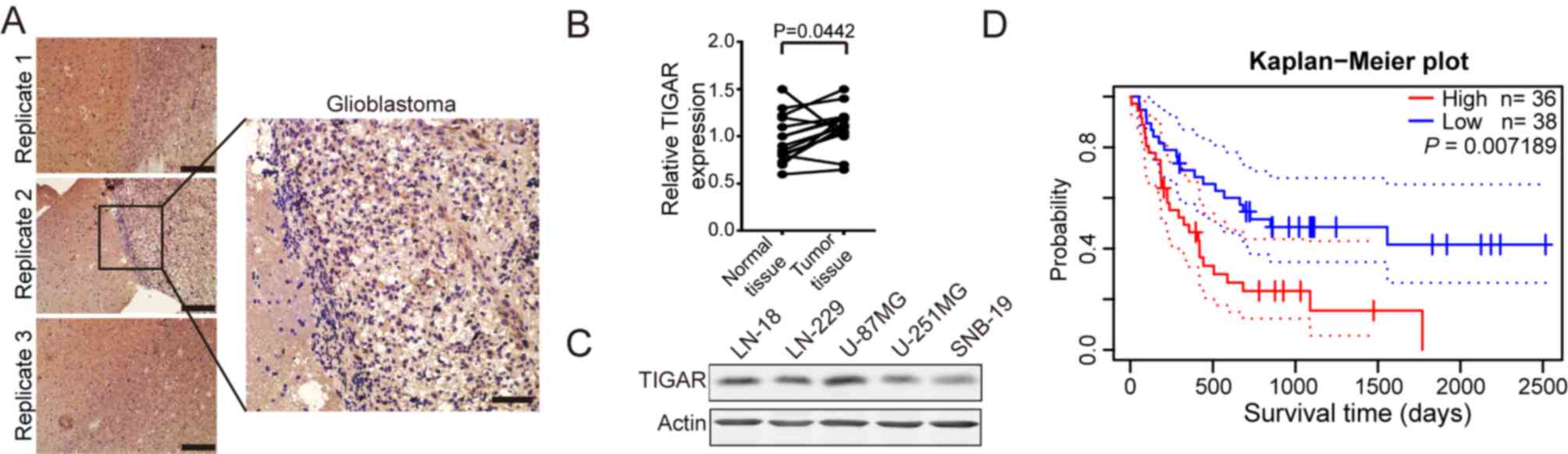
TIGAR is overexpressed in glioblastoma
tissues and positively associated with poor prognosis
It has been reported that TIGAR is upregulated in
several types of tumor (5–9). To determine whether TIGAR was
upregulated in glioblastoma, glioblastoma sections were obtained
and subjected to immunohistochemistry. As shown in Fig. 1A and B, TIGAR was significantly
overexpressed in glioblastoma tissues compared with paired
normal-appearing tissues. TIGAR expression in glioblastoma cell
lines, including LN-18, LN-229, U-87MG, U-251MG and SNB-19, was
detected. The results revealed that TIGAR was overexpressed in
U-87MG cells (Fig. 1C). Furthermore,
PrognoScan-based Kaplan-Meier survival analysis (11) revealed an association between
elevated TIGAR expression levels and shorter surv]ival duration in
the 74 patients with glioblastoma from the database. (Fig. 1D). These results indicated that TIGAR
was overexpressed in glioblastoma, and that its expression was
negatively associated with survival time.
TIGAR maintains NADPH to alleviate
oxidative stress in U-87MG cells
It has been reported that TIGAR promotes NADPH
generation through PPP (3). The
ratio of NADPH to NADP+ in TIGAR-knockdown U-87MG cells
was measured. As shown in Fig. 2A,
TIGAR content was decreased in TIGAR-knockdown cells. As shown in
Fig. 2B, TIGAR knockdown
significantly decreased NADPH content. Glutathione reductase
catalyzes the NADPH-driven reduction of oxidized glutathione (GSSG)
to GSH (12). Therefore, the ratio
of GSH to GSSG was measured in TIGAR-knockdown U-87MG cells. The
conversion of GSH from GSSG was significantly reduced in
TIGAR-knockdown cells (Fig. 2C).
NADPH and GSH are involved in cellular antioxidant responses.
Levels of MDA (13), a byproduct of
nonenzymatic lipid peroxidation and a principal marker of oxidative
stress, were assessed. It has been reported that NADPH addition can
protect against DNA damage in TIGAR-knockdown cells (14). Similarly, in the present study, it
was observed that TIGAR knockdown significantly increased MDA
levels in groups without NADPH treatment, and NADPH inhibited MDA
content in TIGAR-knockdown U-87MG cells compared with negative
control U-87MG cells although these changes were not significant
(Fig. 2D). Additionally, TIGAR
knockdown increased intracellular ROS in U-87MG cells compared with
the negative control U-87MG cells, as detected by higher
fluorescence following incubation with DCFH-DA. The elevated ROS
levels in TIGAR-knockdown U-87MG cells were attenuated by the
addition of NADPH (Fig. 2E and F).
These results suggested that TIGAR maintained the NADPH level to
protect U-87MG cells from oxidative stress.
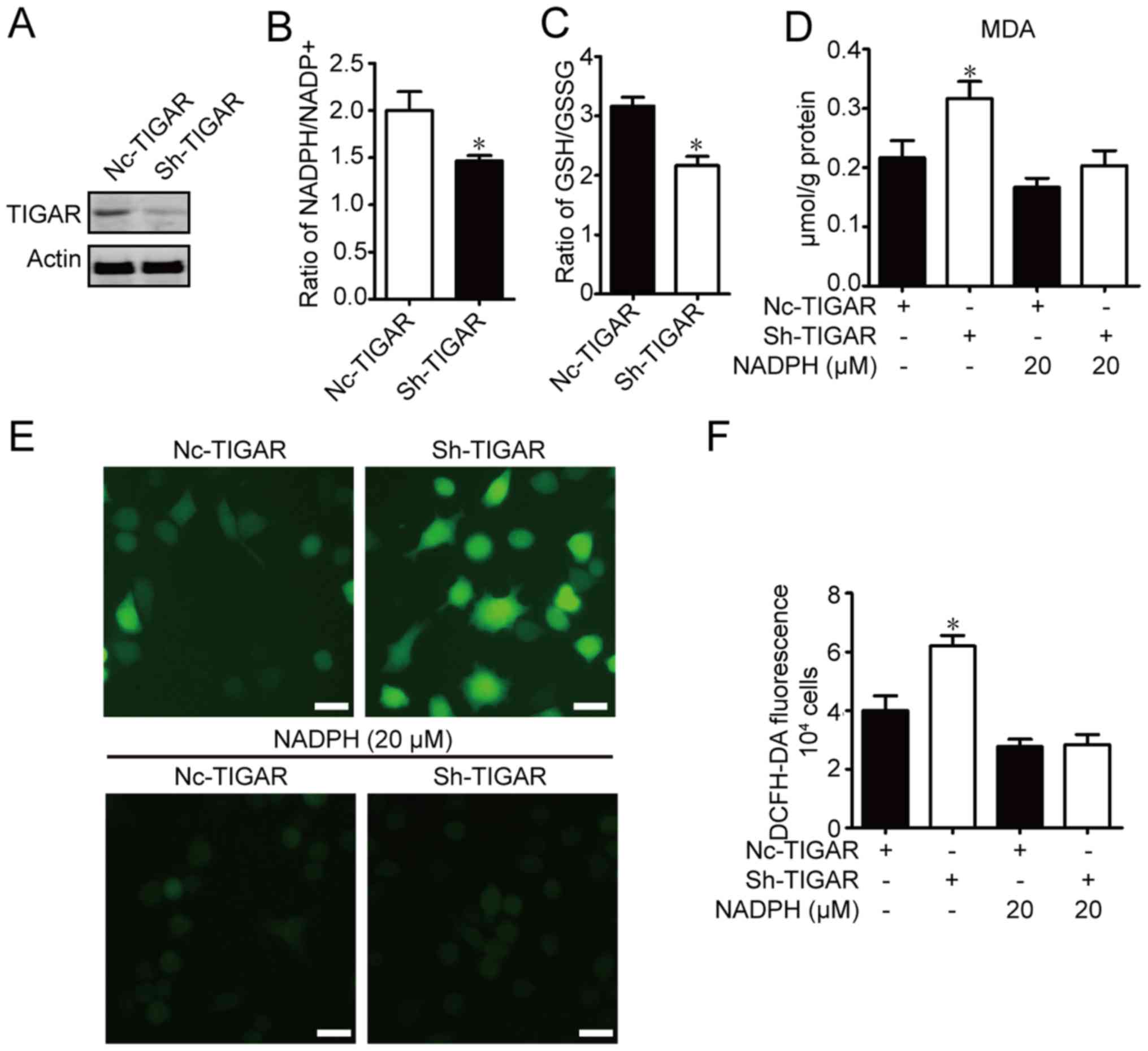 | Figure 2.TIGAR attenuates oxidative stress
through NADPH in U-87MG glioma cells. U-87MG cells were infected
with NC-TIGAR or sh-TIGAR, and were cultured for >2 weeks in the
presence of puromycin (2 µg/ml) prior to further experiments. (A)
TIGAR expression was measured using western blot assay. Ratios of
(B) NADPH/NADP+ and (C) GSH/GSSG were measured in
U-87MG/NC cells and U-87MG/sh-TIGAR cells. (D) MDA contents were
measured in U-87MG/NC, U-87MG/sh-TIGAR, NADPH-pretreated U-87MG/NC
and U-87MG/sh-TIGAR cells. (E) Intracellular ROS levels in
U-87MG/NC, U-87MG/sh-TIGAR, NADPH-pretreated U-87MG/NC and
U-87MG/sh-TIGAR cells were monitored using DCFH-DA under a
fluorescence microscope. Scale bar, 20 µm. (F) Intracellular ROS
levels in U-87MG/NC, U-87MG/sh-TIGAR, NADPH-pretreated U-87MG/NC
and U-87MG/sh-TIGAR cells were determined using DCFH-DA on a
microplate reader. The histogram shows the mean ROS production.
Data of the triple experiments are presented as the means ±
standard deviation. *P<0.05 vs. NC-TIGAR group according
to an unpaired t-test/one-way analysis of variance. GSH,
glutathione; GSSG, oxidized glutathione; MDA, malondialdehyde;
NADP+, nicotinamide adenine dinucleotide phosphate;
NADPH, reduced NADP+; NC, negative control shRNA; ROS,
reactive oxygen species; sh, short hairpin RNA; TIGAR, TP53 induced
glycolysis regulatory phosphatase. |
TIGAR promotes proliferation and
inhibits apoptosis in U-87MG cells
To test the effect of TIGAR in glioblastoma, U-87MG
cells were transfected with pcDNA3.1-TIGAR, and viability rates
were measured. As shown in Fig.
3A-B, TIGAR content was overexpressed after transfection, and
TIGAR overexpression significantly increased U-87MG cell viability,
and the numbers of cell clones in TIGAR-overexpressing U-87MG cells
were higher than in control U-87MG cells (Fig. 3C). The viability of TIGAR-knockdown
U-87MG cells was measured, and cell viability was decreased in
TIGAR-knockdown U-87MG cells. To ascertain whether the decreased
viability of TIGAR knockdown cells was associated with oxidative
stress, exogenous NADPH was added. The results revealed that added
NADPH did not increase cell viability in TIGAR-knockdown U-87MG
cells (Fig. 3D). Additionally, the
formation of cell clones was inhibited in TIGAR-knockdown U-87MG
cells, and additional NADPH did not promote cell growth (Fig. 3E). TMZ-based therapy is the standard
of care for patients with glioblastoma, and resistance to TMZ in
glioblastoma is a universal phenomenon (15). TIGAR knockdown promoted the
TMZ-induced decrease in Bcl2 and increase in BAX protein expression
levels, which was indicative of an increase in apoptosis.
Furthermore, NADPH significantly alleviated apoptosis in the
TMZ-treated U-87MG cells and TIGAR-knockdown U-87MG cells (Fig. 3F). The results indicated that TIGAR
promoted viability, and decreased TMZ-induced apoptosis through
NADPH-mediated antioxidative activity in U-87MG cells.
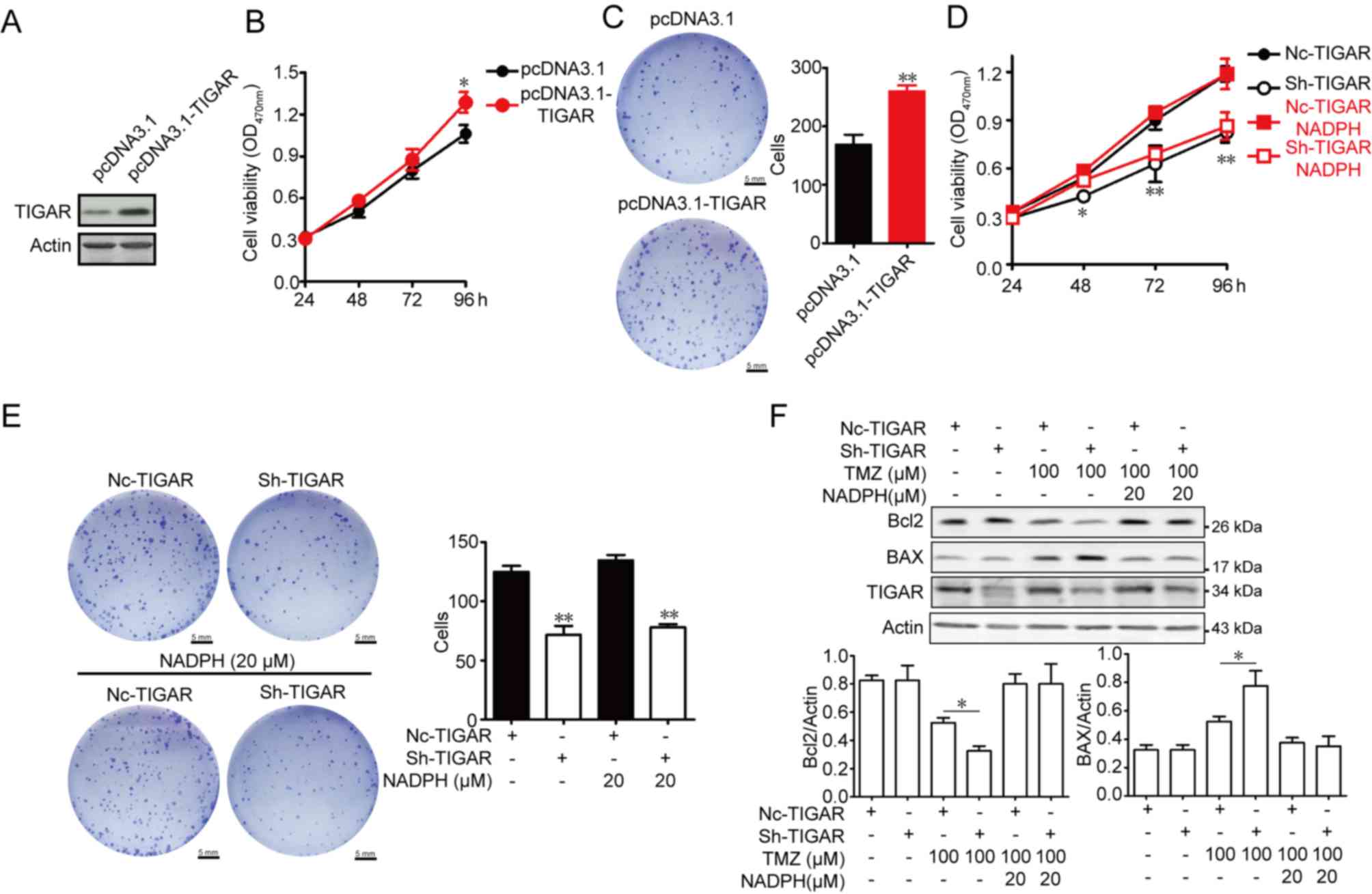 | Figure 3.Effect of TIGAR on viability and
apoptosis in U-87MG glioma cells. (A) TIGAR content was increased
after transfection. (B) Overexpression of TIGAR promoted viability
in U-87MG cells, as measured by am MTT assay (n=5). (C) Cells (200)
transfected with pcDNA3.1 and pcDNA3.1-TIGAR were seeded into
6-well plates and cultured for 14 days. Colony formation was
detected using crystal violet staining. The colony number was
counted (n=3). Scale bar, 5 mm. (D) NADPH treatment of
TIGAR-knockdown U-87MG cells had no effect on viability (n=5). (E)
NADPH treatment of TIGAR-knockdown U-87MG cells had no effect on
colony formation (n=3). Scale bar, 5 mm. (F) NADPH (20 µM) addition
decreased the expression levels of BAX protein and increased Bcl2
protein expression in 100 µM TMZ-treated TIGAR knockdown U-87MG
cells. β-actin was used as an internal control. Bar graphs depict
semi-quantitative analysis of Bcl2 and BAX expression (n=3). Data
are expressed as the means ± standard deviation. *P<0.05,
**P<0.01 vs. pcDNA3.1 or NC-TIGAR. BAX, Bcl2-associated X
protein; Bcl2, B cell lymphoma 2; NADPH, reduced nicotinamide
adenine dinucleotide phosphate; NC, negative control shRNA; OD,
optical density; sh, short hairpin RNA; TIGAR, TP53 induced
glycolysis regulatory phosphatase; TMZ, temozolomide. |
TIGAR promotes metastasis in U-87MG
cells
Glioblastoma is an aggressive intracranial tumor
(16). The number of migratory cells
in the Transwell assay was significantly lower in TIGAR-knockdown
U-87MG cells, and addition of NADPH did not promote cell migration
(Fig. 4A). Additionally, the number
of invasive cells was significantly reduced in TIGAR-knockdown
U-87MG cells; addition of NADPH failed to promote cell invasion in
U-87MG cells (Fig. 4B). The
migration and invasion of cancer cells are associated with the
epithelial-mesenchymal transition (EMT) (17). Therefore, expression levels of EMT
indicators, including N-cadherin, snail, E-cadherin, α-SMA and
vimentin, were assessed. As shown in Fig. 4C, significantly increased E-cadherin,
and decreased N-cadherin, α-SMA, snail and vimentin expression
levels were observed in TIGAR-knockdown cells, indicating that
metastasis was decreased in TIGAR-knockdown U-87MG cells. Notably,
the addition of NADPH had no effect on the metastasis of
TIGAR-knockdown U-87MG cells, which highlighted the pro-metastatic
effect of TIGAR beyond NADPH production in glioblastoma.
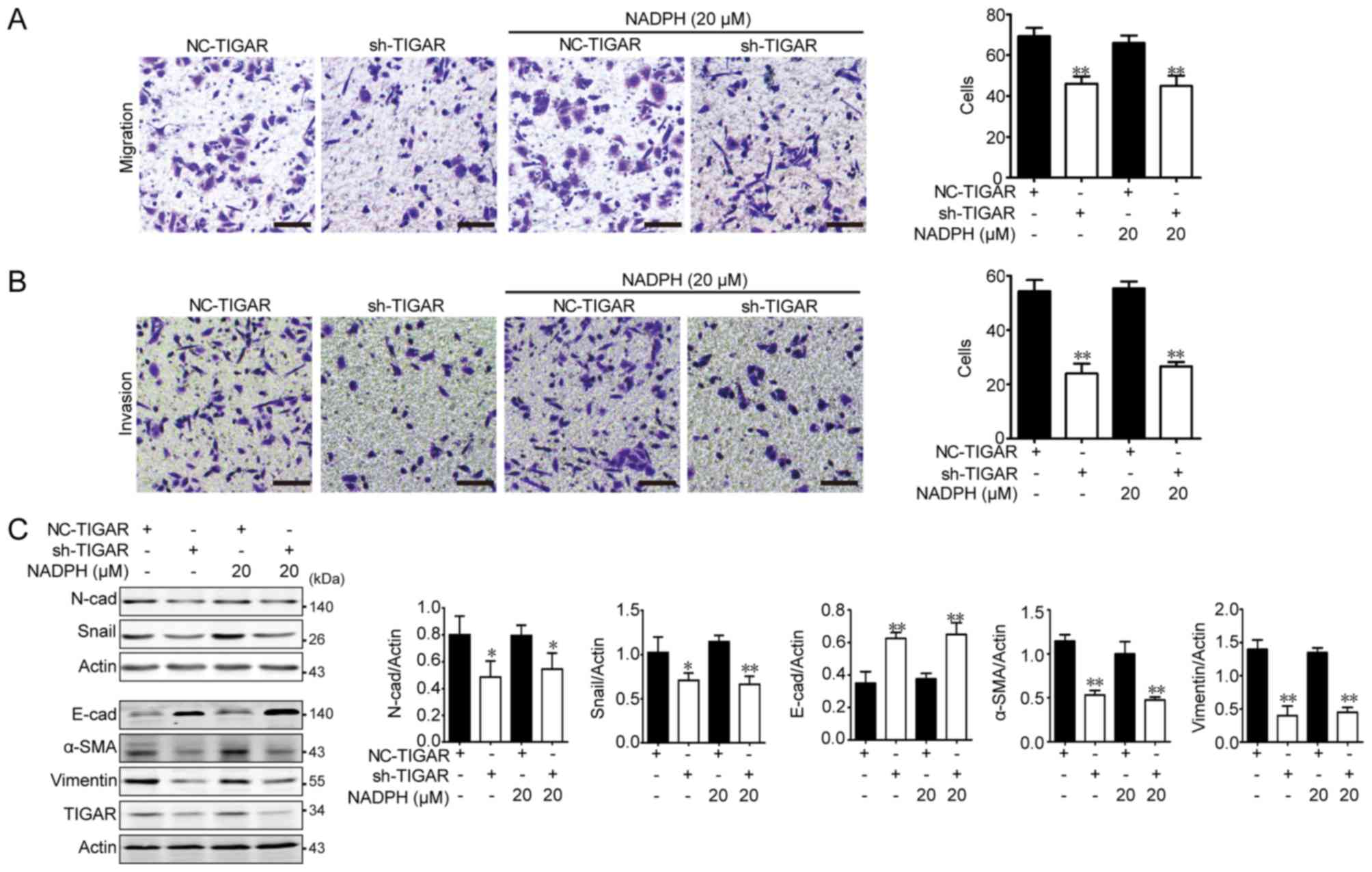 | Figure 4.Effect of TIGAR on migration,
invasion and epithelial-mesenchymal transition in U-87MG glioma
cells. U-87MG/NC and U-87MG/sh-TIGAR cells were stimulated with
NADPH for 24 h, and 5×104 cells in serum-free medium
were seeded into the upper chamber of Transwell for 12 h. (A)
Migration and (B) invasion were decreased in TIGAR-knockdown cells,
and NADPH addition had no effect on migration and invasion in
TIGAR-knockdown U-87MG cells (n=3). Scale bar, 50 µm. (C) Knockdown
of TIGAR increased E-cad, and decreased N-cad, α-SMA, Snail and
vimentin protein expression. Added NADPH had no effect on the
expression of these proteins. Bar graphs depict semi-quantitative
analysis of western blotting for each protein (n=3). *P<0.05,
**P<0.01 vs. NC-TIGAR group. Data are presented as the means ±
standard deviation. α-SMA, α-smooth muscle actin; E-cad, epithelial
cadherin; NADPH, reduced nicotinamide adenine dinucleotide
phosphate; N-cad, neural cadherin; NC, negative control; sh, short
hairpin RNA; TIGAR, TP53 induced glycolysis regulatory
phosphatase. |
TIGAR promotes AKT phosphorylation and
interacts with AKT in U-87MG cells
The PI3K/AKT signaling pathway is activated in
glioblastoma (18). The activation
of AKT promotes cancer growth and metastasis and inhibits autophagy
and apoptosis (19). Therefore,
PI3K/AKT activation was assessed in TIGAR-knockdown and
TIGAR-overexpressing U-87MG cells. As shown in Fig. 5A, no significant differences between
total PI3K and p-PI3K levels were identified in TIGAR-knockdown and
TIGAR-overexpressing U-87MG cells compared with their respective
controls, whereas p-AKT was significantly decreased in
TIGAR-knockdown cells and increased in overexpressing cells. To
determine whether TIGAR promoted AKT activation in U-87MG cells,
TIGAR was transfected into U-87MG cells in a dose-dependent manner.
The present study demonstrated that p-AKT/AKT markedly increased as
TIGAR expression increased (Fig.
5B). Additionally, Co-IP was performed to verify the
interaction between TIGAR and AKT. As shown in Fig. 5C, TIGAR interacted with AKT in U-87MG
cells. These results indicated that TIGAR promoted AKT activation
and interacted with AKT in glioblastoma.
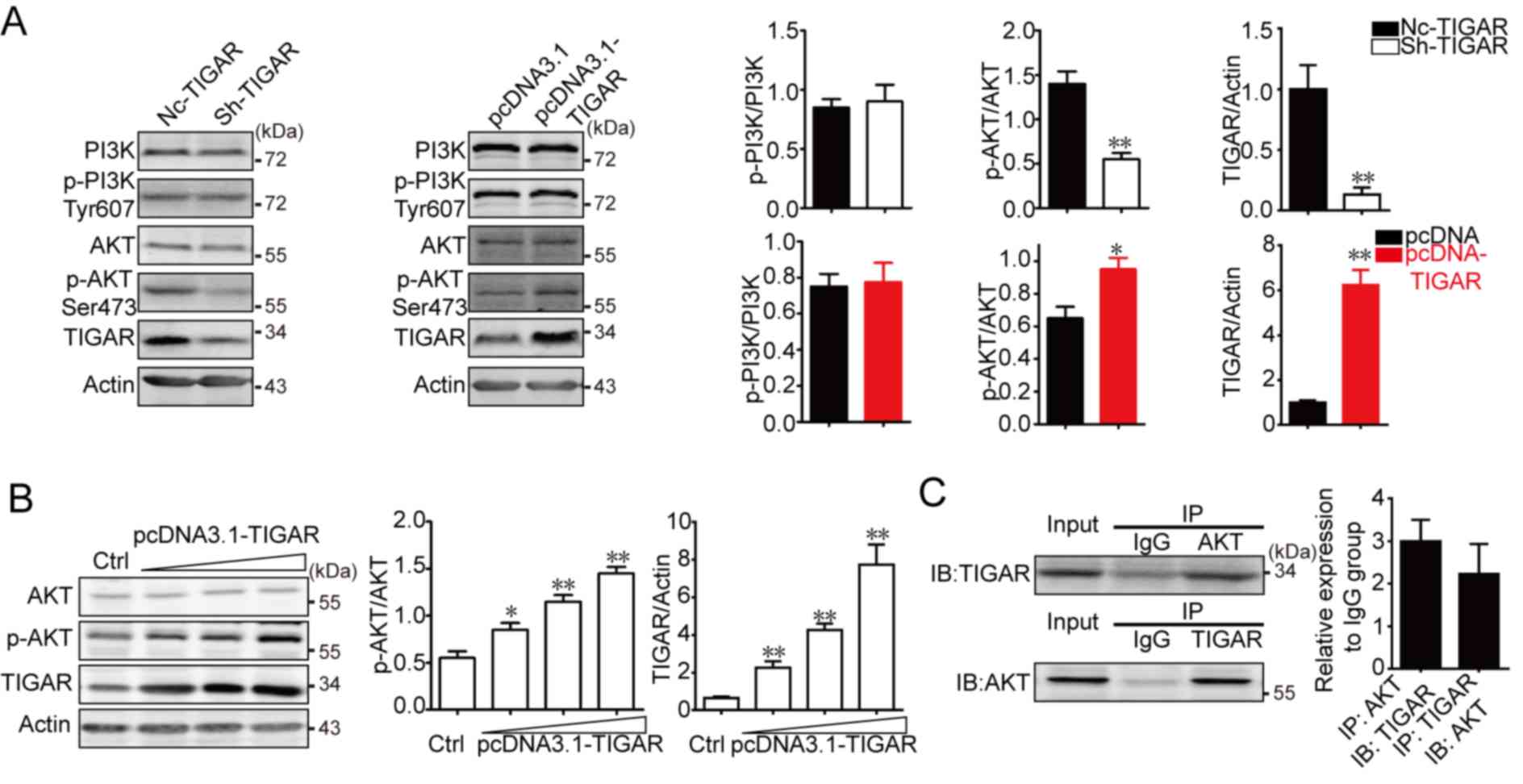 | Figure 5.TIGAR promotes AKT activation and
interacts with AKT in U-87 glioma cells. (A) U-87MG cells were
infected with sh-TIGAR to downregulate TIGAR expression or
transfected with pcDNA3.1-TIGAR to increase TIGAR expression. PI3K,
p-PI3K, AKT and p-AKT were measured in U-87MG/NC, U-87MG/sh-TIGAR,
U-87MG/pcDNA3.1 and U-87MG/pcDNA3.1-TIGAR cells. TIGAR promoted the
phosphorylation of AKT. β-actin was used as an internal control.
Bar graphs depict semi-quantitative analysis of protein expression
levels. (B) U-87MG cells cultured in 6-well plates were transfected
with 1, 2 or 3 µg pcDNA3.1-TIGAR. TIGAR promoted the
phosphorylation of AKT in a dose dependent manner, which was
assessed using western blotting. Bar graphs depict
semi-quantitative analysis for p-AKT and TIGAR. (C) Physical
interactions between TIGAR and AKT in U-87MG cells were confirmed
using a co-immunoprecipitation assay. Bar graph depicts
semi-quantitative analysis for TIGAR and AKT when compared with the
IgG group. Data of the triple experiments are presented as the
means ± standard deviation. *P<0.05, **P<0.01 vs.
NC-TIGAR or pcDNA3.1. AKT, protein kinase B; Ctrl, control; IgG,
immunoglobulin G; NC, negative control; p, phosphorylated; PI3K,
phosphoinositide 3-kinase; sh, short hairpin RNA; TIGAR, TP53
induced glycolysis regulatory phosphatase. |
Discussion
Glioma is a type of tumor that occurs in the brain,
and 75% of gliomas are astrocytomas (20). Glioblastoma represents the highest
grade of astrocytoma, and the overall 5-year survival rate is only
5% (21). Furthermore, glioblastoma
is associated with migration, invasion and chemotherapy resistance
(22).
TMZ, a second-generation oral alkylating agent that
causes DNA damage via methylation of the O(6) position of guanine,
is commonly used to treat human malignant glioma.
O(6)-methylguanine-DNA methyltransferase mitigates the
effectiveness of TMZ and has been used as a marker to predict the
efficacy of TMZ treatment (23). In
addition, DNA damage response activates p53. However, the high
incidence of TP53 mutations leads to p53 loss the function to
prevent tumor formation in cancers (24). As a well-known tumor suppressor, the
prevailing function of p53 is the transcriptional control of target
genes that regulate numerous cellular processes, including the cell
cycle, apoptosis, autophagy and metabolism (25). TMZ treatment induces p53 activation
and subsequent upregulation of p21, Noxa and BAX (26).
As a downstream target protein of p53, TIGAR
expression is dependent on p53, whereas p73, p63, hypoxia inducible
factor 1 and Sp1 transcription factor also promote TIGAR expression
(27,28). A number of studies have demonstrated
that chemotherapy is accompanied by increased TIGAR expression, and
TIGAR-derived NADPH protects cancer cells from chemotherapy-induced
damage (29,30). Additionally, nuclear localized TIGAR
exhibits antioxidant properties, and provides ribose-5-phosphate
for DNA repair following epirubicin treatment (31). TIGAR is localized in the cytoplasm,
endoplasmic reticulum, and the mitochondrial membrane and matrix.
In addition to nuclear translocation, TIGAR may translocate to
mitochondria under hypoxia in cancer (28). Following cerebral ischemia
reperfusion in mice, increased levels of TIGAR are observed in the
mitochondrial membrane and matrix, where TIGAR maintains the
mitochondrial membrane potential under oxidative stress conditions
(32). Therefore, effects of TIGAR
beyond oxidation resistance, and the cellular distribution of
TIGAR, require further investigation.
The PI3K/AKT signaling pathway serves an important
role in the occurrence and development of tumors. This pathway can
be activated by various factors, including platelet-derived growth
factor, epidermal growth factor and insulin-like growth factor, to
promote cell proliferation, differentiation, adhesion and
migration, and inhibit apoptosis. Tumor biology studies have mainly
concentrated on Class IA PI3K, which is composed of a heterodimer
comprising a p110 catalytic subunit and a p85 regulatory subunit
(33,34). PI3K binds to the upstream tyrosine
receptor kinase via the SH2 region of p85, causing activation of
PI3K. Additionally, PI3K is activated by G protein-coupled receptor
(GPCR)-activated Ras upon stimulation with the GPCR ligand cyclic
adenosine monophosphate (35). Once
PI3K is activated, its activated substrates phosphatidylinositol
(PI) 4,5-bisphosphate (PI(4,5)P2) and PI (3,4,5)-trisphosphate (PIP3) act as
second messengers to activate and form a signaling cascade complex,
leading to the phosphorylation of AKT.
PI3K phosphorylates AKT at Thr308 and Ser473 to
activate AKT. PI3K catalyzes the phosphorylation of PI 4-phosphate
and PI(4,5)P2 at their third positions, and converts
them into PI(3,4)P2 and PIP3 to recruit AKT
(36). AKT can be directly activated
by PI(3,4)P2. However, PIP3 activates
phosphoinositide-dependent kinase (PDK)-1 to phosphorylate AKT at
Thr308, and AKT is further phosphorylated at Ser473 and fully
activated in the presence of PDK-2. The tumor suppressor PTEN
reverses the transformation of PI(4,5)P2 to
PIP3 in the PI3K/AKT pathway, and maintains a low level
of PIP3, thereby inhibiting the phosphorylation of AKT.
Additionally, it has been confirmed that mammalian target of
rapamycin complex 2 (mTORC2) phosphorylates AKT at Ser473, and that
this modification requires PIP3 (37).
Activated AKT is involved in the occurrence and
development of glioblastoma; the PI3K-AKT-mTOR pathway is
frequently activated in glioblastoma to regulate cancer survival
(38). Additionally, AKT contributes
to glioblastoma formation through the recruitment of existing mRNAs
to polysomes (39). The direct and
indirect inhibition of AKT activity promotes apoptosis and
suppresses glioblastoma growth (40,41).
Due to the small sample size, the association
between TIGAR expression and survival time could not be calculated
for the patients included in the present study; therefore, the
association was analyzed using PrognoScan. As TIGAR is
overexpressed in glioblastoma, the role of TIGAR in oxidative
stress was investigated. NADPH levels decreased and ROS indicators
increased in the TIGAR-knockdown U-87MG glioblastoma cell line.
Furthermore, cell viability, TMZ-induced apoptosis, migration,
invasion and EMT were investigated in TIGAR-knockdown U-87MG cells.
The results demonstrated that TIGAR-mediated antioxidative effects
inhibited apoptosis but did not affect viability, migration,
invasion or EMT. In addition, TIGAR promoted AKT phosphorylation in
a dose-dependent manner and interacted with AKT in U-87MG cells. It
has been reported that PFK/FB4, an enzyme similar to TIGAR,
phosphorylates nuclear receptor coactivator 3 at Ser857 to enhance
its transcriptional activity and promote breast cancer growth and
metastasis (42). The manner in
which TIGAR, as a phosphatase, promotes AKT phosphorylation
requires further investigation.
In conclusion, the results of the present study
demonstrated that TIGAR inhibited apoptosis and promoted
proliferation, migration and invasion in glioblastoma through
NADPH-mediated antioxidative effects and activation of AKT.
Therefore, TIGAR may be considered as a potential therapeutic
target in glioblastoma.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZT and ZH performed the experiments. ZT analyzed the
data. ZH designed the experiments and wrote the manuscript.
Ethics approval and consent to
participate
The use of glioblastoma sections was approved by the
Ethics Committee of Hunan Cancer Hospital and Affiliated Cancer
Hospital of Xiangya School of Medicine (no. 20180104; Changsha,
China). All patients provided informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Taillibert S, Kanner A, Read W,
Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K,
et al: Effect of tumor-treating fields plus maintenance
temozolomide vs maintenance temozolomide alone on survival in
patients with glioblastoma: A randomized clinical trial. JAMA.
318:2306–2316. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bensaad K, Tsuruta A, Selak MA, Vidal MN,
Nakano K, Bartrons R, Gottlieb E and Vousden KH: TIGAR, a
p53-inducible regulator of glycolysis and apoptosis. Cell.
126:107–120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Green DR and Chipuk JE: P53 and
metabolism: Inside the TIGAR. Cell. 126:30–32. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Agnoletto C, Melloni E, Casciano F,
Rigolin GM, Rimondi E, Celeghini C, Brunelli L, Cuneo A, Secchiero
P and Zauli G: Sodium dichloroacetate exhibits anti-leukemic
activity in B-chronic lymphocytic leukemia (B-CLL) and synergizes
with the p53 activator Nutlin-3. Oncotarget. 5:4347–4360. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou X, Xie W, Li Q, Zhang Y, Zhang J,
Zhao X, Liu J and Huang G: TIGAR is correlated with maximal
standardized uptake value on FDG-PET and survival in non-small cell
lung cancer. PLoS One. 8:e805762013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ko YH, Domingo-Vidal M, Roche M, Lin Z,
Whitaker-Menezes D, Seifert E, Capparelli C, Tuluc M, Birbe RC,
Tassone P, et al: TIGAR metabolically reprograms carcinoma and
stromal cells in breast cancer. J Biol Chem. 116:7402092016.
|
|
8
|
Zou S, Gu Z, Ni P, Liu X, Wang J and Fan
Q: SP1 plays a pivotal role for basal activity of TIGAR promoter in
liver cancer cell lines. Mol Cell Biochem. 359:17–23. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheung EC, Athineos D, Lee P, Ridgway RA,
Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ and Vousden KH:
TIGAR is required for efficient intestinal regeneration and
tumorigenesis. Dev Cell. 25:463–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee P, Vousden KH and Cheung EC: TIGAR,
TIGAR, burning bright. Cancer Metab. 2:12014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mizuno H, Kitada K, Nakai K and Sarai A:
PrognoScan: A new database for meta-analysis of the prognostic
value of genes. BMC Med Genomics. 2:182009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Griffith OW: Determination of glutathione
and glutathione disulfide using glutathione reductase and
2-vinylpyridine. Anal Biochem. 106:207–212. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nielsen F, Mikkelsen BB, Nielsen JB,
Andersen HR and Grandjean P: Plasma malondialdehyde as biomarker
for oxidative stress: Reference interval and effects of life-style
factors. Clin Chem. 43:1209–1214. 1997.PubMed/NCBI
|
|
14
|
Xie JM, Li B, Yu HP, Gao QG, Li W, Wu HR
and Qin ZH: TIGAR has a dual role in cancer cell survival through
regulating apoptosis and autophagy. Cancer Res. 74:5127–5138. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee SY: Temozolomide resistance in
glioblastoma multiforme. Genes Dis. 3:198–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zeng WF, Navaratne K, Prayson RA and Weil
RJ: Aurora B expression correlates with aggressive behaviour in
glioblastoma multiforme. J Clin Pathol. 60:218–221. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heerboth S, Housman G, Leary M, Longacre
M, Byler S, Lapinska K, Willbanks A and Sarkar S: EMT and tumor
metastasis. Clin Transl Med. 4:62015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Wu C, Chen N, Gu H, Yen A, Cao L,
Wang E and Wang L: PI3K/Akt/mTOR signaling pathway and targeted
therapy for glioblastoma. Oncotarget. 7:33440–33450.
2016.PubMed/NCBI
|
|
19
|
Agarwal E, Brattain MG and Chowdhury S:
Cell survival and metastasis regulation by Akt signaling in
colorectal cancer. Cell Signal. 25:1711–1719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gladson CL, Prayson RA and Liu WM: The
pathobiology of glioma tumors. Annu Rev Pathol. 5:33–50. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Delgado-Lopez PD and Corrales-Garcia EM:
Survival in glioblastoma: A review on the impact of treatment
modalities. Clin Transl Oncol. 18:1062–1071. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie Q, Mittal S and Berens ME: Targeting
adaptive glioblastoma: An overview of proliferation and invasion.
Neuro Oncol. 16:1575–1584. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Zhang M, Gan H, Wang H, Lee JH,
Fang D, Kitange GJ, He L, Hu Z, Parney IF, et al: A novel enhancer
regulates MGMT expression and promotes temozolomide resistance in
glioblastoma. Nat Commun. 9:29492018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Blagosklonny MV: Loss of function and p53
protein stabilization. Oncogene. 15:1889–1893. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fischer M: Census and evaluation of p53
target genes. Oncogene. 36:3943–3956. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang WB, Wang Z, Shu F, Jin YH, Liu HY,
Wang QJ and Yang Y: Activation of AMP-activated protein kinase by
temozolomide contributes to apoptosis in glioblastoma cells via p53
activation and mTORC1 inhibition. J Biol Chem. 285:40461–40471.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee P, Hock A, Vousden K and Cheung E:
P53-and p73-independent activation of TIGAR expression in
vivo. Cell Death Dis. 6:e18422015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cheung EC, Ludwig RL and Vousden KH:
Mitochondrial localization of TIGAR under hypoxia stimulates HK2
and lowers ROS and cell death. Proc Natl Acad Sci USA.
109:20491–20496. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Y, Chen F, Tai G, Wang J, Shang J,
Zhang B, Wang P, Huang B, Du J, Yu J, et al: TIGAR knockdown
radiosensitizes TrxR1-overexpressing glioma in vitro and
in vivo via inhibiting Trx1 nuclear transport. Sci Rep.
7:429282017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang H, Gu C, Yu J, Wang Z, Yuan X, Yang
L, Wang J, Jia Y, Liu J and Liu F: Radiosensitization of glioma
cells by TP53-induced glycolysis and apoptosis regulator knockdown
is dependent on thioredoxin-1 nuclear translocation. Free Radic
Biol Med. 69:239–248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu HP, Xie JM, Li B, Sun YH, Gao QG, Ding
ZH, Wu HR and Qin ZH: TIGAR regulates DNA damage and repair through
pentosephosphate pathway and Cdk5-ATM pathway. Sci Rep. 5:98532015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li M, Sun M, Cao L, Gu JH, Ge J, Chen J,
Han R, Qin YY, Zhou ZP, Ding Y and Qin ZH: A TIGAR-regulated
metabolic pathway is critical for protection of brain ischemia. J
Neurosci. 34:7458–7471. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oda K, Stokoe D, Taketani Y and McCormick
F: High frequency of coexistent mutations of PIK3CA and PTEN genes
in endometrial carcinoma. Cancer Res. 65:10669–10673. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: Rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vanhaesebroeck B, Guillermet-Guibert J,
Graupera M and Bilanges B: The emerging mechanisms of
isoform-specific PI3K signalling. Nat Rev Mol Cell Biol.
11:329–341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao L and Vogt PK: Class I PI3K in
oncogenic cellular transformation. Oncogene. 27:5486–5496. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ebner M, Sinkovics B, Szczygiel M, Ribeiro
DW and Yudushkin I: Localization of mTORC2 activity inside cells. J
Cell Biol. 216:343–353. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fan QW and Weiss WA: Autophagy and Akt
promote survival in glioma. Autophagy. 7:536–538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rajasekhar VK, Viale A, Socci ND, Wiedmann
M, Hu X and Holland EC: Oncogenic Ras and Akt signaling contribute
to glioblastoma formation by differential recruitment of existing
mRNAs to polysomes. Mol Cell. 12:889–901. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gao T, Furnari F and Newton AC: PHLPP: A
phosphatase that directly dephosphorylates Akt, promotes apoptosis,
and suppresses tumor growth. Mol Cell. 18:13–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Assad Kahn S, Costa SL, Gholamin S, Nitta
RT, Dubois LG, Fève M, Zeniou M, Coelho PL, El-Habr E, Cadusseau J,
et al: The anti-hypertensive drug prazosin inhibits glioblastoma
growth via the PKCdelta-dependent inhibition of the AKT pathway.
EMBO Mol Med. 8:511–526. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dasgupta S, Rajapakshe K, Zhu B, Nikolai
BC, Yi P, Putluri N, Choi JM, Jung SY, Coarfa C, Westbrook TF, et
al: Metabolic enzyme PFKFB4 activates transcriptional coactivator
SRC-3 to drive breast cancer. Nature. 556:249–254. 2018. View Article : Google Scholar : PubMed/NCBI
|