Introduction
Acute myeloid leukemia (AML) is a myeloid stem cell
tumor characterized by abnormal myeloid cell proliferation, which
inhibits the viability of normal cells. M2 is a subtype of AML, as
classified by the French-American-British classification (1), constituting 10–15% of AML cases. One of
the hallmarks of M2 is the formation of a fusion protein, AML1-ETO,
due to a translocation of chromosome 8 to chromosome 21.
Chemotherapy and hematopoietic stem cell transplantation are the
main treatments of AML. However, to date, the therapeutic effects
are insufficient. For patients <60 years old, the 5-year overall
survival (OS) rate is ~40%. However, for the majority of patients
with AML (>60 years old), the 5-year OS rate is only ~10–20%
(2,3). Furthermore, for patients with M2 AML,
clinical studies have shown that the median survival time was <2
years, with a 5-year OS rate of <40% (4). Therefore, the development of novel and
effective therapies for AML is urgently required, particularly for
patients with type M2.
The human P53 gene is located at 17P13.1. It
is considered to be a classical tumor suppressor gene and is
closely associated with human tumorigenesis (5,6). The p53
protein (a product of P53 gene expression) acts as a
transcription factor that mediates DNA repair and induces cell
cycle arrest and apoptosis, thereby inhibiting the formation of
tumor cells (7,8). However, the P53 gene can be
mutated, rearranged or deleted. This can cause it to lose its
anticancer function and become an oncogene, promoting the formation
of tumors (9,10). Animal experiments have demonstrated
that P53-deficient mice spontaneously develop cancer. It was
also revealed that >50% of patients with cancer carry mutations
in the P53 gene (11).
Similarly, in hematological malignancies, the P53 mutant
occurs in 11.1% of cases, according to version R15 of the
International Agency for Research on Cancer database (12). The overexpression of cellular mutant
P53 in therapy-related myelodysplastic syndrome and AML
(13) was found to be present in ~63
and 75% of patients, respectively, and was associated with poorer
OS (14). In a mouse model of
P53-deficiency, re-inducing the expression of P53
caused a decrease in tumor volume, indicating that P53
remains effective against established tumors (15). A number of studies have also
demonstrated that restoring the function of mutant P53 can
reactive its antitumor effect (16,17).
Restoring mutant P53 may be a hopeful treatment for
hematological tumors.
Natural isothiocyanates exist in all types of
cruciferae, including broccoli, cabbage and watercress. PHI is an
isothiocyanate derivative, which is known to be an apoptotic
inducer and cell proliferation inhibitor (18). Our previous study demonstrated that
this compound suppressed proliferation of the leukemia HL-60 cell
line by inducing apoptosis in vitro and in vivo.
However, it spared normal cells and tissues (19,20). PHI
exerted an effect on PC3 prostate cancer cells by inhibiting the
Akt pathway (21). Furthermore, it
inhibited cell proliferation and induced apoptosis in
hepatocellular carcinoma cells by inducing the accumulation of
acetylated histones H3 and H4, possibly resulting in the loss of
function of P15 (22,23). However, the effect of PHI on other
hematological tumors is unknown, and whether the P53 pathway
is involved in the PHI mechanism is also unclear.
In the present study, the effect of PHI was
evaluated in several hematological tumor cell lines. Cell
proliferation, cycle and apoptosis were investigated. The mechanism
of PHI in inhibiting the growth of Kasumi-1 M2 cells was examined,
with a focus on the P53 pathway. The results revealed that
PHI inhibited M2 cell growth by inducing cell apoptosis and cell
cycle arrest, involving the restoration of mutant P53 and
reactivation of the P53 signaling pathway.
Materials and methods
Chemicals and reagents
All reagents and chemicals were purchased from
Sigma-Aldrich (Merck KGaA), unless stated otherwise. PHI was from
Abcam, fetal bovine serum from Invitrogen (Thermo Fisher
Scientific, Inc.) and RPMI-1640 medium from Gibco (Thermo Fisher
Scientific, Inc.). Primary antibodies against cleaved caspase 3
(cat. no. #9664), cleaved caspase 8 (cat. no. #9748), cleaved
caspase 9 (cat. no. #7237), cleaved poly (ADP-ribose) polymerase
(PARP; cat. no. #5625), p21 (cat. no. #2947), p53 (cat. no. #2527),
Bcl-2 (cat. no. #4223), Bax (cat. no. #5023), Fas (cat. no. #8023),
α-tubulin (cat. no. #3873) and β-actin (cat. no. #3700) were
purchased from Cell Signaling Technology, Inc. The horseradish
peroxidase-conjugated secondary antibodies (goat anti-rabbit IgG
and goat anti-mouse IgG) and pifithrin-α were purchased from
Beyotime Institute of Biotechnology. The methylcellulose medium was
purchased from Stemcell Technologies, Inc.
Cell culture
The human Kasumi-1 and SKNO-1 AML M2 cell lines,
HL-60, NB4 and HT93 AML M3 cell lines, MV4-11 and THP-1 AML M5 cell
lines, HEL AML M6 cells, KG-1 AML cells, K-562 and MEG-01 chronic
myeloid leukemia cell lines, U266 myeloma cells, Raji Burkitt
lymphoma cells, Jurkat acute lymphoblastic leukemia cells, and
MOLT-4 and U-937 histiocytic lymphoma cell lines were purchased
from the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). The cells were cultured in RPMI-1640 medium with
10–20% fetal bovine serum at 37°C with 5% CO2.
Cell proliferation assay
Cell proliferation was assessed using a Cell
Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies,
Inc.). The 16 types of hematological tumor cell were seeded at
2×104 cells/well in 96-well plates and incubated with
increasing concentrations of PHI (5, 10, 20 and 40 µmol/l) at 37°C
for 24 h in an incubator containing 5% CO2; the Kasumi-1
and SKNO-1 cells were further incubated with 10 µmol/l PHI for 12,
24, 48 and 72 h. The cells in the control group were incubated with
medium containing the same volume of dimethyl sulfoxide (DMSO). The
CCK-8 solution (10 µl) was then added to each well, and the plates
were incubated for 1 h at 37°C. The optical density of cells at 450
nm was measured in a microplate reader (Thermo Fisher Scientific,
Inc.). The half maximal inhibitory concentration (IC50)
of PHI was calculated using SPSS version 13.0 (SPSS, Inc.).
Cell apoptosis detection
The Kasumi-1 and SKNO-1 cells were seeded at
2×106 cells/well in 6-well plates and treated with PHI
at concentrations of 5, 10, 20 and 40 µmol/l for 24 h. The same
volume of DMSO was used as a control. The cells were incubated at
37°C in an incubator containing 5% CO2. After being
harvested, the cells were washed in PBS and resuspended in 200 µl
binding buffer. The cells were then incubated with 5 µl Annexin
V-FITC solution and 5 µl propidium iodide (PI), both from BD
Biosciences, at room temperature for 15 min in the dark. The
apoptosis of cells was analyzed by flow cytometry. Data were
analyzed using FlowJo 10.4.1 (FlowJo LLC). All experiments were
performed in triplicate.
Cell cycle analysis
The Kasumi-1 cells were seeded at 2×106
cells/well in 6-well plates and incubated with increasing
concentrations of PHI (1.25, 2.5 and 5 µmol/l) at 37°C for 24 h in
an incubator containing 5% CO2. The same volume of DMSO
was used as a control. After being harvested and washed in PBS, the
cells were fixed using precooled 70% methanol at 4°C for 2 h. The
cells were then washed in PBS and stained with PI (50 mg/ml) at
37°C for 30 min. Cell cycle analysis was performed using flow
cytometry. Data were analyzed using FlowJo 10.4.1 (FlowJo LLC). All
experiments were performed in triplicate.
Colony-formation assay
The Kasumi-1 cells (500 cells/well) were seeded into
6-well plates and cultured in semi-solid RPMI-1640 medium
consisting of 1% methylcellulose and 20% FBS with PHI (5, 10, 20
and 40 µmol/l) at 37°C for 2 weeks. The same volume of DMSO was
used as the control. The numbers of clones were counted under a
light microscope. Images of the clones were captured using a camera
without magnification. All experiments were performed in
triplicate.
Establishment of an AML xenograft
mouse model and PHI treatment
Male athymic nude mice (BALB/c) aged 4–6 weeks and
weighing 18–22 g were purchased from Shanghai SLAC Laboratory
Animal Co., Ltd. and were housed under specific pathogen-free
conditions. A total of 8 mice in the study were in rooms with
12:12-h dark/light cycle (lights on at 07:00), housed with
wood-chip bedding and given ad libitum access to food and
water. There is a maximum of 5 mice per cage. And the room
temperature for mouse housing is maintained to be 22°C. 8 mice were
grafted with 1×107 Kasumi-1 cells via subcutaneous
injection into the right flank. The xenograft model was
established, and tumors were continually measured with digital
calipers until they reached a volume of >0.5 cm3. The
mice were then randomized into two cohorts (n=4 each): Group A,
injected intraperitoneally with 200 µl PHI (40 µmol/l) once a day;
and Group B (control), injected intraperitoneally with 200 µl of
DMSO once a day. Daily measurements of tumors and animal weight
were performed. The tumor volume was calculated as ab2/2
(a, long diameter; b, short diameter). No animals presented with
multiple tumors. Following a 14-day treatment period, the mice were
sacrificed by cervical dislocation. Death of the mice was confirmed
by ascertaining cardiac and respiratory arrest. Necropsy was then
performed and the xenograft tumor was excised.
Hematoxylin and eosin (H&E)
staining
The xenograft samples were fixed in 4% formaldehyde
in PBS for 6 h at 37°C and then embedded in paraffin. The paraffin
sections (4–6 µm) were adhered to glass slides pretreated with
0.01% aqueous solution of poly-L-lysin. The slides were then
stained with hematoxylin for 40 sec and with eosin for 30 sec. The
tissue slides were examined under a light microscope following
mounting with mounting medium.
Detection of xenograft apoptosis using
a TUNEL assay
Apoptosis of the xenograft cells was detected using
a TUNEL assay with an in situ cell death detection kit
(Wuhan Boster Biological Technology, Ltd.). Following treatment
with protease K, the sections of xenograft tissue were treated with
dUTP tagged with digoxin for 2 h under a reaction of terminal
deoxynucleotidyl transferase, in order to promote transfer to the
3′-OH end of the DNA fragment. The streptavidin-biotin complex was
added for 30 min. Diaminobenzidine (DAB) was then used for
staining. Cells exhibiting yellow nuclei were positive, as examined
under a light microscope. The apoptotic cells were counted at ×400
magnification, and the apoptotic index (AI) was calculated using
the following formula: AI = number of apoptotic cells/numbers of
total cells ×100%. All experiments were performed in
triplicate.
Treatment with pifithrin-α
The Kasumi-1 cells were divided into four groups:
PHI group, cells treated with 10 µmol/l PHI for 24 h; pifithrin-α +
PHI group, cells treated with 10 µmol/l pifithrin-α for 30 min, and
then 10 µmol/l PHI for 24 h; pifithrin-α group, cells treated with
10 µmol/l pifithrin-α for 24 h; and control group, cells treated
with an equal volume of DMSO for 24 h. The grouped Kasumi-1 cell
were seeded at 5×106 cells/well in 10 cm culture dishes
and incubated at 37°C with 5% CO2.
Western blot analysis
The cells were lysed in RIPA buffer containing 150
mM NaCl, 50 mM Tris-base, 5 mM EDTA, 1% NP-40, 25% deoxycholate (pH
7.4), according to methods previously described (24). The protein concentrations of the
lysates were detected using a Bradford protein assay. The proteins
(20 µg) were separated on a 10 or 12% gel by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred onto
PVDF membranes (Merck KGaA). Following blocking with 5% skimmed
milk at 37°C for 1 h, the membrane was incubated with primary
antibodies (1:1,000 dilution) overnight at 4°C. The membrane was
then incubated with horseradish peroxidase-conjugated secondary
antibodies (1:500 dilution) for 2 h at 37°C. The signals were
detected using an enhanced chemiluminescence kit (Thermo Fisher
Scientific, Inc.). The intensity of the bands was quantified with
ImageJ software (version 1.520; National Institutes of Health).
β-actin and α-tubulin served as internal controls.
Statistical analysis
Student's t-test was used for the statistical
analysis between the control and treated groups. The statistical
differences between multiple groups were detected using one-way
analysis of variance followed by Tukey's post hoc test. The
comparative data are expressed as the mean ± SD of at least three
independent experiments. P<0.05 was considered to indicate a
statistically significant difference.
Results
PHI inhibits the proliferation of
hematological tumor cell lines
A total of 16 hematological tumor cell lines were
treated with 10, 20 and 40 µmol/l PHI for 24 h. Compared with the
control, cell proliferation was inhibited in a dose-dependent
manner in all hematological tumor cells. (Fig. 1A and B). The results demonstrated
that Kasumi-1 and SKNO-1 cells were more sensitive to PHI than the
other cell lines, providing the lowest IC50 values
(7.45±0.89 and 7.86±0.77 µmol/l, respectively; both P<0.05,
compared with the other cell line). No significant difference was
observed between the IC50 values of the two cells types
(P>0.05) (Fig. 1C).
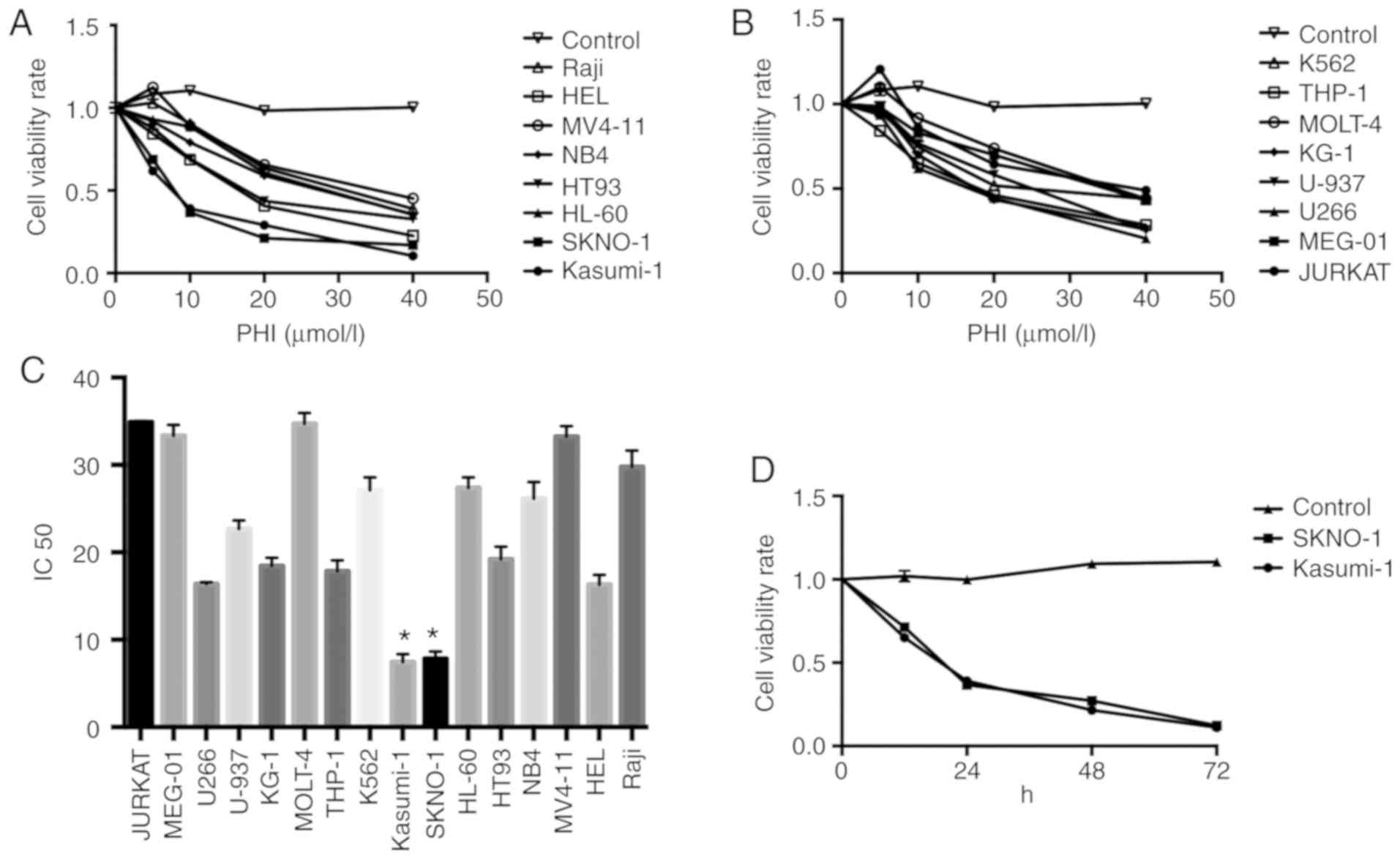 | Figure 1.PHI inhibits the proliferation of
hematological tumor cell lines. Following treatment with 10, 20 and
40 µmol/l PHI for 24 h, the cell viability rate of (A) Raji, HEL,
MV4-11, NB4, HT93, HL-60, SKNO-1, Kasumi and (B) K562, THP-1,
MOLT-4, KG-1, U-937, U266, MEG-01 and Jurkat hematological tumor
cell lines were measured. (C) The IC50 of the 16 cell
lines is presented in the histogram. Compared with the other cell
lines, Kasumi-1 and SKNO-1 cells exhibited the lowest
IC50. *P<0.05. (D) Cell viability rates of Kasumi-1
and SKNO-1 cells following treatment with 10 µmol/l PHI for 12, 24,
48 and 72 h. PHI inhibited the viability of Kasumi-1 and SKNO-1
cells in a time-dependent manner. P<0.05. PHI, phenylhexyl
isothiocyanate; IC50, half maximal inhibitory
concentration. |
Subsequently, the Kasumi-1 and SKNO-1 cells were
treated with 10 µmol/l PHI for 12, 24, 48 and 72 h. The viability
rates of the Kasumi-1 cells were 68.3±1.1, 35.6±1.1, 21.3±2.2 and
12.1±2.1%, respectively, at these time points. For the SKNO-1
cells, the viability rates were 71.2±1.6, 36.3±1.6, 22.6±2.1 and
12.7±2.5%, respectively, indicating a time-dependent effect
(P<0.05). No significant difference was noted between the
Kasumi-1 and SKNO-1 groups (P>0.05) (Fig. 1D).
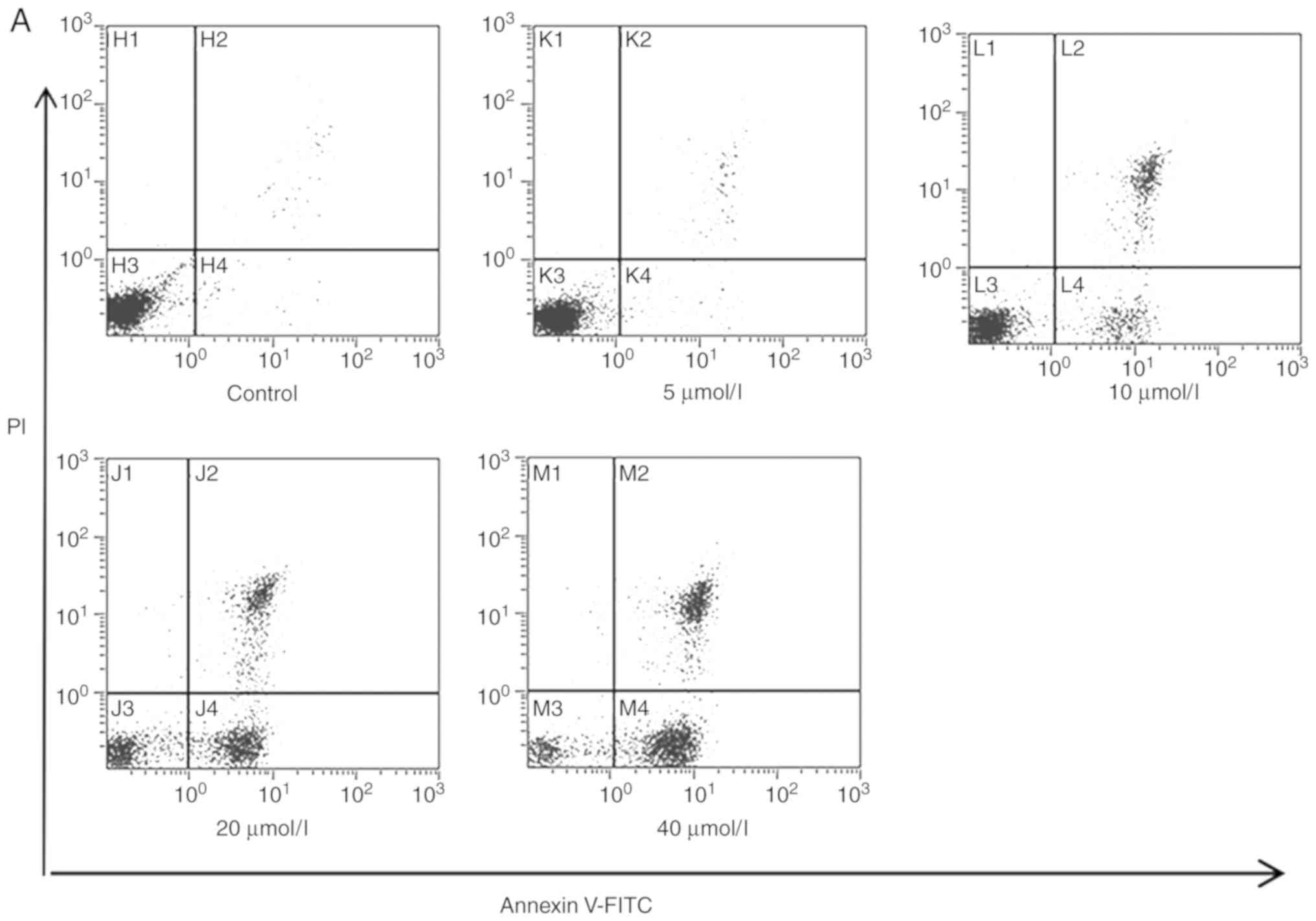
PHI induces Kasumi-1 and SKNO-1 cell
apoptosis
Following treatment of the Kasumi-1 and SKNO-1 cells
with 0, 5, 10, 20 and 40 µmol/l PHI for 24 h, cell apoptosis was
observed to increase in a dose-dependent manner. In the Kasumi-1
group, the early apoptotic rates were 1.3±0.03, 3.1±0.13,
34.2±0.98, 41.2±1.12 and 60.5±1.7%, respectively, following
exposure to the aforementioned concentrations of PHI (Fig. 2A and C). In the SKNO-1 cells, the
early apoptotic rates were 2.5±0.05, 4.5±0.12, 10.7±0.82, 59.8±2.3
and 69.7±2.15%, respectively (P<0.05; Fig. 2B and D).
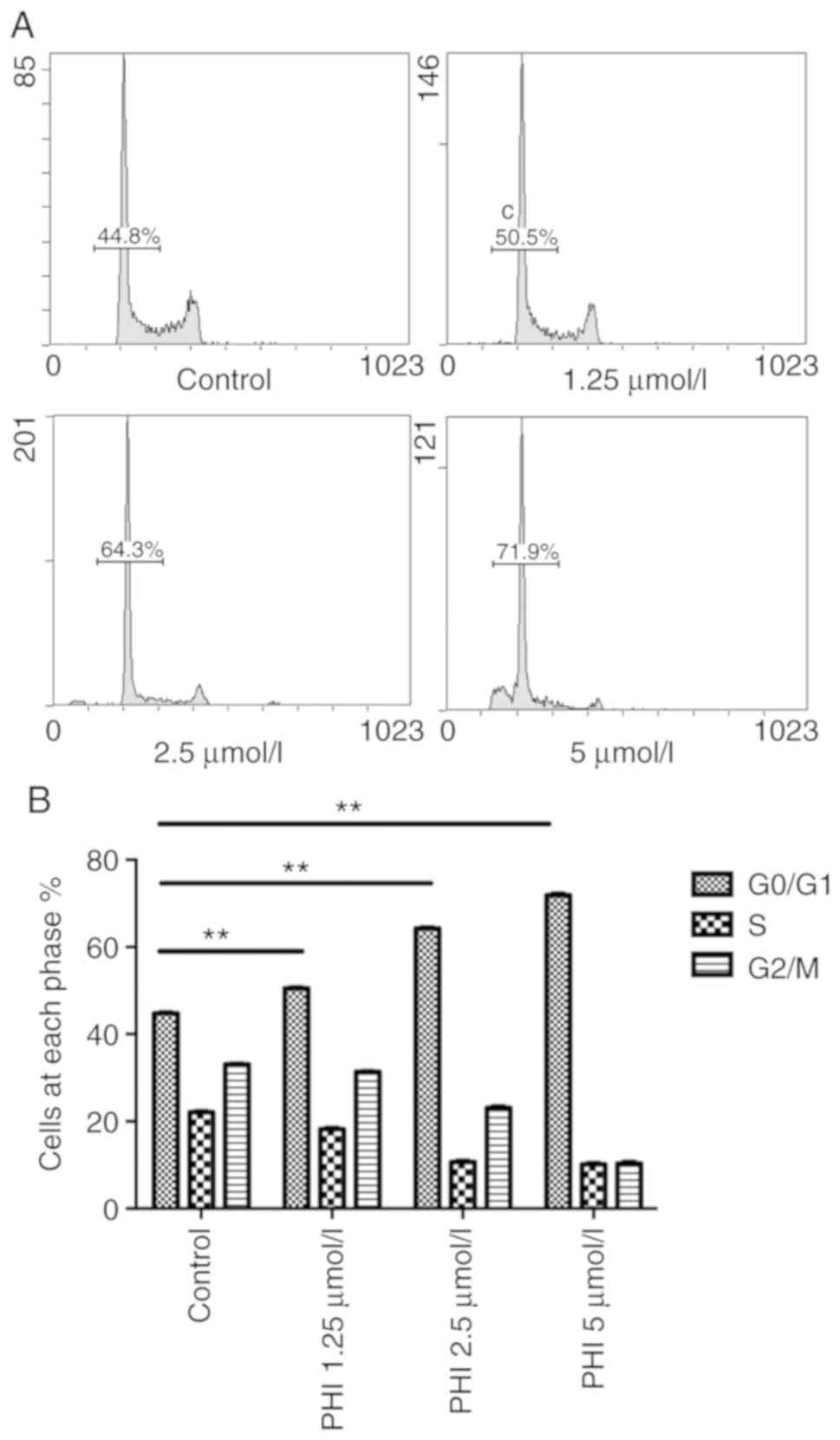
PHI induces cell cycle arrest at the
G0/G1 phase
Following treatment of the Kasumi-1 cells with PHI,
cell cycle was arrested at the G0/G1 phase.
The percentage of cells in the G0/G1 phase
was 44.8±1.2, 50.5±1.8, 64.3±3.1 and 71.9±1.6% following exposure
to 0, 1.25, 2.5 and 5 µmol/l PHI, respectively (Fig. 3A). Compared with the control, the
increases in cell numbers in this cell cycle phase were
statistically significant (all P<0.01; Fig. 3B). The lowest percentage of cells in
the S phase and the peak in apoptosis were observed at the
concentration of 5 µmol/l PHI.
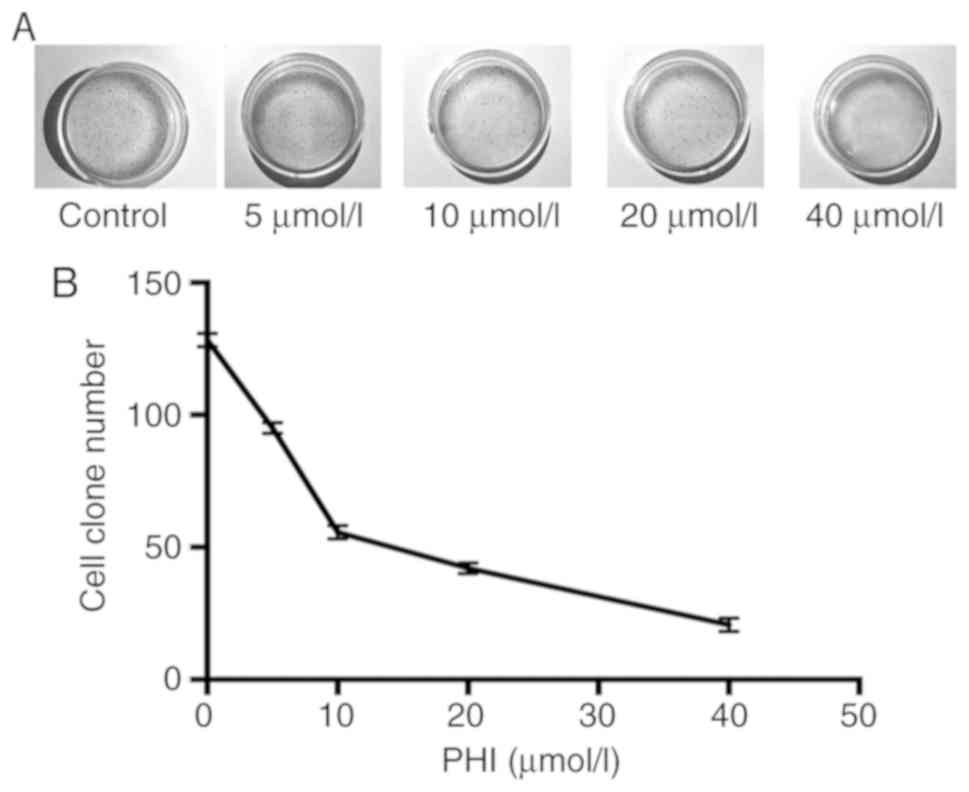
PHI inhibits colony formation in
Kasumi-1 cells
The colony-formation ability of Kasumi-1 cells was
inhibited following treatment with PHI. The numbers of cell
colonies were 92±3, 52±4, 38±4 and 21±2 following treatment with 5,
10, 20 and 40 µmol/l PHI (Fig. 4A and
B). Compared with the control (130±6 colonies), the decrease in
colony-formation ability was statistically significant
(P<0.01).
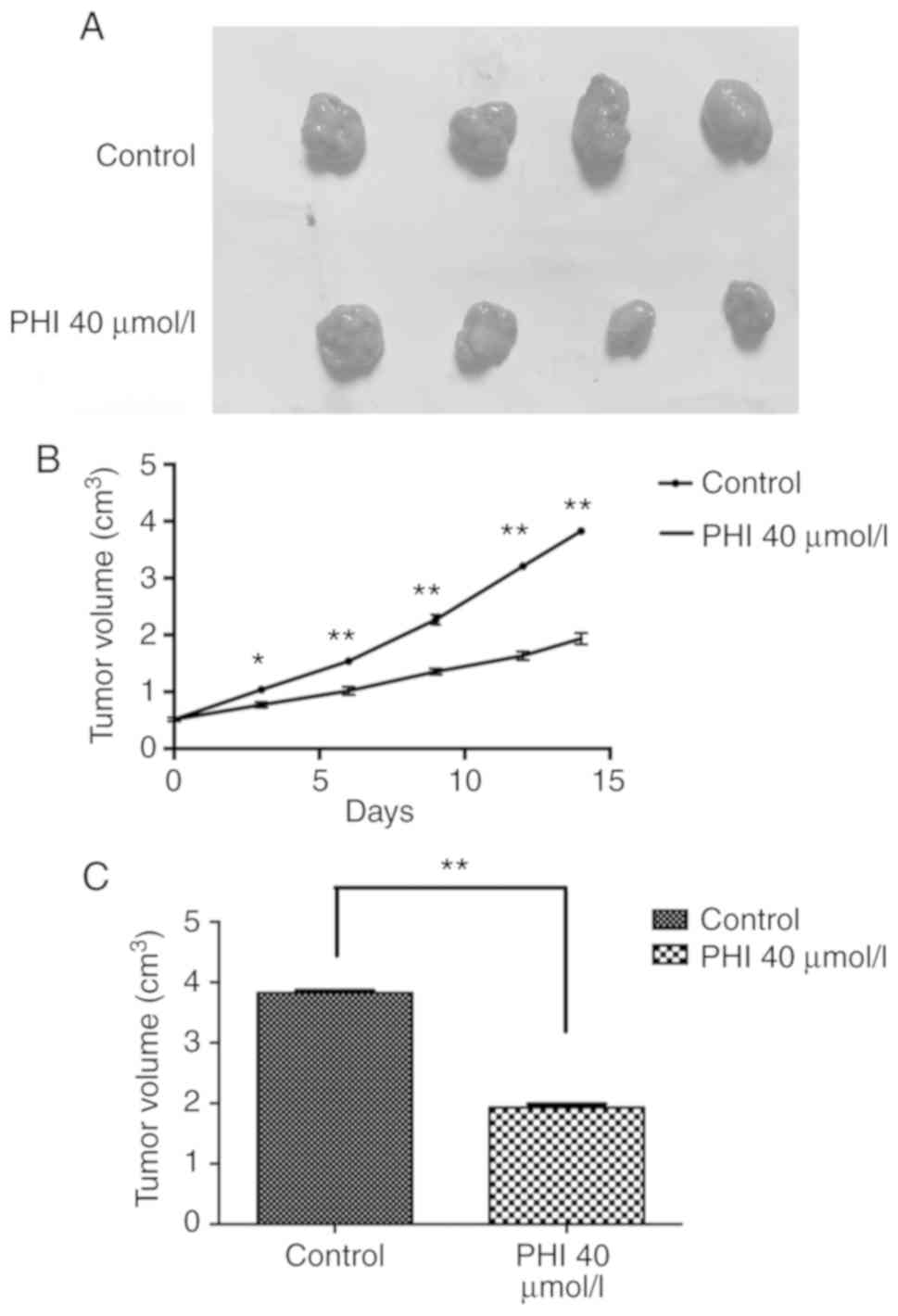
PHI inhibits the growth of Kasumi-1
×enografts
During treatment with PHI, no obvious differences
were observed in the feeding behavior, mental state or weight of
the mice between the control and PHI groups. The tumor volumes were
0.77±0.12, 1.02±0.14, 1.35±0.11, 1.65±0.21 and 1.91±0.18
cm3 on days 3, 6, 9, 12 and 14, respectively, following
administration of 40 µmol/l PHI. The tumor volumes in the control
group were 1.05±0.15, 1.55±0.12, 2.28±0.18, 3.20±0.12 and 3.82±0.12
cm3, respectively, at these time points. From day 3
onward, a smaller size of xenograft tumor was observed in the PHI
group compared with that in the control (all P<0.05; Fig. 5A-C).
PHI induces apoptosis of Kasumi-1
cells in vivo
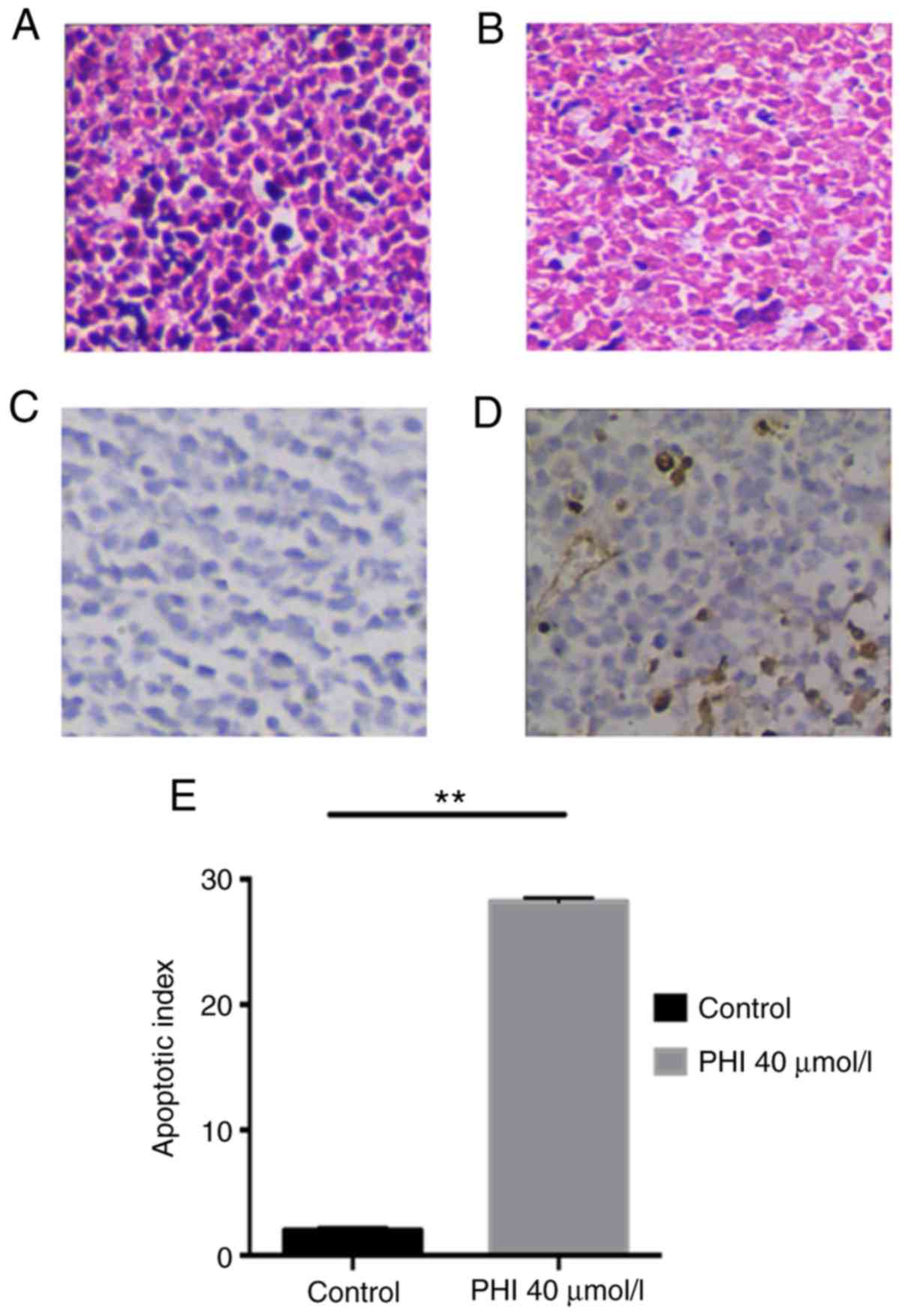
Following 14 days of treatment, the xenograft tumor
was removed and sliced for staining. Using microscopy and H&E
staining, the tumors of the control group were observed to have
grown markedly in a solid trabecular or nested arrangement; the
tumor cells exhibited a large, polygonal or oval shape, and
hyperchromatic nuclei were visible (Fig.
6A). Following exposure to 40 µmol/l PHI, the tumor cells
shrunk and were loosely arranged. Irregular liquefactive necrosis,
karyopyknosis or even loss of nuclei, and connective tissue
proliferation were observed (Fig.
6B). A TUNEL assay revealed that the AI was 28.5±0.5 in the PHI
group, but apoptosis was absent in the controls. The difference
between the two groups was significant (P<0.01; Fig. 6C-E).
PHI induces apoptosis by activating mitochondrial
and death receptor pathways in Kasumi-1 cells. Following treatment
of the Kasumi-1 cells with 5, 10, 20 and 40 µmol/l PHI for 24 h,
the expression levels of apoptosis-associated proteins cleaved
caspases 3, 8 and 9, and cleaved PARP, were increased in a
dose-dependent manner. The expression of cleaved caspase 9 began to
rise at the PHI concentration of 10 µmol/l. However, the expression
of cleaved caspases 3 and 8 and PARP began to rise at 5 µmol/l PHI
(Fig. 7A and C). Treatment with this
compound downregulated Bcl-2, and upregulated Bax, increasing the
Bax/Bcl-2 ratio (Fig. S1). The
expression levels of p53, Fas and p21 were also increased following
exposure to PHI. All of these changes occurred in a dose-dependent
manner (Fig. 7B and D).
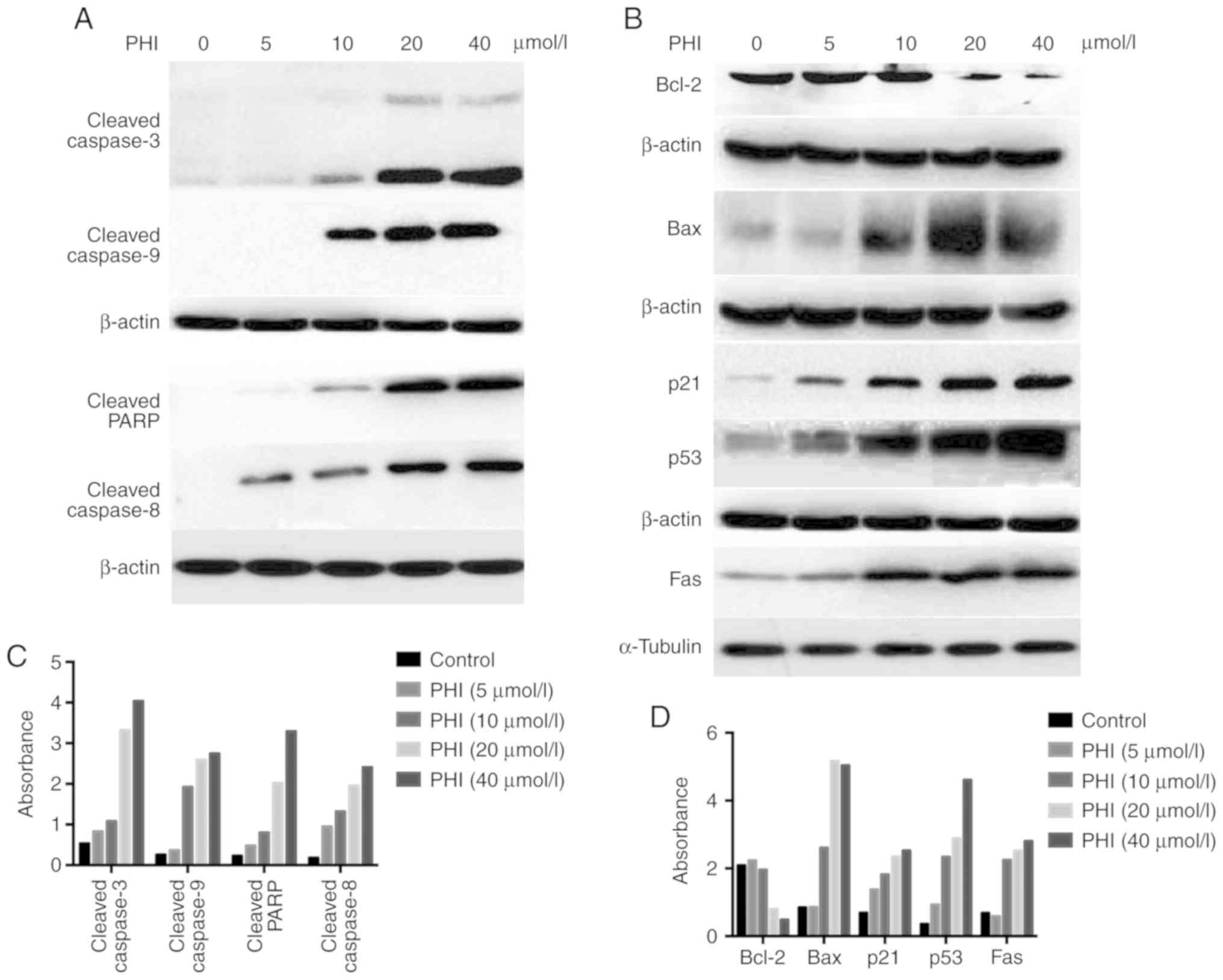 | Figure 7.PHI regulates the expression of
apoptosis-associated proteins. Following treatment with 5, 10, 20
or 40 µmol/l PHI for 24 h, apoptosis-associated proteins in
Kasumi-1 cells were detected by western blotting. The absorbance
values of the protein bands for (A) cleaved caspases 3, 6, and 9
and PARP and for (B) Bcl-2, Bax, p21, p53 and Fas are shown in the
histograms. (C) Expression of cleaved caspase 9 began to rise at
the concentration of 10 µmol/l, and the expression of cleaved
caspases 3 and 8, and PARP began to rise at 5 µmol/l. (D) PHI
downregulated Bcl-2 and upregulated Bax, resulting in an increased
ratio of Bax/Bcl-2. The expression of p53, Fas and p21 were also
increased following exposure to PHI. All changes occurred in a
dose-dependent manner. PHI, phenylhexyl isothiocyanate; PARP, poly
(ADP-ribose) polymerase. |
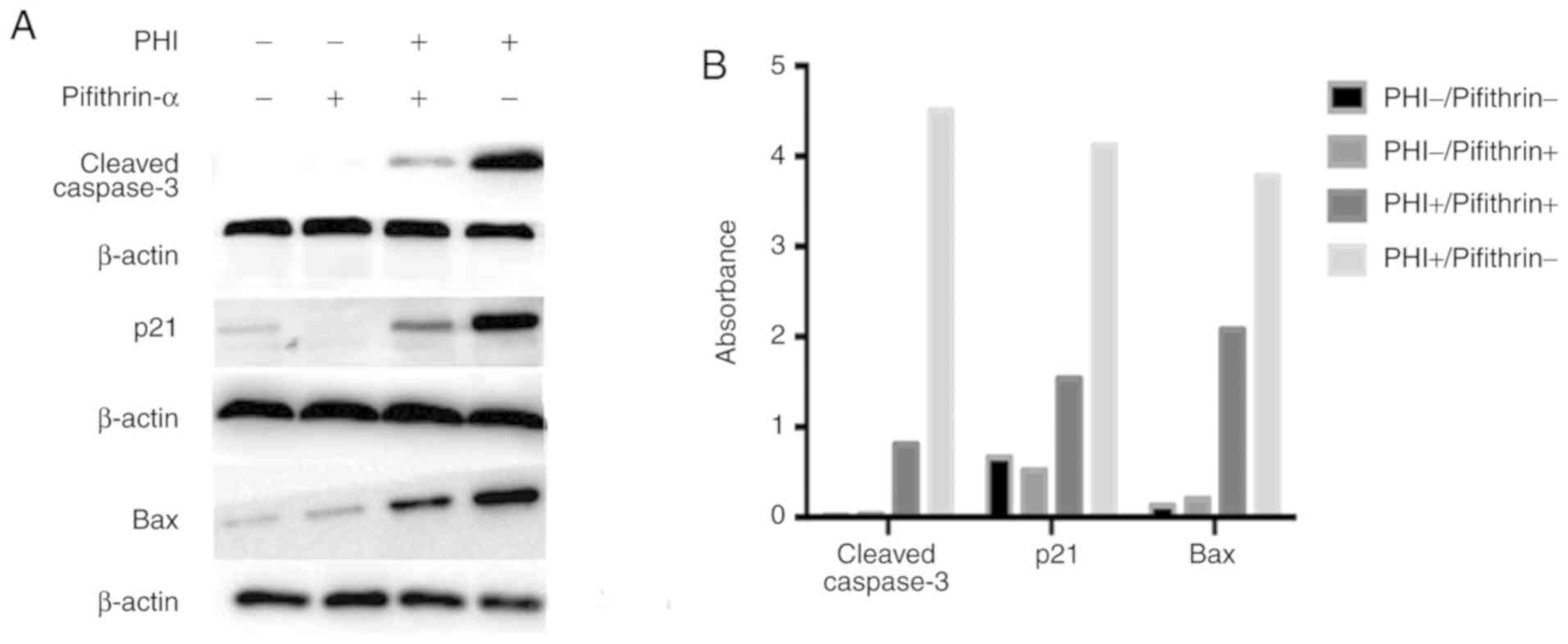
PHI restores mutant P53 and leads to cell apoptosis.
To further confirm whether P53 is key in the PHI-induced
apoptosis in Kasumi-1 cells, pifithrin-α (a p53 inhibitor) was used
to investigate PHI-induced changes in p21, Bax (two downstream
factors of P53) and cleaved caspase 3. Compared with the
control group, the pifithrin-α group had no effect on the
activation of cleaved caspase 3, but had marginally decreased
expression levels of p21, and no obvious difference in the
expression of Bax. Treatment in the PHI group significantly
improved the expression levels of cleaved caspase 3, p21 and Bax.
Compared with the PHI group, the pifithrin-α + PHI group exhibited
notable increases in the expression of cleaved caspase 3, p21 and
Bax (Fig. 8A and B).
Discussion
Previously, PHI has been demonstrated to affect
HL-60 leukemia, PC3 prostate cancer and hepatocellular carcinoma
cell lines. In the present study, PHI was demonstrated to exhibit a
more marked inhibitory effect on M2 cell lines compared with other
types of hematological tumor cell. This compound induced cell cycle
arrest at the G0/G1 phase and inhibited
colony formation in the M2 cells. Further evidence revealed that
PHI induced the apoptosis of M2 cells in vitro and in
vivo.
The pathways of apoptosis mainly include the
mitochondrial and the receptor signaling pathways. In the
mitochondrial pathway, numerous proteins, including Bcl-2, Bax,
induced myeloid leukemia cell differentiation protein Mcl-1 and
BH3-interacting domain death agonist, are activated. Subsequently,
mitochondria release cytochrome c, which activates caspases
9 and 3 (25). Reactive oxygen
species (ROS) have been tightly linked to activation of the
mitochondrial pathway. ROS, including H2O2
and superoxide, can cause the release of cytochrome c from
mitochondria and the induction of apoptosis through the
mitochondrial pathway (26). The
death receptor pathway involves Fas and tumor necrosis factor
receptor superfamily member 10B, and activates caspases, including
caspases 8 and 10 (27). Caspase-3
is further activated; this is a protein-cutting enzyme that cleaves
a series of important enzymes, including PARP, eventually leading
to cell apoptosis (28). The present
data showed that PHI led to the cleavage of caspases 8 and 9,
resulting in the cleavage of caspase 3 and PARP. This indicates
that PHI induced apoptosis through the mitochondrial and death
receptor pathways. Furthermore, treatment enhanced the expression
of Fas, indicating that the Fas/Fas ligand apoptotic pathway may
also be involved in PHI-induced apoptosis. To determine whether ROS
is involved in the mitochondrial pathway, further investigations
will be performed in the future.
The P53 gene is a classic tumor-suppressor
gene, and the p53 protein is the product of its translation. Its
main function is to detect the integrity of the cell genome and
repair DNA damage (29). If cells
cannot be repaired, P53 can be activated by various
signaling pathways and helps the cell to remain stable without
degradation. Furthermore, it can enhance the transcription of
downstream genes, including cyclin-dependent kinase inhibitor
P21 and apoptotic protein BAX. This further induces
cell cycle arrest, cell apoptosis and aging, and inhibits
angiogenesis (30).
P53 mutations are common in hematological
malignancies (12–14). The p53 protein is composed of an
N-terminal transcriptional activation region, an intermediate DNA
binding region and a C-terminal polymer region. Mutations mainly
occur in the DNA-binding domain of the gene, and the majority of
these are missense mutations. Hot-spot mutations include R175,
G245, R248, R249, R273 and R282. These missense mutations usually
result in three-dimensional structural changes in the protein,
altering its DNA-binding capacity and resulting in p53 losing its
function as a transcription factor, and thus its anticancer effect
(31). This is an important factor
in tumorigenesis; therefore, reactivating wild-type p53 and
restoring the function of mutant p53 are important directions in
cancer therapy research. At present, certain drugs are available
targeting mutant p53. These can restore the DNA-binding ability of
the mutant protein, delete the mutated sequence or inhibit
downstream pathways. R248Q is a hot-spot mutation that lies within
the DNA-binding domain of the p53 protein. This leads to the
inhibition of p53 transcriptional activity and eventually results
in the loss of function of P53. Targeted drugs for the
P53 R248Q mutation have been developed. Recently, it has
been reported that phenethyl isothiocyanate, a derivative of PHI,
can reactivate and repair mutant p53 and inhibit tumor growth
(32).
In the present study, the Kasumi-1 and SKNO-1 cells
used carry the R248Q mutation, causing p53 to lose its function as
a transcription factor. Therefore, the P53 pathway was
inactive. However, the present data showed that the expression of
total p53 protein increased following exposure to PHI. Furthermore,
PHI upregulated the expression of Bax and p21, which are downstream
proteins of P53. Therefore, PHI may have the ability to
repair the mutation, recover the function of P53 and reactivate its
pathway. To further demonstrate whether the apoptosis induced by
PHI is associated with P53, a p53 inhibitor (pifithrin-α)
was used. Compared with the controls, pifithrin-α had no effect on
the levels of cleaved caspase 3 or Bax, and marginally decreased
the expression levels of p21. The PHI group exhibited significantly
higher levels of these three proteins. Compared with the PHI group,
the pifithrin-α + PHI group demonstrated near reversal of these
expression levels. This demonstrated that the expression of
apoptotic proteins induced by PHI was reversed when the p53
inhibitor was used, resulting in the inhibition of apoptosis. These
data provide evidence that PHI induced apoptosis by reactivating
the p53 signaling pathway in Kasumi-1 cells.
Taken together, the results of the present study
demonstrated that PHI had a specific and notable inhibitory effect
on M2 cell lines (Kasumi-1 and SKNO-1) in vivo and in
vitro. PHI inhibited cell arrest at the
G0/G1 phase of the cell cycle. Additionally,
PHI inhibited cell proliferation and induced apoptosis by restoring
mutant p53 in Kasumi-1 cells, reactivating the transcription of
downstream genes of P53, including BAX and
P21. Overall, this compound may lead to cell cycle
inhibition, gene repair and apoptosis.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Deipei Wu
and Professor Suning Chen (The First Affiliated Hospital of Soochow
University, Suzhou, China) for their support and technical
advice.
Funding
This study was partly funded by the Key Medical
Innovations Project Science Research Foundation of Health Bureau of
Fujian Provincial Health (grant no. 2012-CX-32), the Science
Foundation of Fujian Province (grant no. 2016J01484) and the
Startup Fund for Scientific Research, Fujian Medical University
(grant no. 2016QH089).
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XM conceived and designed the experiments; YZ
performed the experiments; YH and YZ analyzed the data; YZ wrote
the manuscript. All authors have read and approved this
manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Ethics
Committee of Zhangzhou Affiliated Hospital of Fujian Medical
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PHI
|
phenylhexyl isothiocyanate
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
dUTP nick end labeling
|
|
AML
|
acute myeloid leukemia
|
|
OS
|
overall survival
|
|
CCK-8
|
Cell Counting Kit-8
|
|
DMSO
|
dimethyl sulfoxide
|
|
IC50
|
half maximal inhibitory
concentration
|
|
H&E
|
hematoxylin and eosin
|
|
AI
|
apoptotic index
|
|
PI
|
propidium iodide
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Bennett JM, Catovsky D, Daniel MT,
Flandrin G, Galton DA, Gralnick HR and Sultan C: Proposals for the
classification of the acute leukaemias French-American-British
(FAB) Co-operative Group. Br J Haematol. 33:451–458. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Döhner H, Estey EH, Amadori S, Appelbaum
FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson
RA, et al: Diagnosis and management of acute myeloid leukemia in
adults: Recommendations from an international expert panel, on
behalf of the European LeukemiaNet. Blood. 115:453–474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dohner H, Weisdorf DJ and Bloomfield CD:
Acute Myeloid Leukemia. N Engl J Med. 373:1136–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferrara F and Del Vecchio L: Acute myeloid
leukemia with t(8;21)/AML1/ETO: A distinct biological and clinical
entity. Haematologica. 87:306–319. 2002.PubMed/NCBI
|
|
5
|
Isobe M, Emanuel BS, Givol D, Oren M and
Croce CM: Localization of gene for human p53 tumour antigen to band
17p13. Nature. 320:84–85. 1986. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gualberto A, Aldape K, Kozakiewicz K and
Tlsty TD: An oncogenic form of p53 confers a dominant,
gain-of-function phenotype that disrupts spindle checkpoint
control. Proc Natl Acad Sci USA. 95:5166–5171. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carson DA and Lois A: Cancer progression
and p53. Lancet. 346:1009–1011. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Amundson SA, Myers TG and Fornace AJ Jr:
Roles for p53 in growth arrest and apoptosis: Putting on the brakes
after genotoxic stress. Oncogene. 17:3287–3299. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kirsch DG and Kastan MB: Tumor-suppressor
p53: Implications for tumor development and prognosis. J Clin
Oncol. 16:3158–3168. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Toshinori O and Akira N: Role of p53 in
cell death and human cancers. Cancers. 3:994–1013. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: Lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Orazi A, Cattoretti G, Heerema NA, Sozzi
G, John K and Neiman RS: Frequent p53 overexpression in therapy
related myelodysplastic syndromes and acute myeloid leukemias: An
immunohistochemical study of bone marrow biopsies. Mod Pathol.
6:5211993.PubMed/NCBI
|
|
14
|
Cleven AH, Nardi V, Ok CY, Goswami M, Dal
Cin P, Zheng Z, Iafrate AJ, Abdul Hamid MA, Wang SA and Hasserjian
RP: High p53 protein expression in therapy-related myeloid
neoplasms is associated with adverse karyotype and poor outcome.
Mod Pathol. 28:552–563. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lane DP, Cheok CF and Lain S: p53-based
cancer therapy. Cold Spring Harb Perspect Biol. 2:a0012222010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Di Agostino S, Cortese G, Monti O,
Dell'Orso S, Sacchi A, Eisenstein M, Citro G, Strano S and Blandino
G: The disruption of the protein complex mutantp53/p73 increases
selectively the response of tumor cells to anticancer drugs. Cell
Cycle. 7:3440–3447. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang S, Zhou L, Hong B, van den Heuvel
AP, Prabhu VV, Warfel NA, Kline CL, Dicker DT, Kopelovich L and
El-Deiry WS: Small-Molecule NSC59984 restores p53 pathway signaling
and antitumor effects against colorectal cancer via p73 activation
and degradation of mutant p53. Cancer Res. 75:3842–3852. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chiao JW, Wu H, Ramaswamy G, Conaway CC,
Chung FL, Wang L and Liu D: Ingestion of an isothiocyanate
metabolite from cruciferous vegetables inhibits growth of human
prostate cancer cell xenografts by apoptosis and cell cycle arrest.
Carcinogenesis. 25:1403–1408. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu L, Liu D, Ma X, Beklemishev A, Seiter
K, Ahmed T and Chiao JW: The phenylhexyl isothiocyanate induces
apoptosis and inhibits leukemia cell growth in vivo. Oncol Rep.
16:1363–1367. 2006.PubMed/NCBI
|
|
20
|
Ma X, Fang Y, Beklemisheva A, Dai W, Feng
J, Ahmed T, Liu D and Chiao JW: Phenylhexyl isothiocyanate inhibits
histone deacetylases and remodels chromatins to induce growth
arrest in human leukemia cells. Int J Oncol. 28:1287–1293.
2006.PubMed/NCBI
|
|
21
|
Zhuang Z, Huang Y, Ma X, et al: Histone
Methylation and Acetylation Modulated by PHI in Prostate Cancer PC3
Cell Line Acta Medicinae Universitatis Scientiae Et Technologiae
Huazhong 06. 2012.
|
|
22
|
Lai YD, Ma XD, Huang YQ, Xu XN, Wang XZ,
Chiao DJ and Liu D: Modulation of histone acetylation and induction
of apoptosis in SMMC-7721 cells by phenylhexyl isothiocyanate.
Zhonghua Zhong Liu Za Zhi. 32:8042010.(In Chinese). PubMed/NCBI
|
|
23
|
Jiang S, Ma X, Huang Y, Xu Y, Zheng R and
Chiao JW: Reactivating aberrantly hypermethylated p15 gene in
leukemic T cells by a phenylhexyl isothiocyanate mediated
inter-active mechanism on DNA and chromatin. J Hematol Oncol.
3:482010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yong Z, Ma X, Huang Y, Hong L and Chiao J:
Effect of phenylhexyl isothiocyanate on aberrant histone H3
methylation in primary human acute leukemia. J Hematol Oncol.
5:362012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Del PG, Venditti A, Del Principe MI,
Maurillo L, Buccisano F, Tamburini A, Cox MC, Franchi A, Bruno A,
Mazzone C, et al: Amount of spontaneous apoptosis detected by
Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML).
Blood. 101:2125–2131. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Redza-Dutordoir M and Averill-Bates DA:
Activation of apoptosis signalling pathways by reactive oxygen
species. Biochim Biophys Acta. 1863:2977–2992. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kischkel FC, Lawrence DA, Tinel A, LeBlanc
H, Virmani A, Schow P, Gazdar A, Blenis J, Arnott D and Ashkenazi
A: Death receptor recruitment of endogenous caspase-10 and
apoptosis initiation in the absence of caspase-8. J Biol Chem.
276:466392001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boulares AH, Yakovlev AG, Ivanova V,
Stoica BA, Wang G, Iyer S and Smulson M: Role of poly(ADP-ribose)
polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP
mutant increases rates of apoptosis in transfected cells. J Biol
Chem. 274:22932–22940. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shaulsky G, Ben-Ze'Ev A and Rotter V:
Subcellular distribution of the p53 protein during the cell cycle
of Balb/c 3T3 cells. Oncogene. 5:1707–1711. 1990.PubMed/NCBI
|
|
30
|
Liao N, Sun L, Chen J, Zhong J, Zhang Y
and Zhang R: A novel polysaccharide conjugate from Bullacta exarata
induces G1-Phase arrest and apoptosis in human hepatocellular
carcinoma HepG2 cells. Molecules. 22:3842017. View Article : Google Scholar
|
|
31
|
Parrales A and Iwakuma T: Targeting
oncogenic mutant p53 for Cancer Therapy. Front Oncol. 5:2882015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aggarwal M, Saxena R, Sinclair E, Fu Y,
Jacobs A, Dyba M, Wang X, Cruz I, Berry D, Kallakury B, et al:
Reactivation of mutant p53 by a dietary-related compound phenethyl
isothiocyanate inhibits tumor growth. Cell Death Differ.
23:1615–1627. 2016. View Article : Google Scholar : PubMed/NCBI
|