Introduction
Anthracyclines are a group of highly effective
chemotherapeutic agents used in the treatment and management of
various cancer types (1). The
anthracycline doxorubicin (DOX) is a potent and broad-spectrum
antineoplastic agent used to treat patients with leukemia,
lymphoma, sarcoma or breast cancer (2). However, cardiovascular complications
are well-documented side-effects of anthracyclines. DOX can impair
the pumping function of the left ventricle and lead to dilated
cardiomyopathy and congestive heart failure (HF), resulting in the
termination of the treatment and worsening the prognosis of the
patient (2,3). Furthermore, the risk of developing
subsequent cardiotoxicity increases with the dose of DOX received.
A previous study demonstrated that patients who received a
cumulative dose of 400 mg/m2 DOX exhibted a 5% increase
in the risk of HF. This risk was further increased by 26 and 48%
when doses of 550 and 700 mg/m2 DOX were administered,
respectively (4). From a clinical
pathology point of view, DOX-treated patients can exhibit extensive
cardiac remodeling, including vast cytoplasmic vacuolization,
sarcoplasmic reticulum swelling and myofibrillar disarray (5). To the best of our knowledge, currently
no drug has been confirmed to effectively prevent DOX-induced
cardiotoxicity without diminishing its antitumor activity, which is
predominantly due to a lack of understanding regarding the
mechanisms underlying DOX-induced pathology.
The broad mechanisms underlying DOX-induced
cardiotoxicity are the following: i) oxidative stress, ii) DNA
damage and its subsequent effects, and iii) autophagy dysfunction
or impaired autophagic flux (1).
Autophagy, which has gradually been recognized as a physiological
process essential for maintaining cellular homeostasis, is not
limited to a single signaling pathway (1,4). It is a
dynamic, multistep biological process that can be regulated by
numerous factors at a number of stages and overlaps with other
cellular processes (6). The
regulatory pathways of autophagy remain to be fully elucidated and
a number of studies have reported conflicting evidence regarding
the role of DOX in the regulation of autophagy in cardiac cells
(7,8). Therefore, it is essential to
investigate these regulatory pathways, as well as other mechanisms
associated with autophagy, to improve understanding of the role of
autophagy in DOX-induced cardiotoxicity and assist with the
development of drugs to prevent this pathological condition.
Interactions between autophagy and
apoptosis
Programmed cell death (PCD) refers to a process in
which cells follow a defined intracellular program and commit
suicide in response to physiological or pathological conditions
(9). Based on the death mechanisms,
PCD can be divided into caspase-independent and caspase-dependent
cell death, the former including autophagic programmed cell death
(9). The present section focuses on
autophagy, which is the death of numerous organelles, apoptosis,
which is the death of the whole cell, and their association.
Autophagy is a catabolic process involving the
degradation of cellular components (1,10).
Autophagy is controlled by autophagy-related genes (ATGs) and
lysosomal proteolytic mechanisms, which are activated in response
to different types of metabolic stress, including hypoxia, reactive
oxygen species (ROS) accumulation and nutritional or energetic
deficiency (11,12). The five core steps of autophagy
include initiation, nucleation, expansion, maturation, and
degradation (13). Each of these
steps have corresponding regulators, including serine-threonine
kinase Unc-51-line kinase-1 (ULK1), ATG13 and focal adhesion kinase
family interacting protein of 200 kD (FIP200) complexes, which are
important for the promotion of autophagy (Fig. 1). Adenosine monophosphate-activated
protein kinase (AMPK) and mammalian target of rapamycin (mTOR)
signaling pathways constitute the regulatory core of autophagy;
however, it is also regulated by a number of other signaling
pathways and mediators, including Beclin-1, B-cell lymphoma 1
(Bcl-2), phosphoinositide-3-kinase (PI3K) and p53 proteins
(14,15). Consistently, numerous studies have
demonstrated that the AMPK and mTOR signaling pathways promote and
inhibit autophagy, respectively (1,16). mTOR
is regulated by major upstream signals in the PI3K-Akt-mTOR
pathway. In more detail, intracellular or extracellular stress can
activate the PI3K receptor, which catalyzes the conversion of
phosphatidylinositol 4,5-biphosphate into phosphatidylinositol
3,4,5,-triphosphate (PIP3), which is a catalytic reaction that can
be reversed by phosphatase and tensin homolog. In co-operation with
phosphoinositide-dependent kinase-1, PIP3 then further activates
Akt. In turn, activated Akt increases the activity of Ras homolog
enriched in brain, which upregulates mTOR activity, and inhibits
autophagy initiation by promoting ULK1 phosphorylation (17,18)
(Fig. 2).
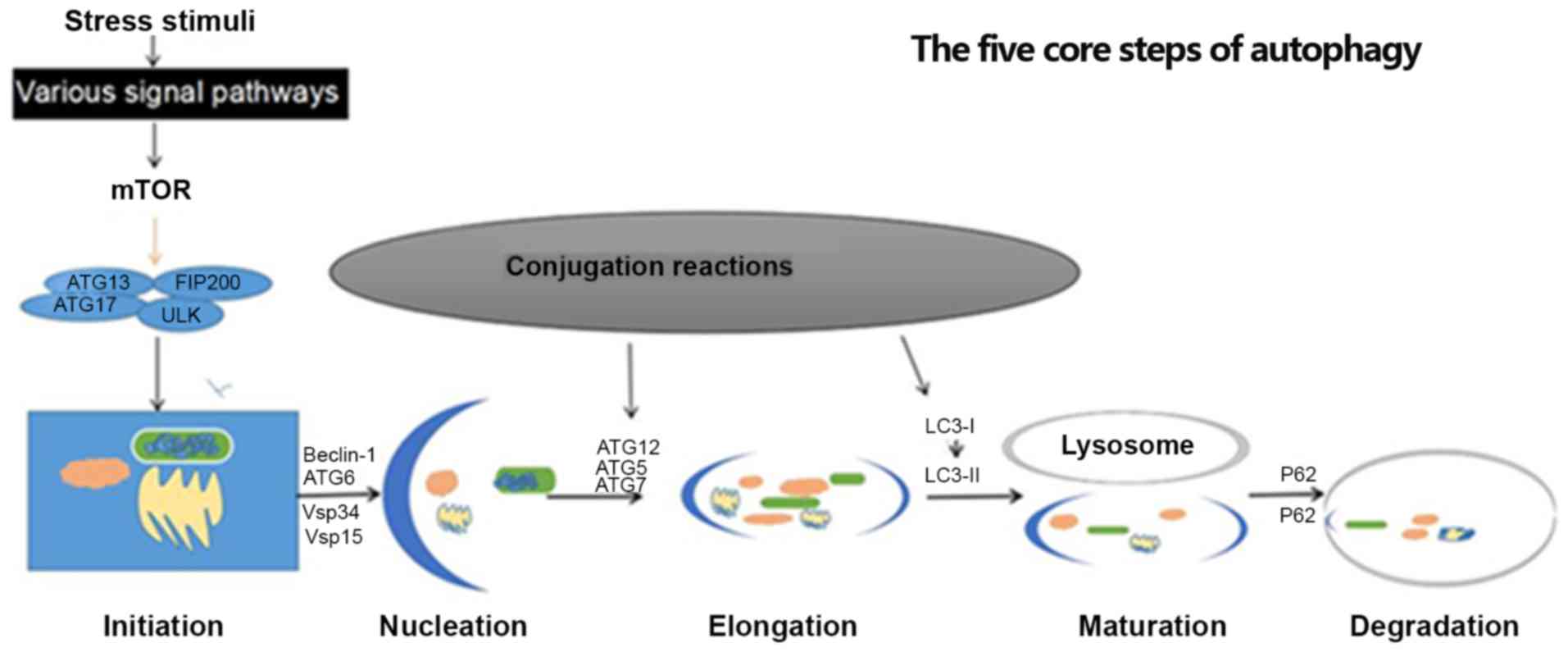 | Figure 1.Five core steps of autophagy:
Initiation, nucleation, elongation, maturation and degradation.
This process is induced by various stress stimuli, including
inhibition of mTOR. In initiation, the organelles to be degraded
are gradually wrapped. Subsequently, an autophagosome is formed and
cytosolic components are sequestered and characterized by a
LC3-II-positive double membrane structure. In the final step,
cytosolic components are degraded in an autolysosome. ATG,
autophagy related gene; LC3, light chain 3; mTOR, mammalian target
of rapamycin; ULK1, serine-threonine kinase Unc-51-line kinase-1;
Vsp, vacuolar protein sorting; FIP200, focal adhesion kinase family
interacting protein of 200 kD. |
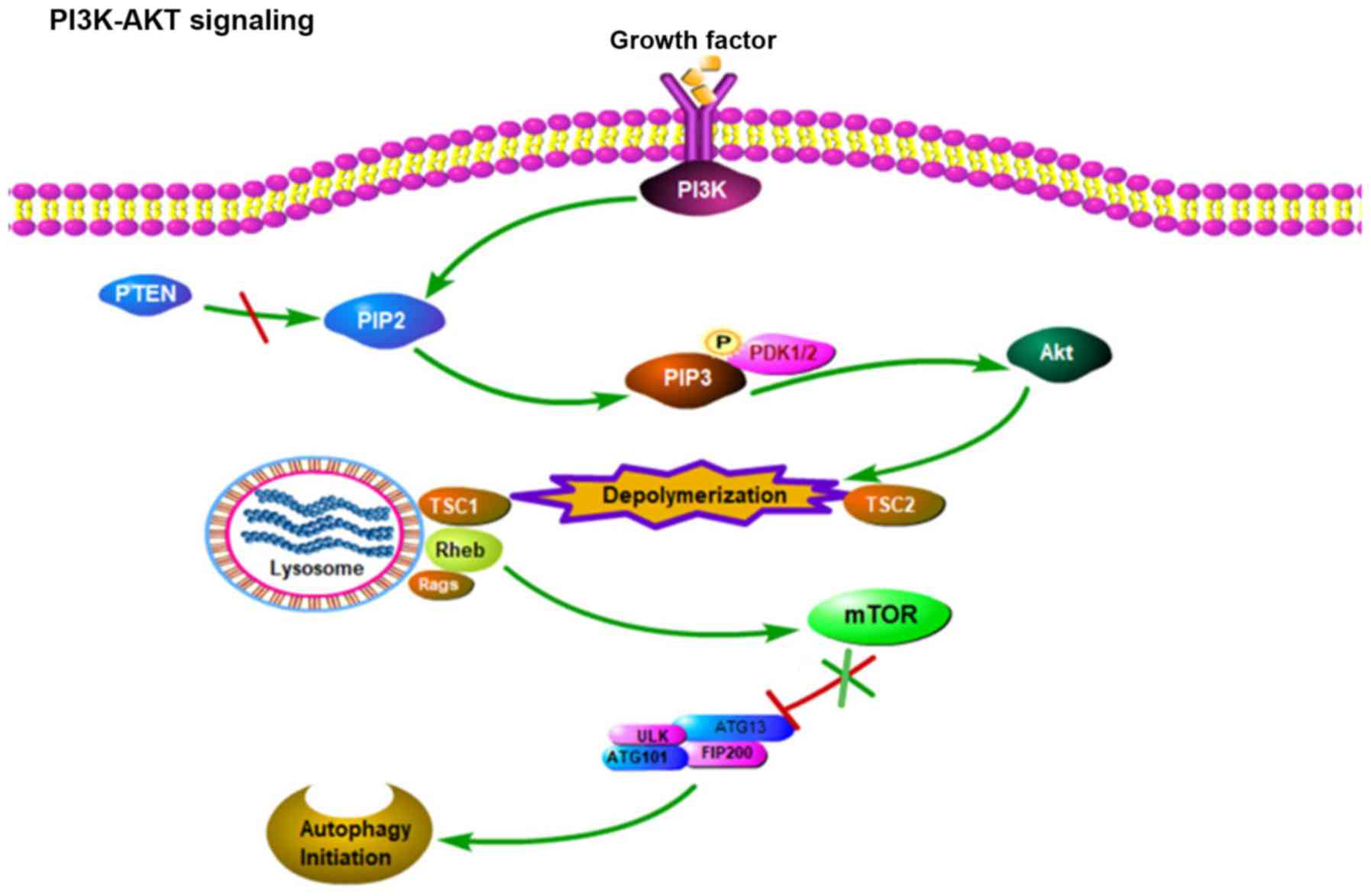 | Figure 2.PI3K-AKT signal path diagram:
Intracellular and extracellular factors activate the receptor of
PI3K, which in turn catalyzes the conversion of PIP2 to PIP3, a
catalytic reaction that is reversed by PTEN. PIP3 further activates
Akt with the cooperation of PDK1/2. Activated Akt increases the
activity of Rheb, which in turn upregulates mTOR activity, which
inhibits autophagy initiation by promoting ULK1 phosphorylation.
PI3K, phosphoinositide-3-kinase; PIP2, phosphatidylinositol
4,5-biphosphate; PIP3, phosphatidylinositol 3,4,5,-triphosphate;
PTEN, phosphatase and tensin homolog; PDK1,
phosphoinositide-dependent kinase 1 and 2; TSC, tuberous sclerosis
1; Rheb, Ras homolog enriched in brain; Rags,
recombination-activating genes; mTOR, mammalian target of
rapamycin; ATG, autophagy related gene; ULK, serine-threonine
kinase Unc-51-line kinase; FIP200, focal adhesion kinase family
interacting protein of 200 kD; P, phosphorylated. |
Conversely, a number of studies have demonstrated
that when the body lacks nutrients or energy, an increased
adenosine monophosphate/ATP ratio can activate AMPK, thereby
inhibiting mTOR, weakening the inhibition of ULK1 and finally
initiating autophagy (15,16). As a sensor in autophagy regulation,
mTOR receives information from different upstream signal molecules
and accordingly regulates downstream signal molecules, including
p70S6K, eukaryotic translation initiation factor 4E-binding protein
1, ULK1, lipin-1 and peroxisome proliferator-activated receptor γ
coactivator 1α, which produce their own biological effects
(19). The absolute increases and
decreases in mTOR activity are therefore determined by the ratio of
various factors or stimuli. In other words, autophagy regulation
may involve co-occurring activation and inhibition of mTOR,
depending on which of the respective regulatory factors
predominate. Autophagy regulation is indeed a complicated process.
While a basal level of autophagy is necessary to maintain cellular
homeostasis, autophagic cell death without nuclear pyknosis is
initiated when autophagy gets overactivated and exceeds a certain
threshold (10,20).
Apoptosis similarly serves a fundamental role in
maintaining cellular homeostasis and is also regulated by Bcl-2 and
p53 genes, which are closely associated with autophagy (19,21).
Autophagy and apoptosis respond to similar types of stress and
signals, including a high intracellular Ca2+
concentration, ROS accumulation, toxins, growth factors and
hormonal stimulation (22).
Furthermore, the same proteins can promote autophagy and apoptosis
mechanisms, in their respective processes. For example, under
cellular stress conditions, autophagy-related 5 (ATG5), a protein
necessary for the formation of the autophagy precursor, is cleaved
by a cysteine protease and serves a key role in the initiation of
apoptosis. Notably, under autophagy-inducing conditions, Beclin-1,
which normally binds to Bcl-2 and inhibits autophagy, is
competitively displaced and subsequently promotes the autophagic
process (21). Altogether, the
promotion and inhibition of autophagy and apoptosis contribute to
the difference between cell death and survival. In the case of an
increased proportion of apoptosis, caspases, the primary mediators
of apoptosis, can cleave numerous key autophagy proteins, for
example, caspase-3 and −8 can cleave PI3K and ATG3, respectively
(21). Strengthening this process
will inevitably lead to cell destruction. On the other hand,
resveratrol can induce autophagy via the kinase triad, AMPK, mTOR
and ULK1, resulting in a decreased proportion of apoptosis
(23–25).
Given that the regulatory pathways of autophagy are
complex and intertwined with those of apoptosis, it remains
challenging to elucidate the precise role of autophagy under drug
treatments conditions, including DOX.
Complex role of autophagy in
doxorubicin-induced cardiotoxicity
The autophagic process involves the formation of a
structure sealed by a lipid bilayer membrane called an
autophagosome (26). This
autophagosome can fuse with a lysosome to form an autolysosome,
which can degrade damaged and redundant organelles or invading
microorganisms (1,10). To date, to the best of our knowledge,
38 ATGs have been identified to regulate different stages of
autophagy in yeast. In mammals, part of this network is conserved,
but a number of additional genes are involved in the regulation of
autophagy (27).
The function of ATG products, particularly their
capacity to interact with other autophagy regulators at different
stages of the process, is predominantly achieved by proteolysis and
protein post-translational modifications, including
phosphorylation, glycosylation, ubiquitination, methylation,
acetylation and lipidation (27).
For example, in mammals, ATG13 and FIP200 are involved in the
formation of a complex required for the initiation of autophagy. By
recruiting the Beclin-1 and vacuolar protein sorting 34/15
proteins, ATG13 is involved in the formation and regulation of a
class III PI3K complex essential for the nucleation step (28). During the expansion step, ATG3, ATG4,
ATG7, ATG10 and ATG16 serve important roles in promoting the
formation of two essential ubiquitin-like (Ubl) conjugation
systems, ATG12-ATG5 and light chain 3
(LC3)-phosphatidylethanolamine (27,29).
These two systems contribute to the amplification of phagocytic
cells. In addition to the Ubl conjugation systems, the
ATG9-mediated cycling systems have been demonstrated to contribute
to the elongation of the phagophore and numerous other ATG products
also function in the late steps of autophagy (27). In the later steps of maturation and
degradation, the autophagosomes move to and fuse with lysosomes in
a kinesin-dependent manner (13).
Subsequently, the cargo is degraded into macromolecules and
released into the cytoplasm to be reused. A number of cytokines are
also involved in the regulation of this process (10,16).
However, the role of ATG genes in this context remains to be fully
investigated.
DOX treatment has adverse effects on multiple
tissues and organs of the body, and primarily affects the heart due
to induced DNA damage, swelling of the sarcoplasmic reticulum,
cytoplasmic vacuolization, intracellular calcium imbalance and
myofibrillar disarray, which are observed in microscopic
observations of the pathological structures (5). In addition to DOX itself, the
pathological changes also cause autophagy dysfunction via the
inhibition and activation of the AMPK and p38-mitogen-activated
protein kinase (MAPK) pathways, respectively (30). Notably, numerous studies (7,8,25,31) have
presented conflicting conclusions, which suggests that autophagy
may serve a dual role in DOX-induced cardiotoxicity.
Light chain 3 (LC3), a central protein in the
autophagy pathway, contributes to substrate selection and
autophagosome biogenesis. By inhibiting the Beclin-2 protein, DOX
can increase the protein expression level of LC3, which leads to
cardiomyocyte death (32).
Furthermore, in vitro assays have demonstrated that DOX can
upregulate the pro-autophagy protein Beclin-1 and induce
cardiomyocyte apoptosis (7). This
indicates that the upregulation of the level of autophagy serves an
adverse role in DOX-induced cardiotoxicity. Inconsistently, it has
also been demonstrated that strengthening the level of autophagy
via the AMPK/mTOR/ULK1 signaling pathway can improve the survival
of cardiomyocytes (25).
Furthermore, aspalathin can attenuate DOX-induced cardiotoxicity by
activating the AMPK pathway, which increases the level of
autophagy, while reducing the extent of p53/mTOR/p62 signaling
(8). In summary, these data suggest
an opposite conclusion as aforementioned, which is that
upregulating autophagy can attenuate DOX-induced cardiotoxicity.
Furthermore, DOX has also been demonstrated to inhibit autophagy by
activating the E2F transcription factor 1 (E2F1)/mTOR complex and
further induces apoptosis by activating the E2F1/AMPKα2 pathway
(31). The conflicting results of
these studies are most likely due to the observation of different,
intertwined pathways in each set of experiments. It is plausible
that the differences in the conclusions of these studies may come
from differences in the experimental reagents, animal models and
other experimental conditions, including the dose of DOX used.
Therefore, it is conceivable that DOX inhibits autophagy in a mouse
model, whereas, under different conditions, an increase of
autophagy is observed in a rat model.
Therefore, the present study proposes that the
apparent complexity of the role of autophagy in DOX-induced
cardiotoxicity is due to the existing associations between
autophagy and other physiological and pathological processes
(33).
Oxidative stress and autophagy in
doxorubicin-induced cardiotoxicity
The large accumulation of ROS caused by DOX, a
process that predominantly occurs in the mitochondria and
cytoplasm, has long been recognized as the main source of cardiac
damage. Under DOX treatment conditions, hydrogen peroxide and
superoxide radicals are formed following the induction of
endogenous oxidases. This process ultimately leads to the
production of the highly reactive and toxic hydroxyl radical by the
iron-catalyzed Haber-Weiss reaction (1,34). These
radicals can react with proteins, lipids, DNA molecules and
endogenous anti-oxidants, which causes protein oxidation, lipid
peroxidation, DNA damage and antioxidant depletion, respectively
(33). Furthermore, ROS can result
in the activation of the AMPK/mTOR signaling pathway or ULK1
kinase, which increases autophagy (35). Indeed, previous studies have reported
that treating cells with the ROS scavenger N-acetylcysteine can
reduce ROS production and autophagy (36,37).
Notably, it is well documented that ROS, like other external
stimuli, including hypoxia, starvation and tumor necrosis factor-α
(TNF-α), can affect the autophagy processes (38). For example, ATG4 is an autophagic
protease with an active cysteine residue regulated by intracellular
redox reactions. In a reducing environment, ATG4 can serve its
unique role in promoting the formation of autophagosomes. However,
in DOX-induced cardiotoxicity conditions, ATG4 protease activity is
inhibited by the oxidization of cysteine residues (39). These observations illustrate the
complex and close association between autophagy and ROS.
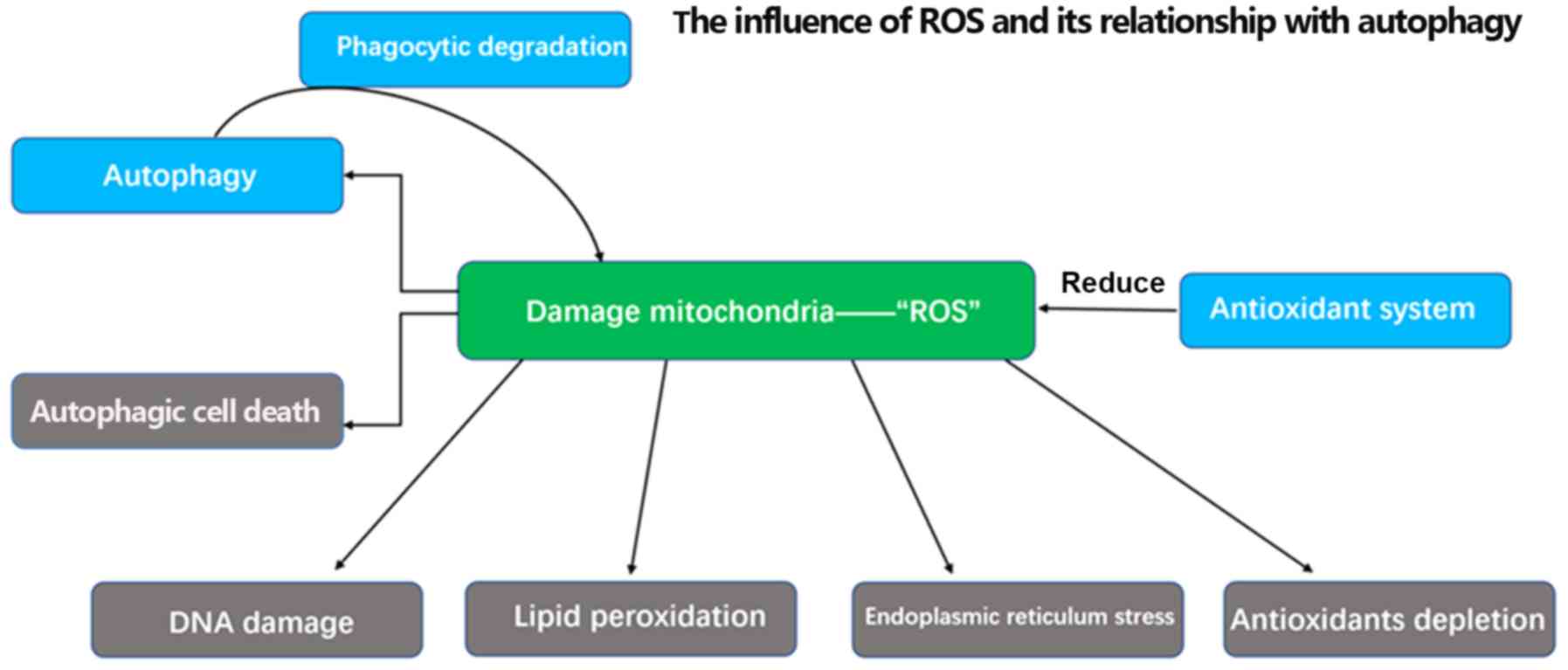
It is worth noting that overwhelming levels of ROS
can trigger autophagic cell death (Fig.
3). Furthermore, previous studies have demonstrated that redox
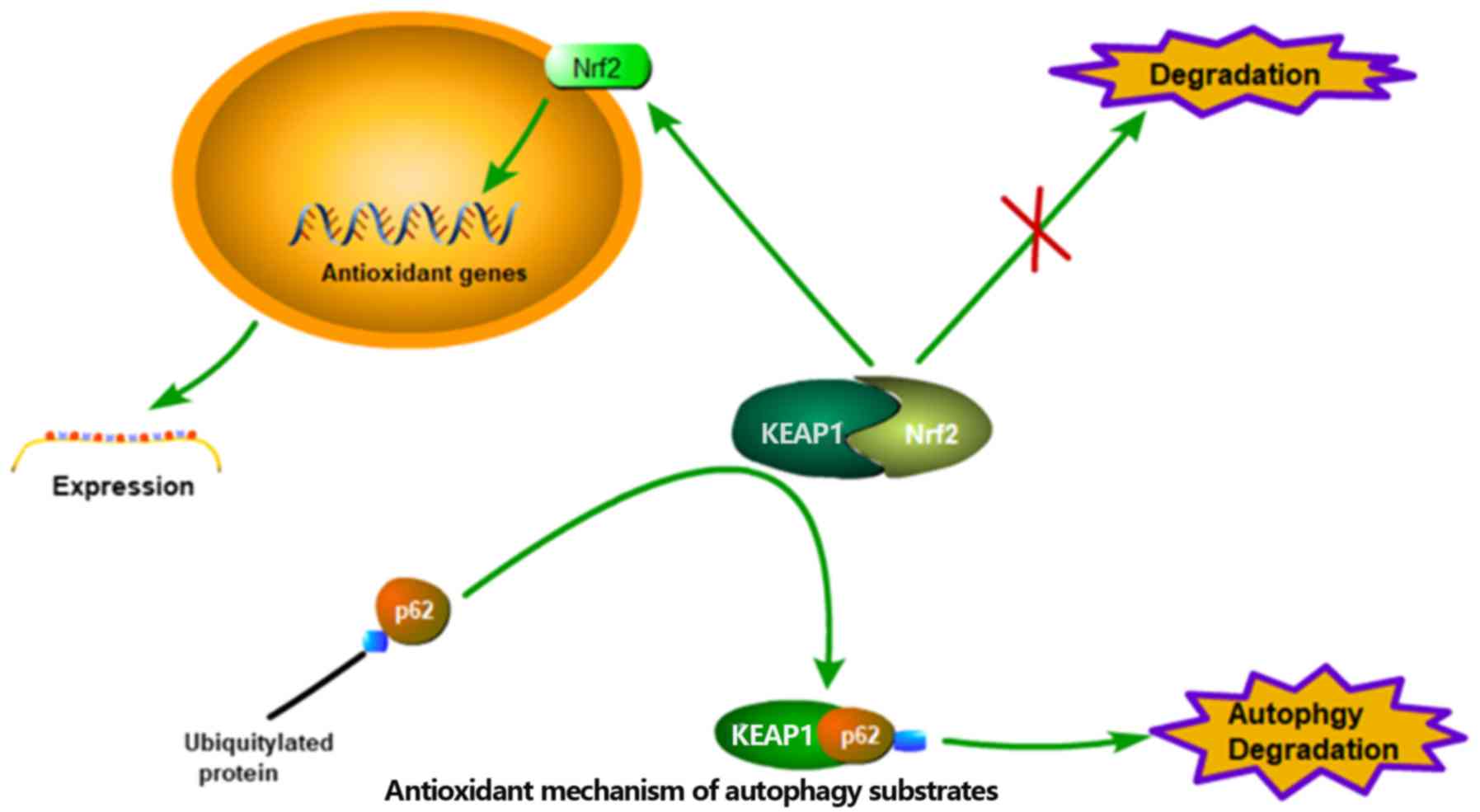
signaling is not the only mechanism that links autophagy and ROS.
When bound to ubiquitylated protein aggregates, p62, an autophagy
substrate used as a reporter of the degradation process in
autophagosomes, can undergo phosphorylation on ser351 in a
redox-independent manner. Phosphorylated p62 then serves an
important regulatory role in the dissociation of the Kelch-like
ECH-associated protein 1-nuclear factor-like 2 (Nrf2) complex,
allowing Nrf2 to translocate to the nucleus and act as a
transcription factor instead of being degraded. Subsequently,
nuclear Nrf2 can transactivate anti-oxidation genes, driving their
expression and ultimately attenuating the intracellular ROS levels
(Fig. 4) (40–42).
DNA damage and autophagy in
doxorubicin-induced cardiotoxicity
With a number of studies (43–48)
being published in the domain of onco-cardiology, the associations
between DOX and DNA damage are becoming increasingly apparent.
Previous studies have suggested that DOX, in a topoisomerase
IIβ-dependent manner, induces the formation of DNA double-strand
breaks (DSBs), which can lead to ROS formation, mitochondriopathy
and apoptosis of cardiomyocytes (43,44).
However, other studies investigating the molecular basis of
anthracycline-induced cardiotoxicity, using knockout mice, have
ruled out a direct involvement of topoisomerase IIβ (43–46).
Additionally, it has been suggested that DNA damage can initiate
autophagy through multiple pathways. In the context of DOX-induced
DNA damage, the activity of the p53 tumor suppressor gene is
enhanced, and the sestrin 1 and sestrin 2 proteins can subsequently
activate AMPK, thereby contributing to the initiation step of
autophagy (47). Furthermore, in a
different pathway, the p53 protein can also activate liver kinase
B1 and thus induce autophagy via AMPK (48).
Notably, autophagy has a significant impact on the
DNA damage response, by affecting the homologous recombination (HR)
and non-homologous end joining (NHEJ) pathways, which are the two
main DSB repair pathways (49).
Previous studies have demonstrated that autophagy-deficient cells
exhibit markedly diminished phosphorylation levels of checkpoint
kinase 1, a critical mediator kinase in the HR pathway (50,51).
Consequently, these autophagy-deficient cells become highly
dependent on the NHEJ pathway to perform DSB repair. However, in
the long run, this continued dependence on NHEJ results in loss of
nucleotides and gross chromosomal rearrangements, including
translocations, and eventually causes a detrimental accumulation of
DNA damage leading to cell death. As early as 2007, a study had
established an association between autophagy and DNA damage. When
the expression of BECN1, the gene coding for the autophagy protein
Beclin 1 in mammals, is downregulated or completely abrogated, the
autophagic survival response to DNA damage is severely impaired,
which allows the cells to die by apoptosis (52). The combined data of numerous studies
have led to the conclusion that stress-induced autophagy limits the
amount of DNA damage and thus contributes to maintaining genome
integrity. In the future, continuous investigations of the
associations between autophagy and DNA damage will assist with the
development of new therapeutic strategies.
Inflammation and autophagy in
doxorubicin-induced cardiotoxicity
Previous studies have reported that acute and
chronic myocarditis, inflammatory diseases, occur in a
dose-dependent manner in patients receiving DOX treatment (4). Inflammation is a factor reported to be
involved in DOX-induced cardiotoxicity and results from the
combined action of numerous regulatory mechanisms (53). Indeed, in patients with cancer who
exhibit DOX-associated adverse effects, a large number of
cytokines, including interleukin-1 and TNF-α, are released, which
triggers various signaling pathways, including the nuclear
factor-κB (NF-κB) and p38-MAPK pathways, and the subsequent
inflammatory cascades (54,55).
These inflammatory signaling pathways are understood
to have close associations with the autophagy pathway. NF-κB acts
as a key regulator of the inflammatory response and is kept in the
cytoplasm in an inactive state by the inhibitor of κB protein. IκB
kinase (IKK) interacts with TGF-β-activated kinase 1 and its
cofactors TAK1 and MAP3K7-binding protein 2 and 3 to form a complex
that degrades IκBα. Activated NF-κB can then translocate to the
nucleus and induce an inflammatory response (53,54,56).
Notably, the TAK1 cofactors transforming growth factor-β-activated
kinase 1-binding protein 2/3 can also bind to Beclin-1 and induce
autophagy (57). Therefore, the
IKK-NF-κB pathway and part of the autophagy pathway are in an
interdependent equilibrium association. Furthermore, under
autophagy-deficient conditions, p62 overexpression activates IKK in
human keratinocytes in a toll-like receptor-dependent manner,
thereby activating NF-κB and triggering an inflammatory response.
Conversely, p62 is degraded to inhibit the NF-κB signaling pathway
when autophagy is upregulated (58).
In the context of DOX chemotherapy, high levels of
NF-κB-p65 phosphorylation contribute to the nitric oxide-induced
phosphorylation/activation of AMPK. In turn, the AMPK/mTOR
signaling pathway can enhance autophagy, which inhibits the NF-κB
signaling pathway and downregulates subsequent inflammatory
cytokine responses (54). Therefore,
upregulation of autophagy can serve an anti-inflammatory role. Of
note, similar effects have been observed in a study associated with
the treatment of rheumatoid arthritis (59).
In summary, these findings clearly demonstrate that
autophagy serves an anti-inflammatory role and can exhibit
additional beneficial effects in the context of doxorubicin-induced
cardiotoxicity.
Conclusions
Doxorubicin causes cardiotoxicity through multiple
mechanisms, including DNA damage, ROS accumulation, lipid
peroxidation, energy deficiency, mitochondrial destruction,
lysosomal destruction, calcium disorder and autophagy dysfunction
(1,4). These mechanisms are not entirely
independent, as they occur simultaneously and interact with each
other (1,4). The exact role of autophagy is not fully
understood, due to a large number of associated signaling pathways,
the association between regulatory factors and the overlap with
other mechanisms (10,16). Apart from the effects on autophagy of
ROS, DNA damage and inflammation, total calcium signaling can also
affect the proximal and distal steps of the autophagic flux
(60).
Furthermore, it is difficult for a single reporter
to reflect the status of autophagy accurately and the majority of
studies do not adequately consider the influence of external
factors in their experimental setup. This has led to contradicting
conclusions regarding the role of autophagy in doxorubicin-induced
cardiotoxicity. Instead of monitoring a single indicator, the use
of multiple methods to assess the status of autophagy would further
our understanding of its entire regulatory process and assist with
understanding how DOX induces changes in the regulation of
autophagy.
Although the role of autophagy in DOX-induced
cardiotoxicity remains controversial, numerous studies have
reported that autophagy appears to be beneficial, by reducing the
level of ROS, promoting the repair of damaged DNA, counteracting
the effects of inflammation effects and protecting cardiomyocytes
from apoptosis (23,40,41,49,58).
As the number of studies continues to increase,
there are numerous multi-faceted issues to resolve, including: i)
the presence of other types of autophagy, including
chaperone-mediated autophagy, which exhibit an unknown effect on
cardiomyocytes; ii) the associations between doses or
concentrations of DOX and the level of autophagy; and iii) how to
modulate autophagy to attenuate cardiotoxicity without triggering
autophagic cell death. Addressing these questions is not only
essential to assist with the selection of drugs to antagonize the
cardiotoxicity induced by chemotherapeutic drugs but also to avoid
resistance to chemotherapeutic drugs and ultimately prevent and
treat cancer.
Acknowledgements
Not applicable.
Funding
This study was funded by the National Natural
Science Foundation of China (grant no. 81300115)
Availability of data and materials
Not applicable.
Authors' contributions
BX, LH and XC drafted the article and revised the
manuscript. SM and PZ participated in the manuscript's revision and
gave some suggestions for important content. LS proposed concepts,
revised the article and obtained funding. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shabalala S, Muller CJF, Louw J and
Johnson R: Polyphenols, autophagy and doxorubicin-induced
cardiotoxicity. Life Sci. 180:160–170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Damiani RM, Moura DJ, Viau CM, Caceres RA,
Henriques JAP and Saffi J: Pathways of cardiac toxicity: Comparison
between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch
Toxicol. 90:2063–2076. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chatterjee K, Zhang J, Honbo N and
Karliner JS: Doxorubicin cardiomyopathy. Cardiology. 115:155–162.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li DL and Hill JA: Cardiomyocyte autophagy
and cancer chemotherapy. J Mol Cell Cardiol. 71:54–61. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mitry MA and Edwards JG: Doxorubicin
induced heart failure: Phenotype and molecular mechanisms. Int J
Cardiol Heart Vasc. 10:17–24. 2016.PubMed/NCBI
|
|
6
|
Yang KC, Sathiyaseelan P, Ho C and Gorski
SM: Evolution of tools and methods for monitoring autophagic flux
in mammalian cells. Biochem Soc Trans. 46:97–110. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu X, Chen K, Kobayashi S, Timm D and
Liang Q: Resveratrol attenuates doxorubicin-induced cardiomyocyte
death via inhibition of p70 S6 kinase 1-mediated autophagy. J
Pharmacol Exp Ther. 341:183–195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johnson R, Shabalala S, Louw J, Kappo AP
and Muller CJF: Aspalathin Reverts Doxorubicin-Induced
Cardiotoxicity through Increased Autophagy and Decreased Expression
of p53/mTOR/p62 Signaling. Molecules. 22:E15892017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fuchs Y and Steller H: Programmed cell
death in animal development and disease. Cell. 147:742–758. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ryter SW, Mizumura K and Choi AM: The
impact of autophagy on cell death modalities. Int J Cell Biol.
2014:5026762014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nishida K, Yamaguchi O and Otsu K:
Crosstalk between autophagy and apoptosis in heart disease. Circ
Res. 103:343–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eskelinen EL and Saftig P: Autophagy: A
lysosomal degradation pathway with a central role in health and
disease. Biochim Biophys Acta. 1793:664–673. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu B, Cheng Y, Liu Q, Bao JK and Yang JM:
Autophagic pathways as new targets for cancer drug development.
Acta Pharmacol Sin. 31:1154–1164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Heras-Sandoval D, Pérez-Rojas JM,
Hernández-Damián J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fasolo A and Sessa C: Targeting mTOR
pathways in human malignancies. Curr Pharm Des. 18:2766–2777. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Toyama EQ, Herzig S, Courchet J, Lewis TL
Jr, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, et
al: Metabolism. AMP-activated protein kinase mediates mitochondrial
fission in response to energy stress. Science. 351:275–281. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wong PM, Feng Y, Wang J, Shi R and Jiang
X: Regulation of autophagy by coordinated action of mTORC1 and
protein phosphatase 2A. Nat Commun. 6:80482015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hung CM, Garcia-Haro L, Sparks CA and
Guertin DA: mTOR-dependent cell survival mechanisms. Cold Spring
Harb Perspect Biol. 4:a0087712012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Su M, Mei Y and Sinha S: Role of the
Crosstalk between Autophagy and Apoptosis in Cancer. J Oncol.
2013:1027352013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cooper KF: Till Death Do Us Part: The
Marriage of Autophagy and Apoptosis. Oxid Med Cell Longev.
2018:47012752018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gump JM and Thorburn A: Autophagy and
apoptosis: What is the connection? Trends Cell Biol. 21:387–392.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen S, Ren Q, Zhang J, Ye Y, Zhang Z, Xu
Y, Guo M, Ji H, Xu C, Gu C, et al: N-acetyl-L-cysteine protects
against cadmium-induced neuronal apoptosis by inhibiting
ROS-dependent activation of Akt/mTOR pathway in mouse brain.
Neuropathol Appl Neurobiol. 40:759–777. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu J, Zheng C, Chen J, Luo J, Su B, Huang
Y, Su W, Li Z and Cui T: Ghrelin protects human umbilical vein
endothelial cells against high glucose-induced apoptosis via
mTOR/P70S6K signaling pathway. Peptides. 52:23–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gu J, Hu W, Song ZP, Chen YG, Zhang DD and
Wang CQ: Resveratrol-induced autophagy promotes survival and
attenuates doxorubicin-induced cardiotoxicity. Int Immunopharmacol.
32:1–7. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reggiori F: 1. Membrane origin for
autophagy. Curr Top Dev Biol. 74:1–30. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xie Y, Kang R, Sun X, Zhong M, Huang J,
Klionsky DJ and Tang D: Posttranslational modification of
autophagy-related proteins in macroautophagy. Autophagy. 11:28–45.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Russell RC, Tian Y, Yuan H, Park HW, Chang
YY, Kim J, Kim H, Neufeld TP, Dillin A and Guan KL: ULK1 induces
autophagy by phosphorylating Beclin-1 and activating VPS34 lipid
kinase. Nat Cell Biol. 15:741–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakatogawa H: Two ubiquitin-like
conjugation systems that mediate membrane formation during
autophagy. Essays Biochem. 55:39–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang X, Wang XL, Chen HL, Wu D, Chen JX,
Wang XX, Li RL, He JH, Mo L, Cen X, et al: Ghrelin inhibits
doxorubicin cardiotoxicity by inhibiting excessive autophagy
through AMPK and p38-MAPK. Biochem Pharmacol. 88:334–350. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gu J, Fan YQ, Zhang HL, Pan JA, Yu JY,
Zhang JF and Wang CQ: Resveratrol suppresses doxorubicin-induced
cardiotoxicity by disrupting E2F1 mediated autophagy inhibition and
apoptosis promotion. Biochem Pharmacol. 150:202–213. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kobayashi S, Volden P, Timm D, Mao K, Xu X
and Liang Q: Transcription factor GATA4 inhibits
doxorubicin-induced autophagy and cardiomyocyte death. J Biol Chem.
285:793–804. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bartlett JJ, Trivedi PC and Pulinilkunnil
T: Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol
Cell Cardiol. 104:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cappetta D, De Angelis A, Sapio L,
Prezioso L, Illiano M, Quaini F, Rossi F, Berrino L, Naviglio S and
Urbanek K: Oxidative Stress and Cellular Response to Doxorubicin: A
Common Factor in the Complex Milieu of Anthracycline
Cardiotoxicity. Oxid Med Cell Longev. 2017:15210202017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Weerapana E, Wang C, Simon GM, Richter F,
Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D and Cravatt
BF: Quantitative reactivity profiling predicts functional cysteines
in proteomes. Nature. 468:790–795. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li L, Chen Y and Gibson SB:
Starvation-induced autophagy is regulated by mitochondrial reactive
oxygen species leading to AMPK activation. Cell Signal. 25:50–65.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bonet-Ponce L, Saez-Atienzar S, da Casa C,
Sancho-Pelluz J, Barcia JM, Martinez-Gil N, Nava E, Jordan J,
Romero FJ and Galindo MF: Rotenone Induces the Formation of
4-Hydroxynonenal Aggresomes. Role of ROS-Mediated Tubulin
Hyperacetylation and Autophagic Flux Disruption. Mol Neurobiol.
53:6194–6208. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kirkland RA, Adibhatla RM, Hatcher JF and
Franklin JL: Loss of cardiolipin and mitochondria during programmed
neuronal death: Evidence of a role for lipid peroxidation and
autophagy. Neuroscience. 115:587–602. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Scherz-Shouval R, Shvets E, Fass E, Shorer
H, Gil L and Elazar Z: Reactive oxygen species are essential for
autophagy and specifically regulate the activity of Atg4. EMBO J.
26:1749–1760. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dodson M, Redmann M, Rajasekaran NS,
Darley-Usmar V and Zhang J: KEAP1-NRF2 signalling and autophagy in
protection against oxidative and reductive proteotoxicity. Biochem
J. 469:347–355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mizunoe Y, Kobayashi M, Sudo Y, Watanabe
S, Yasukawa H, Natori D, Hoshino A, Negishi A, Okita N, Komatsu M,
et al: Trehalose protects against oxidative stress by regulating
the Keap1-Nrf2 and autophagy pathways. Redox Biol. 15:115–124.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu
YL, Liu LF and Yeh ET: Identification of the molecular basis of
doxorubicin-induced cardiotoxicity. Nat Med. 18:1639–1642. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wallace KB: Doxorubicin-induced cardiac
mitochondrionopathy. Pharmacol Toxicol. 93:105–115. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Capranico G, Tinelli S, Austin CA, Fisher
ML and Zunino F: Different patterns of gene expression of
topoisomerase II isoforms in differentiated tissues during murine
development. Biochim Biophys Acta. 1132:43–48. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Azuma Y, Arnaoutov A and Dasso M: SUMO-2/3
regulates topoisomerase II in mitosis. J Cell Biol. 163:477–487.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang M, Xu Y, Liu J, Ye J, Yuan W, Jiang
H, Wang Z, Jiang H and Wan J: Recent Insights into the Biological
Functions of Sestrins in Health and Disease. Cell Physiol Biochem.
43:1731–1741. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shaw RJ, Kosmatka M, Bardeesy N, Hurley
RL, Witters LA, DePinho RA and Cantley LC: The tumor suppressor
LKB1 kinase directly activates AMP-activated kinase and regulates
apoptosis in response to energy stress. Proc Natl Acad Sci USA.
101:3329–3335. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sørensen CS, Hansen LT, Dziegielewski J,
Syljuåsen RG, Lundin C, Bartek J and Helleday T: The cell-cycle
checkpoint kinase Chk1 is required for mammalian homologous
recombination repair. Nat Cell Biol. 7:195–201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu EY, Xu N, O'Prey J, Lao LY, Joshi S,
Long JS, O'Prey M, Croft DR, Beaumatin F, Baudot AD, et al: Loss of
autophagy causes a synthetic lethal deficiency in DNA repair. Proc
Natl Acad Sci USA. 112:773–778. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Abedin MJ, Wang D, McDonnell MA, Lehmann U
and Kelekar A: Autophagy delays apoptotic death in breast cancer
cells following DNA damage. Cell Death Differ. 14:500–510. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Monkkonen T and Debnath J: Inflammatory
signaling cascades and autophagy in cancer. Autophagy. 14:190–198.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Qing G, Yan P and Xiao G: Hsp90 inhibition
results in autophagy-mediated proteasome-independent degradation of
IkappaB kinase (IKK). Cell Res. 16:895–901. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Salminen A, Hyttinen JMT, Kauppinen A and
Kaarniranta K: Context-Dependent Regulation of Autophagy by
IKK-NF-κB Signaling: Impact on the Aging Process. Int J Cell Biol.
2012:8495412012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Niso-Santano M, Criollo A, Malik SA,
Michaud M, Morselli E, Mariño G, Lachkar S, Galluzzi L, Maiuri MC
and Kroemer G: Direct molecular interactions between Beclin 1 and
the canonical NFκB activation pathway. Autophagy. 8:268–270. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lee HM, Shin DM, Yuk JM, Shi G, Choi DK,
Lee SH, Huang SM, Kim JM, Kim CD, Lee JH, et al: Autophagy
negatively regulates keratinocyte inflammatory responses via
scaffolding protein p62/SQSTM1. J Immunol. 186:1248–1258. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhou HF, Yan H, Hu Y, Springer LE, Yang X,
Wickline SA, Pan D, Lanza GM and Pham CT: Fumagillin prodrug
nanotherapy suppresses macrophage inflammatory response via
endothelial nitric oxide. ACS Nano. 8:7305–7317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fan M, Li Y, Yao C, Liu X, Liu J and Yu B:
DC32, a Dihydroartemisinin Derivative, Ameliorates Collagen-Induced
Arthritis Through an Nrf2-p62-Keap1 Feedback Loop. Front Immunol.
9:27622018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bootman MD, Chehab T, Bultynck G, Parys JB
and Rietdorf K: The regulation of autophagy by calcium signals: Do
we have a consensus? Cell Calcium. 70:32–46. 2018. View Article : Google Scholar : PubMed/NCBI
|