Introduction
Endometrial cancer (EC) is the most common female
gynaecological cancer worldwide with 280,000 new cases per year
(1). Alarmingly, the incidence of EC
is on the rise, particularly in younger women of reproductive age
(2), and this is believed to be
caused by increasing rates of societal obesity predominantly in
developed countries (3). There are
currently no effective early diagnostic screening tests, and
advanced or recurrent EC presents a poor prognosis. EC is broadly
categorized into two major types, and 80–90% of EC are classified
as type I (4). Type I EC are
associated with unopposed oestrogen and endometrial hyperplasia
and, depending on the extent of solid tumour growth, Type I EC can
be further divided into grades based on differentiation status: i)
Grade 1 (G1), well differentiated; ii) grade 2 (G2), moderately
differentiated; and iii) grade 3 (G3), poorly differentiated
(5). Type II EC occurs less
frequently, but is more aggressive than type I, it is not
associated with oestrogen stimulation and arises from a state of
atrophy (5). However, individual EC
tumours can have diverse molecular and genomic profiles; therefore,
treatments based on histological classification alone may not be
effective (6). This limitation
highlights the need for molecular characterization, which may
facilitate the use of personalised therapies (7).
MicroRNAs (miRs) are a class of small (18–22
nucleotides), highly conserved, non-coding RNAs that regulate gene
expression post-transcriptionally (8). The mature form of miRs can act by
destabilising mRNAs or by repressing protein translation, leading
to a downregulation of the target protein products, influencing
their biological functions (8). miR
expression is tightly coordinated and each miR has the ability to
act on numerous target genes, regulating some of the most basic
cellular processes such as proliferation, apoptosis and metabolism
(8).
The miR-29 family includes miR-29-a, -b and -c,
which share an identical seed sequence and mature sequences with
only minor nucleotide differences, and thus share many predicted
targets (9). Members of the miR-29
family are implicated in targeting genes encoding extracellular
matrix proteins, transcription factors, cell cycle proteins,
factors involved in cytokine signalling, phosphatases and proteins
with roles in many other biological processes (9). miR-29-a, -b and -c have all been shown
to be downregulated in EC (10–14);
however, the specific role for miR-29c and its targets in EC growth
and development are unknown.
miR-29c is located and transcribed from chromosome
1q32.2. miR-29c is an established tumour suppressor in many cancer
types including lung, gastric, pancreatic and bladder cancer
(15). miR-29c expression is
elevated in early and mid-secretory phase endometrial epithelium of
women with primary infertility (16). Elevated endometrial miR-29c impairs
endometrial epithelial collagen type IV a1 (COL4A1) production,
likely impairing the adhesive capacity of the luminal epithelium
and contributing to implantation failure and infertility (16). miR-29c also contributes to
endometriosis-induced infertility by affecting endometriosis cell
proliferation, invasion and apoptosis (17).
The aims of the present study were to define miR-29c
expression in endometrioid (type I) EC, to investigate the
functional effects of re-introducing miR-29c expression in EC cell
lines and to identify miR-29c targets involved in cancer
progression in order to pinpoint future potential therapeutic
targets.
Materials and methods
Patient samples
The present study was approved by the Monash Health
Human Research and Ethics Committee (approval no. 06014C) and the
Victorian Cancer Biobank (Melbourne, Australia; project no. 13018).
Informed consent was obtained from each participant at the time of
sample collection (n=35).
EC samples
EC samples were collected as follows from female
patients with the following characteristics. The Victorian Cancer
Biobank provided RNA from type I EC (n=10 samples/grade) or benign
post-menopausal endometrium (n=5) whole tissue. There was no
difference in patient age between the three EC grades (age of
patients with G1, 53.4±3.5 years; age of patients with G2, 63.8±4.2
years; age of patients with G3, 66.1±4.3 years), whereas age
information was not available for patients with benign endometrium.
The patients did not receive chemotherapy, radiotherapy or other
biological targeted therapy prior to surgery. Samples were
collected by the Victoria Cancer Biobank between 2007 and 2014.
EC cell line culture
G1-derived Ishikawa cells were provided by Dr
Nishida (Tsukuba University, Tochigi, Japan) and cultured in
DMEM/F12 medium (Thermo Fisher Scientific, Inc.) supplemented with
10% foetal calf serum (FCS; Invitrogen; Thermo Fisher Scientific,
Inc.). G2-derived HEC1A cells [authenticated by the Monash Health
Translation Precinct (MHTP) Medical Genomics Facility] were
purchased from the American Type Culture Collection (ATCC) and
cultured in McCoy's medium (Thermo Fisher Scientific, Inc.)
supplemented with 10% FCS. G3-derived AN3CA cells (authenticated by
the MHTP Medical Genomics Facility) were purchased from the ATCC
and cultured in DMEM/F12 medium supplemented with 10% FCS. All
cells were cultured in a humidified incubator maintained at 37°C in
an atmosphere with 5% CO2.
Primary epithelial cell isolation and
culture
Endometrial biopsies were collected at curettage
from women with regular menstrual cycles at the proliferative stage
(days 7–13) of the menstrual cycle. The women had no steroid
treatment for ≥2 months prior to tissue collection. The biopsies
were examined by an experienced gynaecological pathologist to
confirm the stage and absence of endometrial dysfunction. Human
endometrial epithelial cells (HEEC) were prepared as previously
described (18). Briefly,
endometrial tissue was digested with collagenase and the suspension
was filtered through nylon mesh to collect endometrial epithelial
glands. The epithelial glands were collected and resuspended in
DMEM/F12 supplemented with 10% FCS and 1% Penicillin-Streptomycin
antibiotic-antimycotic solution (Gibco; Thermo Fisher Scientific,
Inc.) before being serially replated for three times in plastic
culture dishes for 30 min in a humidified incubator with 5%
CO2 at 37°C to allow adherence of contaminating stromal
cells. Non-adherent cells and glands were transferred to 96 or
48-well plates and epithelial cells were allowed to grow out from
glandular structures for 48 h in a humidified incubator with 5%
CO2 at 37°C. A purity of >95% HEEC as previously
described (18) was necessary for
the cells to be used experimentally. Confluent HEEC were cultured
in serum-free DMEM/F12 containing 1% Penicillin-Streptomycin
solution for 16 h prior to RNA extraction.
miR-29c mimic and miR-29c inhibitor
transfection
Ishikawa, HEC1A or AN3CA cells were transfected
according to the manufacturer's protocol, as previously described
(19) using
Lipofectamine® RNAiMAX (Applied Biosystems; Thermo
Fisher Scientific, Inc.) and miR-29c mimic (25 nM; hsa-miR-29c-3p;
seed sequence:
5′-UCUCUUACACAGGCUGACCGAUUUCUCCUGGUGUUCAGAGUCUGUUUUUGUCUAGCACCAUUUGAAAUCGGUUAUGAUGUAGGGGGA-3′;
cat. no. MC10518; Thermo Fisher Scientific, Inc), miR-29c inhibitor
(25 nM; hsa-miR-29c-3p; seed sequence:
5′-AUCUCUUACACAGGCUGACCGAUUUCUCCUGGUGUUCAGAGUCUGUUUUUGUCUAGCACCAUUUGAAAUCGGUUAUGAUGUAGGGGGA-3′;
cat. no. MH10518; Thermo Fisher Scientific, Inc.) for 72 h in a
humidified incubator with 5% CO2 at 37°C. A scrambled
miR sequence (25 nM; cat. no. 4464058; Thermo Fisher Scientific,
Inc.) was used as a control. Transfection efficiency was determined
using reverse transcription-quantitative PCR (RT-qPCR). Further
experiments, including proliferation assay, flow cytometry and
RT-qPCR, were performed 72 h after initial transfection.
xCELLigence real time proliferation
assay
Experiments were carried out using the real-time
cell analyser (RTCA) DP xCELLigence instrument (ACEA Biosciences;
Agilent Technologies GmbH), as previously described (20). The xCELLigence instrument was kept in
a humidified incubator maintained at 37°C with 5% CO2.
Cells were transfected with miR-29c mimic, miR-29c inhibitor or
control for 72 h, and seeded in E-plate 96 (ACEA Biosciences;
Agilent Technologies GmbH) at ~10,000 cells/well in media (DMEM/F12
for Ishikawa and AN3CA, McCoy for HEC1A) supplemented with 5% FCS.
The plates were monitored every 15 min for a total of 72 h. Data
were analysed using RTCA software v1.2, supplied with the
instrument (ACEA Biosciences; Agilent Technologies GmbH) and
exported for statistical analysis.
Flow cytometry and cell cycle
analysis
Ishikawa, HEC1A or AN3CA cells were serum-starved
for 24 h to synchronize populations into G0, before
cells were transfected with miR-29c mimic, inhibitor or scrambled
miR sequence (as described above). Cells were harvested after 72 h,
fixed in 70% ethanol and stained with FxCycle propidium iodide
(PI)/RNase staining solution (Molecular Probes; Thermo Fisher
Scientific, Inc.) and analysed on a BDFACSCanto II flow cytometer.
Cell cycle, apoptosis analysis and model fitting were performed
with FlowJo (Version X, FlowJo LLC, Ashland, OR, USA) (19).
RNA preparation and RT-qPCR
RNA was extracted from cultured cells or human
epithelial endometrial biopsies using Tri Reagent (Sigma-Aldrich;
Merck KGaA) according to the manufacturer's protocol. Genomic DNA
was digested using the DNAfree kit (Ambion; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. RNA
samples concentration, yield and purity were analysed using a
spectrophotometer (Nanodrop Technologies; Thermo Fisher Scientific,
Inc.) at an absorbance ratio of 260/280 nm.
RNA was reverse transcribed using the TaqMan reverse
transcription kit (Applied Biosystems; Thermo Fisher Scientific,
Inc), and specific TaqMan primer sets for miRs (miR-29c-3p; cat.
no. 000587; rnU44, cat. no. 001094; Applied Biosystems; Thermo
Fisher Scientific, Inc.) or with a Moloney Murine Leukemia Virus RT
system (Life Technologies; Thermo Fisher Scientific, Inc.) and
oligo primers (Sigma Aldrich; Merck KGaA) for non-miR genes. qPCR
was performed using TaqMan Fast Universal PCR Master mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) or Power SYBR Green
master mix (Applied Biosystems; Thermo Fisher Scientific, Inc.)
using TaqMan probes (miR-29c-3p; cat. no. 000587; rnU44, cat. no.
001094; Thermo Fisher Scientific, Inc.) or specific primer pairs
(Table I) performed on the ABI
7500HT fast block qPCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) in triplicate (final reaction volume, 10 µl) in
384-well micro optical plates (Applied Biosystems; Thermo Fisher
Scientific, Inc.). A template-free negative control in the presence
of primers and RNase-free water, and RNase-free water only negative
controls were added for each run. The qPCR protocol was as follows:
Initial denaturation at 95°C for 10 min, followed by 40 cycles of
95°C for 15 sec and 60°C for 1 min. miR-29c expression levels were
normalised against the control RNU44 probes. Gene expression was
normalised against 18S ribosomal RNA (18S). Relative expression
levels were calculated using comparative cycle threshold method
(2−ΔΔCq) 6 (21)
according to the manufacturer's protocol, with 18S serving as the
endogenous control for normalization.
 | Table I.microRNA-29c target gene primer
sequences. |
Table I.
microRNA-29c target gene primer
sequences.
| Gene | Gene name | Sequence
(5′→3′) |
|---|
| CAV1 | Caveolin-1 | F:
CAGGGACATCTCTACACC |
|
|
| R:
TCAAAGTCAATCTTGACCAC |
| CDC42 | Cell division cycle
42 | F:
GAACAAACAGAAGCCTATCAC |
|
|
| R:
TTTAGGCCTTTCTGTGTAAG |
| COL4A1 | Collagen type IV
α1 | F:
AAAGGGAGATCAAGGGATAG |
|
|
| R:
TCACCTTTTTCTCCAGGTAG |
| FBN1 | Fibrillin 1 | F:
AAAGATCTTGATGAGTGTGC |
|
|
| R:
GTATGGTGTTGGGTAAATCC |
| HBP1 | HMG-box
transcription factor 1 | F:
GAATGCCTTCATGCTTTTTG |
|
|
| R:
CACACTTATGGCTCTGTTATC |
| ITGB1 | Integrin β 1 | F:
ATTCCCTTTCCTCAGAAGTC |
|
|
| R:
TTTTCTTCCATTTTCCCCTG |
| MCL1 | MCL1 apoptosis
regulator, BCL2 family member | F:
TAGTTAAACAAAGAGGCTGG |
|
|
| R:
ATAAACTGGTTTTGGTGGTG |
| MDM2 | MDM2
proto-oncogene | F:
CAGCAGGAATCATCGGACTCA |
|
|
| R:
ACACAGAGCCAGGCTTTCAT |
| MMP2 | Matrix
metalloproteinase 2 | F:
GTGATCTTGACCAGAATACC |
|
|
| R:
GCCAATGATCCTGTATGTG |
| NUMB | NUMB endocytic
adaptor protein | F:
AGGCTCTTTCCGACCTTTTC |
|
|
| R:
GCTGAAGGCATTGGTGATCT |
| SGK1 |
Serum/glucocorticoid regulated kinase
1 | F:
AGACTACATTAATGGTGGAGAG |
|
|
| R:
ATTTCAGCAGCATAGAAACG |
| SIRT1 | Sirtuin 1 | F:
AAGGAAAACTACTTCGCAAC |
|
|
| R:
GGAACCATGACACACTGAATTATC |
| VEGFA | Vascular
endothelial growth factor A | F:
AATGTGAATGCAGACCAAAG |
|
|
| R:
GACTTATACCGGGATTTCTTG |
| 18S | 18S ribosomal
RNA | F:
GTAACCCGTTGAACCCCATTC |
|
|
| R:
GCCTCACTAAACCATCCAATCG |
Predicted and confirmed miR-29c target genes
analysed in this study were chosen by bioinformatics analysis using
online databases: picTAR (https://pictar.mdc-berlin.de/established 2007),
TarBase (Version 7, http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index)
and TargetScan (Version 7.2, http://www.targetscan.org/vert_72/).
The following miR-29c target genes were analysed
using RT-qPCR: Caveolin 1 (CAV1), cell division cycle 42 (CDC42),
COL41A, fibrillin 1, HMG-box transcription factor 1 (HBP1),
integrin subunit β 1 (ITGB1), NUMB endocytic adaptor protein
(NUMB), MCL1 apoptosis regulator BCL2 family member (MCL1), MDM2
proto-oncogene (MDM2), serum/glucocorticoid regulated kinase 1
(SGK1), sirtuin 1 (SIRT1) and vascular endothelial growth factor A
(VEGFA). Primer sequences are presented in Table I.
Statistical analysis
Statistical analysis was carried out using GraphPad
Prism (v8.0.1; GraphPad Software, Inc.) and data were assessed by
paired Student's t-test for two groups or one-way ANOVA with
Tukey's post-hoc test for multiple groups. Real-time xCELLigence
monitoring and flow cytometry was assessed using repeated measures
two-way ANOVA, with Sidak's post-hoc test. Data are presented as
the mean ± SEM. A minimum of three repeats was required for
statistical analysis to be performed. P<0.05 was considered to
indicate a statistically significant difference.
Results
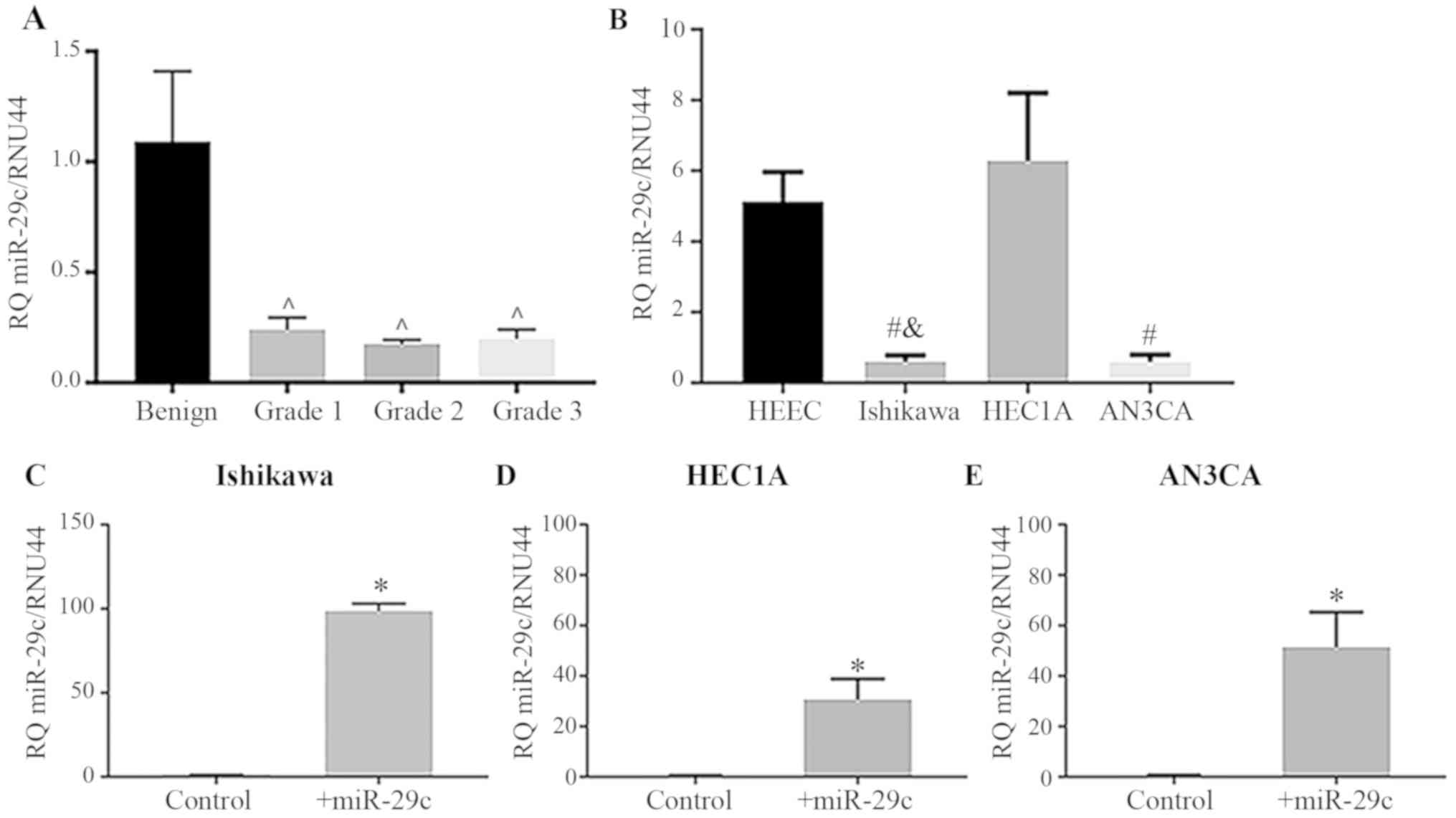
miR-29c expression is downregulated in
human type I endometrioid cancer cell lines
miR-29c expression was significantly reduced in
human G1, 2 and 3 type I endometrioid cancer biopsies compared with
that in the benign endometrium (Fig.
1A; P<0.05). Similarly, miR-29c expression levels were
significantly reduced in the EC epithelial cell lines Ishikawa
(G1), and AN3CA (G3) compared with that in the primary HEECs
(Fig. 1B; P<0.05); however, no
reduction was seen in HEC1A (G2) cells. Confirmation of miR-29c
mimic transfection efficiency in Ishikawa, HEC1A and AN3CA cells
showed that miR-29c was significantly upregulated following miR-29c
mimic overexpression (Fig. 1C-E;
P<0.05).
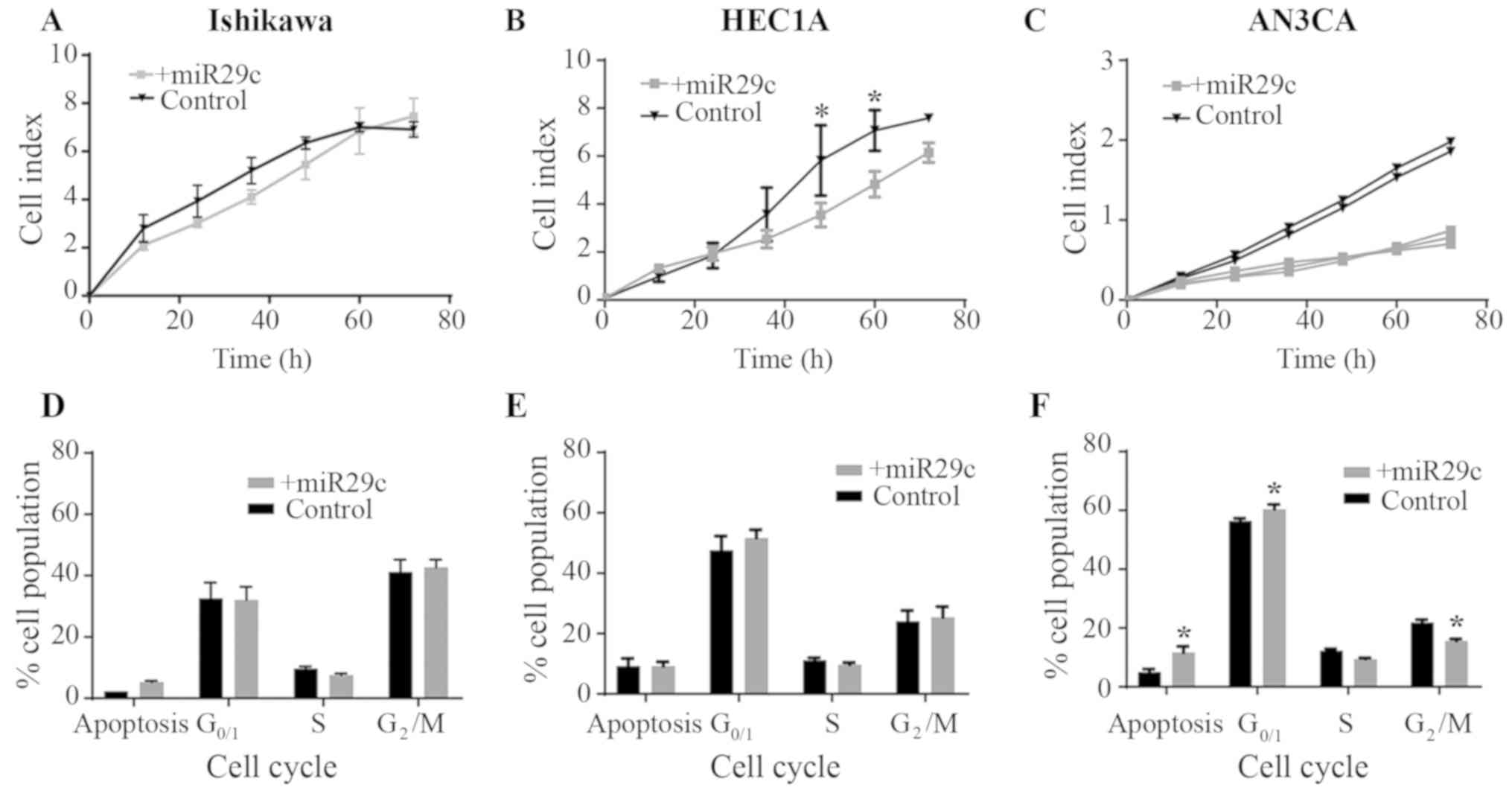
miR-29c overexpression reduces cell
proliferation in G2- and G3-derived EC cell lines
To determine the functional role of miR-29c in EC
cell proliferation, miR-29c was overexpressed in Ishikawa (G1)
HEC1A (G2) and AN3CA (G3) cell lines (Fig. 2A-C). Using the xCELLigence real-time
system, miR-29c mimic overexpression was identified to
significantly reduce HEC1A proliferation (at 48 and 60 h;
P<0.05) compared with that in the control cells (Fig. 2B). Proliferation was also reduced
following miR-29c overexpression in the AN3CA cell line; however,
statistical analyses could not be performed, as there were only two
control samples (Fig. 2C). There was
no effect of miR-29c mimic overexpression on Ishikawa cell
proliferation (Fig. 2A).
To determine the effect of miR-29c overexpression on
cell cycle and the levels of apoptosis in Ishikawa (G1), HEC1A (G2)
and AN3CA (G3) EC cell lines, flow cytometry was performed
following PI/RNase staining. There was no effect of miR-29c
overexpression in Ishikawa or HEC1A cells compared with the
corresponding controls (Fig. 2D and
E). However, in AN3CA cells, miR-29c overexpression increased
the percentage of apoptotic cells and the rate of cells in
G0/G1 phase, and decreased the percentage of
cells in G2/M phase (Fig.
2F; P<0.05).
miR-29c overexpression reduces the
expression of target genes
miRs act by binding to target sequences in the
3′-untranslated regions of multiple genes. miR-29c regulation of
target gene expression is presented in Fig. 3. In the Ishikawa cell line miR-29c
overexpression significantly reduced HBP1, ITGB1, MCL1, MDM2 and
SGK1 expression compared with the scrambled control. HBP1, ITGB1,
MDM2, SIRT1 and VEGFA expression was significantly reduced
following miR-29c overexpression in HEC1A cells compared with that
in the scrambled control group. CDC42, ITGB1, MCL1 and MDM2 were
all significantly reduced in the AN3CA cells following miR-29c
overexpression compared with that in the scrambled control group.
There was no significant effect on CAV1 or NUMB gene expression in
any cell line (data not shown).
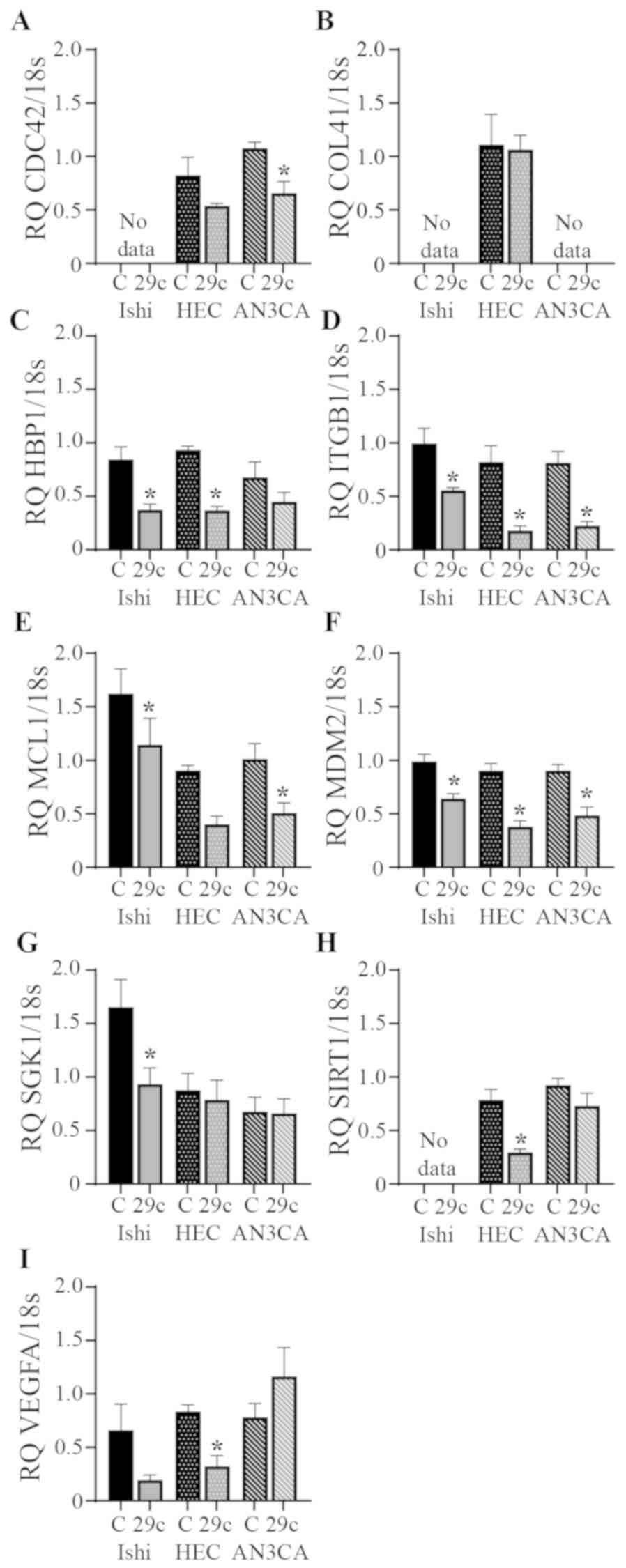 | Figure 3.Effects of miR-29c overexpression on
the expression levels of its predicted target genes. miR-29c
overexpression reduced the expression levels of (A) CDC42, (B)
COL4A1, (C) HBP1, (D) ITGB1, (E) MCL1, (F) MDM2, (G) SGK1, (H)
SIRT1 and (I) VEGFA in Ishikawa, HEC1A and AN3CA endometrial cancer
cell lines, as assessed by reverse transcription-quantitative PCR.
Expression levels of all genes were normalized to 18s. Data are
presented as the mean ± SEM. n=3–6 in each group. *P<0.05 vs.
corresponding control. 29c, miR-29c overexpression; C, control;
Ishi, Ishikawa; HEC, HEC1A; CDC42, cell division cycle 42; COL41A,
collagen type IV α 1 chain; HBP1, HMG-box transcription factor 1;
ITGB1, integrin subunit β 1; MCL1, MCL1 apoptosis regulator, BCL2
family member; MDM2, MDM2 proto-oncogene; SGK1,
serum/glucocorticoid regulated kinase 1; SIRT1, sirtuin 1; VEGFA,
vascular endothelial growth factor A; 18s, 18S ribosomal RNA; RQ,
relative quantification. |
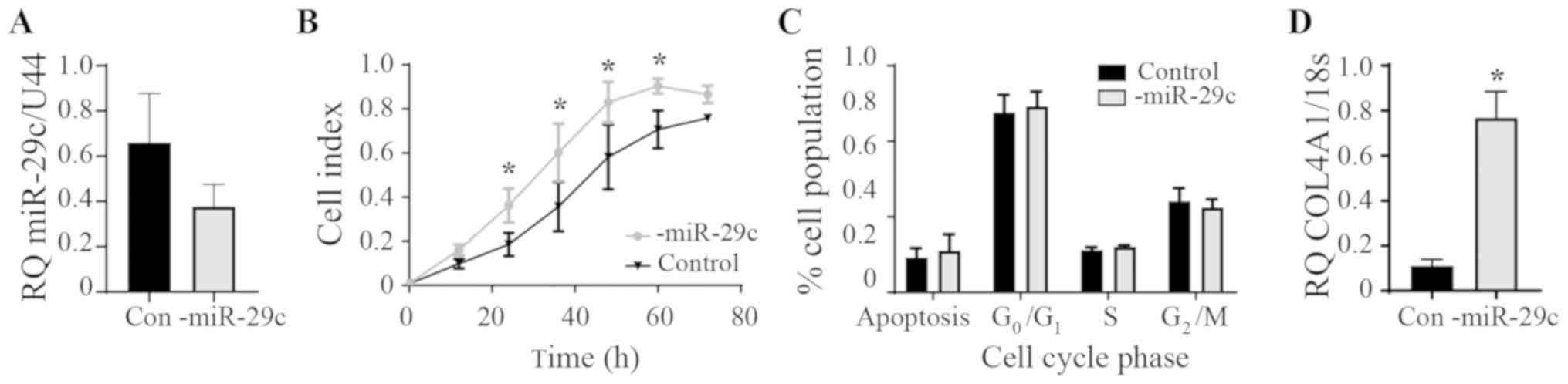
miR-29c inhibition in HEC1A
Endogenous levels of miR-29c expression in the HEC1A
cells were found to be similar to the HEEC control cells and not
reduced as in the Ishikawa and AN3CA cancer cell lines (Fig. 1B). Therefore, the effect of
inhibiting miR-29c in the HEC1A cells was examined (Fig. 4A). The inhibition of miR-29c
expression significantly increased proliferation between 24 and 60
h (P<0.05) compared with that in the control group (Fig. 4B). No differences were seen in the
cell cycle phases upon the inhibition of miR-29c (Fig. 4C). COL4A1 gene expression was
significantly increased following miR-29c inhibition (Fig. 4D). COL4A1 is a known target of
miR-29c; overexpression of miR-29c reduces COL4A1 production in
endothelial (22) and endometrial
epithelial (16) cells. The
expression of the other aforementioned genes identified following
bioinformatics analysis were investigated following miR-29c
inhibition, but only COL4A1 was significantly altered (data not
shown).
Discussion
To the best of our knowledge, the present study is
the first to describe the functional effects and downstream targets
of miR-29c in the EC cell lines Ishikawa (G1), HEC1A (G2) and AN3CA
(G3). It was identified that miR-29c was downregulated in human G1,
G2 and G3 EC tumours compared with benign tissue. Compared with
benign primary human endometrial epithelial cells, the EC cell
lines Ishikawa (G1) and AN3CA (G3) had reduced endogenous miR-29c
levels, whereas HEC1A levels were equivalent to the levels in the
benign primary human endometrial epithelial cells. Exogenous
miR-29c overexpression reduced proliferation of the HEC1A cell
line, increased apoptosis and reduced cell cycle progression in the
AN3CA cell line. Conversely, inhibition of miR-29c in HEC1A cells
significantly increased cell proliferation and COL4A1 expression.
miR-29c target genes, such as ITGB1 and MDM2, which are involved in
adhesion, cell survival and migration (23,24),
were downregulated following miR-29c overexpression, suggesting
that miR-29c may act as a tumour suppressor in EC.
Previous studies showed that miR-29c expression was
downregulated in grades 1–3 type I EC tissue compared with that in
benign tissue from non-malignant endometrium (10,14). The
Ishikawa (G1) and AN3CA (G3) cells also exhibited decreased miR-29c
expression compared with that in isolated human endometrial
epithelial cells. However, the HEC1A (G2) cell line presented
similar endogenous miR-29c levels to the non-cancerous endometrial
epithelial control cells. All three of these cell lines are known
to harbour different mutational profiles (25), which could potentially be involved in
regulating miR-29c expression and may account for differences in
functional responses to specific factors, including miR-29c.
miR-29c expression can be regulated by different factors
influencing its promoter region such as methylation sites (26). Moreover, miR-29c promoter possesses a
p53 consensus responsive element (27) which may downregulate its activity.
Additionally, miR-29c is regulated by ovarian hormones in uterine
leiomyoma smooth muscle cells (28).
miR-29c overexpression reduced cell proliferation in
HEC1A cells and cell cycle progression in the AN3CA cell line.
Supporting a role of miR-29c in repressing endometrial cell growth,
a previous study found that miR-29c overexpression reduced cell
growth in the endometriosis cell line CRL-7566, possibly by
increasing apoptosis (17). In the
present study, the percentage of apoptotic cells was increased only
in AN3CA cells. The time-point selected to collect the cells for
flow cytometry (72 h) may be too late to see an effect on apoptosis
in HEC1A cells. Moreover, the higher endogenous levels of miR-29c
in HEC1A cells may mask the effects of miR-29c overexpression. In
the endometriosis cells, apoptosis was suggested to increase
following repression of c-Jun (17).
c-Jun was not investigated in the present study, but miR-29c
reduced expression of the antiapoptotic protein MCL1 in AN3CA
cells. It would be of interest to determine whether miR-29c is able
to target c-Jun in EC cells, particularly AN3CAs.
It is surprising that no effect on proliferation or
cell cycle progression was found in the Ishikawa cell line;
however, this may be due to the peculiar mutational profile found
in this cell line (25). However,
miR-29c overexpression repressed the expression of various target
genes in the three cell lines investigated. Notably, more detailed
studies investigating the effect of miR-29c on apoptosis and cell
cycle progression in EC are required.
miR-29c predicted and confirmed target genes
involved in cellular growth, adhesion, migration and invasion were
examined by qPCR. The present data suggested that these genes are
regulated by miR-29c; however, luciferase reporter assays are
required to demonstrate direct targeting of these genes by miR-29c.
Interestingly, variation in miR-29c target genes was found between
the three EC cell lines. This phenomenon is likely due to the
variable level of endogenous miR-29c and the specific mutational
profiles within each individual cell line (detailed in 25). For
example, Ishikawa and AN3CA cells exhibit different mutations in
PTEN, leading to the absence of the protein, whereas HEC1A has no
mutations and PTEN protein is present (25). Ishikawa and AN3CA cells also exhibit
mutations in the PI3K subunit PIKR1, whereas HEC1A cells exhibit
mutations in the PI3K subunit PIK3CA or KRAS (25).
In all three EC cell lines restoration of miR-29c
reduced ITGB1 and MDM2 expression levels. ITGB1 is involved in cell
adhesion, metastatic diffusion, cell repair and tumour growth
(23). ITGB1 is a confirmed target
of miR-29c in gastric cancer and their expression is correlated
with clinical outcome (29). ITGB1
has been shown to promote the invasive capacity of endometrial
stromal cells in a previous study investigating endometriosis
(30). MDM2 is an oncogene that
promotes tumour occurrence and development predominantly by
inhibiting the p53 tumour suppressor activity (24). Jiang et al (31) found increased MDM2 mRNA and protein
levels with increasing EC grade.
Numerous genes associated with cell proliferation
were found to be regulated by miR-29c; however, the genes regulated
by miR-29c in each cell line suggested that the mechanism by which
cell proliferation is regulated may be different among cell
lines.
In Ishikawa and HEC1A cells, miR-29c overexpression
reduced the expression of genes associated with cell proliferation.
HBP1 was downregulated by miR-29c in both Ishikawa and HEC1A cells.
HBP1 is a transcriptional repressor and is associated with the
induction of cyclins involved in cellular proliferation (32). HBP1 knockdown arrests cells in
G0/1 phase, decreases those in S phase, but
does not affect apoptosis in neural progenitor cells (33).
SGK1 is a serine/threonine kinase that modulates
cancer cell growth and survival (34). SGK1 expression was reduced following
miR-29c overexpression in Ishikawa cells; however, these cells did
not exhibit significant changes in proliferation. SGK1 is increased
in EC compared with benign endometrial tissue, and its inhibition
via the small molecule inhibitor SI113 results in decreased cell
viability and increased apoptosis and autophagy (34).
SIRT1 is involved in cell proliferation, apoptosis
and metastasis (35). miR-29c
overexpression significantly reduced SIRT1 in HEC1A cells. There
are conflicting results published on the expression of SIRT1 in EC
(36,37). Functionally, SIRT1 enhances EC
growth, progression and chemo-resistance in EC cell lines in
vitro and in vivo (37).
SIRT1 is a confirmed target of miR-29c and its overexpression
recapitulates SIRT1 knockdown and suppresses hepatocellular
carcinoma cell growth (38).
VEGFA is upregulated in many cancer types including
EC, and VEGFA upregulation is associated with proliferation,
migration, differentiation, blood vessel formation and poor
prognosis (39). miR-29c
overexpression reduced VEGFA production in HEC1A cells. VEGFA is
increased in EC, and may play a role in angiogenesis during tumour
growth, and it has been identified as a target of miR-29b (11).
In AN3CA cells, miR-29c reduced the expression
levels of cell cycle and anti-apoptotic proteins, thus influencing
these processes, as assessed by flow cytometry. CDC42 is a
confirmed target of miR-29c (40)
and plays a role in cell cycle regulation, cell-cell adhesion,
migration and cancer progression (41). Following miR-29c overexpression, the
resulting loss of CDC42 in the AN3CA cells coincides with altered
G0/G1 and G2/M phase cell
populations for which CDC42 is known to be required (42). In cervical cancer, CDC42
overexpression promoted migration via the formation of cellular
filopodia (42,43), and overexpression of the miR-29a/b/c
family was able to reduce migration and invasion of glioma cell
lines (U87MG and U251) via the reduction of CDC42 (44). Other studies identified a correlation
between increased CDC42 levels and higher cancer stage (42,45,46).
MCL1 is an antiapoptotic protein. MCL1 is a direct
target of miR-29c, and miR-29c overexpression is able to knockdown
the expression of MCL1 in lymphoma (15). Immunohistochemistry analysis of
high-grade EC tumour samples identified that MCL1 is overexpressed
compared with benign cells (47). In
the present study, a significant reduction in MCL1 expression in
Ishikawa and AN3CA cell lines following miR-29c overexpression was
found, and this reduction was associated with the increased
apoptotic rate seen in the AN3CA cell line.
A number of factors identified in the results of the
present study, including ITGB1, CDC42, SIRT1 and VEGFA, can
regulate EC cell adhesion, migration and invasion; however, these
functions were not directly investigated in the present study. The
role of the other miR-29 family members (a and b) in regulating EC
should also be considered. Similarly to miR-29c, miR-29a regulates
EC cell proliferation and apoptosis (13) via TPX2 microtubule nucleation factor,
a gene which was not investigated in the present study.
Importantly, the therapeutic potential of each miR-29 family member
and their specific roles in EC requires further investigation.
In the present study, the endogenous levels of
miR-29c in HEC1A cells were not reduced, in contrast with the
Ishikawa and AN3CA cells. The expression level of miR-29c in HEC1A
was similar to the levels found in the non-cancerous endometrial
epithelial cells. Downregulation of miR-29c, identified in EC,
increased HEC1A proliferation and COL4A1 expression, but had no
effect on cell cycle. To the best of our knowledge, the present
study is the third study to identify COL4A1 as a target gene of
factors dysregulated in EC; in fact, COL4A1 production is regulated
by Kruppel-like factor 9 in HEC1A cells (48) and Galectin-7 in Ishikawa cells
(49). Collagen IV is the most
abundant structural component of cellular basement membranes,
involved in defining compartments that separate endothelial and
epithelial layers from the underlying mesenchyme (50). Collagen IV is necessary for cell
adhesion and acts by binding to cell surface receptors, mainly
integrin β1 (50). Within the
endometrium, collagen IV abundance changes across the menstrual
cycle and within endometrial compartments (51), and the loss of collagen IV within the
epithelial compartments is associated with primary infertility
(16). The direct role of COL4A1 in
EC remains to be elucidated.
In conclusion, miR-29c was downregulated in EC,
possibly resulting in increased proliferation and COL4A1
production. Following miR-29c overexpression in EC cell lines, a
reduction in proliferation and/or cell cycle progression were
identified in HEC1A and AN3CA cells. miR-29c target genes involved
in adhesion, migration, angiogenesis, proliferation and apoptosis
were also downregulated following miR-29c overexpression,
suggesting that miR-29c may be a potential therapeutic target for
EC. Finally, the precise mechanism by which miR-29c acts is likely
not identical among the three EC cell lines investigated,
highlighting the importance of molecular characterization of
tumours. The present study provided novel insights into specific
treatments for different disease stages of EC.
Acknowledgements
The authors would like to thank Dr Masashi Takamura,
University Hospital of Tokyo (Tokyo, Japan), for primary cell
culture preparations and Ms. Katarzyna Rainczuk, Hudson Institute
of Medical Research (Clayton, Australia), for technical assistance.
These data were presented at the Australian ESA-SRB meeting in
Adelaide, 19–22 August 2018 (abstract no. 61).
Funding
The present study was supported by the Victorian
Government's Operational Infrastructure Support Program. ED was
supported by The National Health and Medical Research Council
(Australia) Senior Research Fellowship (grant no. 550905).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MVS and ED conceived and designed the experiments,
critically analysed the data and reviewed the manuscript. MVS wrote
the manuscript, and contributed to data collection and analysis. EM
analysed the data and wrote the manuscript. MG contributed to data
collection and analysis and manuscript review. KN contributed to
data analysis. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Monash Health
Human Research and Ethics Committee (approval no. 06014C) and the
Victorian Cancer Biobank (project no. 13018). Informed consent was
obtained from each patient.
Patient consent for publication
Participant consent for the publication of research
generated from anonymous samples was obtained.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Soliman PT, Oh JC, Schmeler KM, Sun CC,
Slomovitz BM, Gershenson DM, Burke TW and Lu KH: Risk factors for
young premenopausal women with endometrial cancer. Obstet Gynecol.
105:575–580. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bilyk O, Coatham M, Jewer M and Postovit
LM: Epithelial-to-Mesenchymal Transition in the female reproductive
tract: From normal function to disease pathology. Front Oncol.
7:1452017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hong B, Le Gallo M and Bell DW: The
mutational landscape of endometrial cancer. Curr Opin Genet Dev.
30:25–31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Di Cristofano A and Ellenson LH:
Endometrial carcinoma. Annu Rev Pathol. 2:57–85. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Piulats JM, Guerra E, Gil-Martin M,
Roman-Canal B, Gatius S, Sanz-Pamplona R, Velasco A, Vidal A and
Matias-Guiu X: Molecular approaches for classifying endometrial
carcinoma. Gynecol Oncol. 145:200–207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Arend RC, Jones BA, Martinez A and
Goodfellow P: Endometrial cancer: Molecular markers and management
of advanced stage disease. Gynecol Oncol. 150:569–580. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim VN: MicroRNA biogenesis: Coordinated
cropping and dicing. Nat Rev Mol Cell Biol. 6:376–385. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schmitt MJ, Margue C, Behrmann I and Kreis
S: MiRNA-29: A microRNA family with tumor-suppressing and
immune-modulating properties. Curr Mol Med. 13:572–585. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Castilla MÁ, Moreno-Bueno G, Romero-Pérez
L, Van De Vijver K, Biscuola M, López-García MÁ, Prat J,
Matías-Guiu X, Cano A, Oliva E and Palacios J: Micro-RNA signature
of the epithelial-mesenchymal transition in endometrial
carcinosarcoma. J Pathol. 223:72–80. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen HX, Xu XX, Tan BZ, Zhang Z and Zhou
XD: Micro-RNA-29b inhibits angiogenesis by targeting VEGFA through
the MAPK/ERK and PI3K/Akt signaling pathways in endometrial
carcinoma. Cell Physiol Biochem. 41:933–946. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hiroki E, Akahira J, Suzuki F, Nagase S,
Ito K, Suzuki T, Sasano H and Yaegashi N: Changes in microRNA
expression levels correlate with clinicopathological features and
prognoses in endometrial serous adenocarcinomas. Cancer Sci.
101:241–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang T, Sui D, You D, Yao S, Zhang L,
Wang Y, Zhao J and Zhang Y: miR-29a-5p inhibits proliferation and
invasion and induces apoptosis in endometrial carcinoma via
targeting TPX2. Cell Cycle. 17:1268–1278. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ushakov DS, Dorozhkova AS, Babayants EV,
Ovchinnikov VY, Kushlinskii DN, Adamyan LV, Gulyaeva LF and
Kushlinskii NE: Expression of microRNA potentially regulated by AhR
and CAR in malignant tumors of the endometrium. Bull Exp Biol Med.
165:688–691. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mazzoccoli L, Robaina MC, Apa AG, Bonamino
M, Pinto LW, Queiroga E, Bacchi CE and Klumb CE: MiR-29 silencing
modulates the expression of target genes related to proliferation,
apoptosis and methylation in Burkitt lymphoma cells. J Cancer Res
Clin Oncol. 144:483–497. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Griffiths M, Van Sinderen M, Rainczuk K
and Dimitriadis E: miR-29c overexpression and COL4A1 downregulation
in infertile human endometrium reduces endometrial epithelial cell
adhesive capacity in vitro implying roles in receptivity.
Sci Rep. 9:86442019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Long M, Wan X, La X, Gong X and Cai X:
miR-29c is downregulated in the ectopic endometrium and exerts its
effects on endometrial cell proliferation, apoptosis and invasion
by targeting c-Jun. Int J Mol Med. 35:1119–1125. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marwood M, Visser K, Salamonsen LA and
Dimitriadis E: Interleukin-11 and leukemia inhibitory factor
regulate the adhesion of endometrial epithelial cells: Implications
in fertility regulation. Endocrinology. 150:2915–2923. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Winship A, Van Sinderen M, Rainczuk K and
Dimitriadis E: Therapeutically blocking Interleukin-11
Receptor-alpha enhances doxorubicin cytotoxicity in high grade type
I endometrioid tumours. Oncotarget. 8:22716–22729. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Van Sinderen M, Cuman C, Winship A,
Menkhorst E and Dimitriadis E: The chrondroitin sulfate
proteoglycan (CSPG4) regulates human trophoblast function.
Placenta. 34:907–912. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak K and Schmittgen T: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta DeltaC(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Licholai S, Szczekilk W and Sanak M:
miR-29c-3p is an effective biomarker of abdominal aortic aneurysm
in patients undergoing elective surgery. MicroRNA. 5:124–131. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu Z, Zou L, Gang M, Wu X, Huang F, Feng
T, Li S, Lin Q, He X, Liu Z and Cao X: Integrin b1 is a critical
effector in promoting metastasis and chemo-resistance of esophageal
squamous cell carcinoma. Am J Cancer Res. 7:531–542.
2017.PubMed/NCBI
|
|
24
|
Gupta A, Shah K, Oza MJ and Behl T:
Reactivation of p53 gene by MDM2 inhibitors: a novel therapy for
cancer treatment. Biomed Pharmacother. 109:484–492. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weigelt B, Warne PH, Lambros MB,
Reis-Filho JS and Downward J: PI3K pathway dependencies in
endometrial cancer cell lines. Clin Cancer Res. 19:3533–3544. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bhardwaj A, Singh H, Rajapakshe K,
Tachibana K, Ganesan N, Pan Y, Gunaratne PH, Coarfa C and Bedrosian
I: Regulation of miRNA-29c and its downstream pathways in
preneoplastic progression of triple-negative breast cancer.
Oncotarget. 8:19645–19660. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen G, Zhou T, Li Y, Yu Z and Sun L: p53
target miR-29c-3p suppresses colon cancer cell invasion and
migration through inhibition of PHLDB2. Biochem Biophys Res Commun.
487:90–95. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chuang TD and Khorram O: Mechanisms
underlying aberrant expression of miR-29c in uterine leiomyoma.
Fertil Steril. 105:236–245 e1. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
He B, Xiao YF, Tang B, Wu YY, Hu CJ, Xie
R, Yang X, Yu ST, Dong H, Zhao XY, et al: hTERT mediates gastric
cancer metastasis partially through the indirect targeting of ITGB1
by microRNA-29a. Sci Rep. 6:219552016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J, Gu L, Ni J, Hu P, Hu K and Shi YL:
MiR-183 Regulates ITGB1P Expression and Promotes Invasion of
Endometrial Stromal Cells. Biomed Res Int.
2015:3402182015.PubMed/NCBI
|
|
31
|
Jiang Z, Xu W, Dan G, Liu Y and Xiong J:
P53 and murine double mimute 2 (MDM2) expression changes and
significance in different types of endometrial lesions. Med Sci
Monit. 22:4786–4793. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bollaert E, de Rocca Serra A and Demoulin
JB: The HMG box transcription factor HBP1: A cell cycle inhibitor
at the crossroads of cancer signaling pathways. Cell Mol Life Sci.
76:1529–1539. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
He S, Yang S, Niu M, Zhong Y, Dan Gao,
Zhang Y, Ma H, Xiong W, Zhou M, Zhou Y, et al: HMG-box
transcription factor 1: A positive regulator of the G1/S transition
through the Cyclin-CDK-CDKI molecular network in nasopharyngeal
carcinoma. Cell Death Dis. 9:1002018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Conza D, Mirra P, Cali G, Tortora T,
Insabato L, Fiory F, Schenone S, Amato R, Beguinot F, Perrotti N
and Ulianich L: The SGK1 inhibitor SI113 induces autophagy,
apoptosis, and endoplasmic reticulum stress in endometrial cancer
cells. J Cell Physiol. 232:3735–3743. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Palmirotta R, Cives M, Della-Morte D,
Capuani B, Lauro D, Guadagni F and Silvestris F: Sirtuins and
cancer: role in the epithelial-mesenchymal transition. Oxid Med
Cell Longev. 2016:30314592016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bartosch C, Monteiro-Reis S, Almeida-Rios
D, Vieira R, Castro A, Moutinho M, Rodrigues M, Graça I, Lopes JM
and Jerónimo C: Assessing sirtuin expression in endometrial
carcinoma and non-neoplastic endometrium. Oncotarget. 7:1144–1154.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Asaka R, Miyamoto T, Yamada Y, Ando H,
Mvunta DH, Kobara H and Shiozawa T: Sirtuin 1 promotes the growth
and cisplatin resistance of endometrial carcinoma cells: A novel
therapeutic target. Lab Invest. 95:1363–1373. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bae HJ, Noh JH, Kim JK, Eun JW, Jung KH,
Kim MG, Chang YG, Shen Q, Kim SJ, Park WS, et al: MicroRNA-29c
functions as a tumor suppressor by direct targeting oncogenic SIRT1
in hepatocellular carcinoma. Oncogene. 33:2557–2567. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mahecha AM and Wang H: The influence of
vascular endothelial growth factor-A and matrix metalloproteinase-2
and −9 in angiogenesis, metastasis, and prognosis of endometrial
cancer. Onco Targets Ther. 10:4617–4624. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Park SY, Lee JH, Ha M, Nam JW and Kim VN:
miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat
Struct Mol Biol. 16:23–29. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qadir MI, Parveen A and Ali M: Cdc42: Role
in cancer management. Chem Biol Drug Des. 86:432–439. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ye H, Zhang Y, Geng L and Li Z: Cdc42
expression in cervical cancer and its effects on cervical tumor
invasion and migration. Int J Oncol. 46:757–763. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Feng Y, Fang S and Li M: Expression of
P21-activated kinase 1 and cell division control protein 42 homolog
correlates with clinicopathological features and prognosis in
cervical carcinoma. J Obstet Gynaecol Res. 42:860–869. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shi C, Ren L, Sun C, Yu L, Bian X, Zhou X,
Wen Y, Hua D, Zhao S, Luo W, et al: miR-29a/b/c function as
invasion suppressors for gliomas by targeting CDC42 and predict the
prognosis of patients. Br J Cancer. 117:1036–1047. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang D, Zhang Y, Cheng Y, Hong L, Wang C,
Wei Z, Cai Q and Yan R: High expression of cell division cycle 42
promotes pancratic cancer growth and predicts poor outcome of
pancreatic cancer patients. Dig Dis Sci. 62:958–967. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kamai T, Yamanishi T, Shirataki H, Takagi
K, Asami H, Ito Y and Yoshida K: Overexpression of RhoA, Rac1 and
Cdc42 GTPases is associated with progression in testicular cancer.
Clin Cancer Res. 10:4799–4805. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Konno Y, Dong P, Xiong Y, Suzuki F, Lu J,
Cai M, Watari H, Mitamura T, Hosaka M, Hanley SJ, et al:
MicroRNA-101 targets EZH2, MCL-1 and FOS to suppress proliferation,
invasion and stem cell-like phenotype of aggressive endometrial
cancer cells. Oncotarget. 5:6049–6062. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Simmen F, Su Y, Xiao R, Zeng Z and Simmen
RC: The Kruppel-like factor 9 (KLF9) network in HEC-1-A endometrial
carcinoma cells suggests the carcinogenic potential of
dys-regulated KLF9 expression. Reprod Biol Endocrinol. 6:412008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Menkhorst E, Griffiths M, Van Sinderen M,
Rainczuk K, Niven K and Dimitriadis E: Galectin-7 is elevated in
endometrioid (type I) endometrial cancer and promotes cell
migration. Oncol Lett. 16:4721–4728. 2018.PubMed/NCBI
|
|
50
|
Van Agtmael T and Bruckner-Tuderman L:
Basement membranes and human disease. Cell Tissue Res. 339:167–188.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kelly FD, Tawia SA and Rogers PA:
Immunohistochemical characterization of human endometrial
microvascular basement membrane components during the normal
menstrual cycle. Hum Reprod. 10:268–276. 1995. View Article : Google Scholar : PubMed/NCBI
|