Introduction
Breast cancer (BC), a type of cancer most frequently
diagnosed in females, is a considerable threat to female health
worldwide. In the USA, ~230,000 new cases of BC are diagnosed each
year, of which ~5.6% are women >40 years old (1). Although the surgical methods and drug
regimens used to treat BC are constantly improving, the clinical
outcomes of individual patients remain difficult to predict due, in
part, to a number of clinically associated factors (2,3). In
previous studies, tumor size, tissue grade and lymph node status
have been used to speculate the clinical outcomes of patients
(4,5). However, research has suggested that the
accuracy of these indicators is not satisfactory (6). As a consequence of the development of
sequencing technologies, the search for novel biomarkers has
rapidly accelerated (7). The
expression levels of specific microRNAs (miRNAs/miRs) have been
identified as potential biomarkers for predicting survival rate in
several types of human cancer. Han et al (8) revealed that the upregulation of miR-21
was associated with aggressive advancement and poor prognosis in
patients with cervical cancer. Recent findings have reported that
miR-106b-5p activity may be used to classify tumor protein 53-like
bladder tumors into more- and less-favorable predictive categories
(9). Similarly, Yue et al
(10) revealed that serum miR-205
may be a useful biomarker for the diagnosis of glioma, and a
predictive factor for gliomas of an advanced pathological grade. In
addition, the expression levels of several other RNAs have been
indicated as predictors of survival, including cohesin subunit SA-2
in bladder cancer and high mobility group protein B1 in lung
adenocarcinoma (11,12). Furthermore, long non-coding RNAs
(lncRNAs) HOXA distal transcript antisense RNAand HOX transcript
antisense RNA have been used as novel biomarkers in the diagnosis
of renal cell carcinoma and BC, respectively (13,14).
The findings of the aforementioned studies support
the long-standing use of gene biomarkers in the clinical prediction
of disease course and outcome. However, in these studies,
predictions were based on single-gene biomarkers, which are known
to be less reliable for predicting patient survival than their
multi-gene counterparts (15).
Furthermore, multi-gene indicators may enhance the sensitivity and
specificity of prognosis for tumor patients when compared with
those generated using single biomarker methods. In 2002, van de
Vijver et al (16) reported
the gene-expression profile to be a strong projector of disease
stage in young patients with BC compared with clinical and
histological measures (15), and in
2006, Paik et al (17)
revealed that a 21-gene recurrence score was able to predict the
degree of chemotherapy success in patients with breast cancer. In
addition, the results of a study by Wang et al (18) illustrated that histological grades 1
and 3 could be distinguished with high accuracy from gene
expression levels, determined using RNA-sequencing in patients with
breast cancer.
In the present study, a Cox multiple regression
model was used to assess gene expression in BC samples from The
Cancer Genome Atlas (TCGA; http://www.tcga.org/). Patients with high risk scores
reported shorter survival rates compared with those with low risk
scores, a finding that was further validated using the training and
complete test datasets. Moreover, the risk score is independent of
other clinical variables, and performs better than clinical
information to determine BC prognosis. Risk scores and other
clinical factors were combined to develop a nomogram enabling the
accurate and convenient prediction of the 5- and 10-year survival
rates of patients with BC.
Materials and methods
Data sources and pre-processing
The data of 631 cases of BC were downloaded from
TCGA breast cancer database (TCGA-Breast Invasive Carcinoma), and
included 87 cases in the healthy control group and 544 cases in the
cancer group. Differentially-expressed genes were screened
according to the criteria of P<0.01 and log2-fold change >2.
All data analysis and min-max normalization was performed using
Perl and R scripts. The integrity of the patients' RNA expression
profiles and clinically relevant information (age, sex, stage and
histological type) was an important prerequisite for the selection
of patients. In addition, complete ER, PR information was also a
necessary condition for enrollment. Patients who had previously
been diagnosed with breast cancer or any other cancer were
excluded.
Training data set: Survival analysis
and Cox multiple regression model
Following the identification of
differentially-expressed genes in cancerous and adjacent-healthy
tissues (using the R package edge; http://www.bioconductor.org/packages/release/bioc/html/edge.html),
87 samples that lacked survival data were excluded from the
datasets, and the remaining 544 patients with BC were screened for
subsequent analysis. The 544 patients with BC were randomly divided
into a training dataset (n=365) and a test dataset (n=179) using
scripts written in R (Table I). With
the aim to establish a multi-gene biomarker model of prognosis, the
training dataset was then screened for biomarker genes that were
significantly associated with the prognosis of patients with BC.
The specific steps used were as follows: The association between
differentially-expressed genes and overall survival (OS) in
patients with BC was determined using a univariate Cox proportional
regression model. Genes for which P<0.001 were defined as
significantly associated with prognosis, and Cox multivariate
analysis was subsequently performed for these genes. The
proportional hazard assumption (P=0.806) was tested using Stata
version 15.0 (https://www.stata.com) prior to Cox
proportional regression analysis in the final multivariate model.
Finally, a BC prognostic model was determined using stepwise
regression. The R packages function, coxph and survival were used
to construct a risk score staging model (https://cran.r-project.org/web/packages/survival/index.html).
The risk score formula was as follows:
 | Table I.Clinical characteristics of the
patients with breast cancer in each dataset. |
Table I.
Clinical characteristics of the
patients with breast cancer in each dataset.
| Covariate | Total | Training set | Testing set | P-valuea |
|---|
| N | 544 | 365 | 179 |
|
| Risk score |
|
|
| 0.051 |
|
Low | 265 | 167 | 98 |
|
|
High | 279 | 198 | 81 |
|
| Age (years) |
|
|
| 0.382 |
|
≤65 | 388 | 256 | 132 |
|
|
>65 | 156 | 109 | 47 |
|
| Sex |
|
|
| 0.395 |
|
Male | 6 | 5 | 1 |
|
|
Female | 538 | 360 | 178 |
|
| Stage |
|
|
| 0.826 |
| I | 99 | 69 | 30 |
|
| II | 300 | 201 | 99 |
|
|
III | 136 | 90 | 46 |
|
| IV | 9 | 5 | 4 |
|
| Histological
type |
|
|
| 0.592 |
|
Infiltrating ductal | 397 | 267 | 130 |
|
|
Infiltrating lobular | 94 | 62 | 32 |
|
|
Mixed | 21 | 12 | 9 |
|
|
Others | 32 | 24 | 8 |
|
| Estrogen
receptor |
|
|
| 0.492 |
|
Negative | 122 | 85 | 37 |
|
|
Positive | 422 | 280 | 142 |
|
| Progesterone
receptor |
|
|
| 0.958 |
|
Negative | 171 | 115 | 56 |
|
|
Positive | 373 | 250 | 123 |
|
Risk score=∑inβixxi
Where n indicates the number of prognostic genes
screened, i refers to the relative expression of corresponding
gene. β is the coefficient of the individual gene and x
indicates the relative expression of the gene. If β>0, genes are
negatively correlated with the survival time or survival rate, and
if β<0, genes are considered to be protective. Patients were
categorized into high- and low-risk groups according to the median
risk score (0.95). The risk score of each patient was calculated
using a gene-based risk score prediction model. Additionally, an OS
curve was created using the R package survival. A 2-sided log-rank
test was utilized to determine variations in survival among high-
and low-risk patients. Receiver operating characteristic (ROC)
analysis using the R package survival ROC (19) was used to evaluate the sensitivity
and specificity of the gene-based prognostic model in predicting
medical outcomes.
Authentication of the 7-gene signature
for survival projection in the validation and entire datasets
The predicted performance of the 7 differential gene
model was authenticated using both the validation set and the
entire dataset. Patients in both datasets were grouped according to
the cut-off values of the experimental groups, separating the 2
groups of data into high- and low-risk categories, respectively.
Kaplan-Meier survival curves were generated, and the log-rank test
was performed to reveal alterations in survival time among patients
in both datasets. The ROC curve was generated to assess the
clinical predictive power of 7 differentially-expressed gene
signatures in both datasets.
Development of a novel nomogram
including risk scores
The Cox proportional hazards regression model was
used to determine whether the risk-scoring model was an autonomous
predictive factor for patients with BC. Stratified analysis was
performed to verify whether the 7 differential gene characteristics
were independent prognostic factors for patients with BC, compared
with other clinical variables. In addition, a nomogram was
constructed using the risk scores, age, sex and primary tumor
staging and visualized using the R package rms (20). The accuracy of the model was assessed
using the C-index. All data analysis and processing were conducted
using R software (version 3.4.2; www.r-project.org).
Results
Identification of survival-associated
genes in the training dataset
To identify novel genetic biomarkers associated with
the clinical outcomes of patients with BC, a univariate Cox
proportional hazard regression model was applied to
differently-expressed genes in BC and healthy breast tissues. In
total, 18 genes were found to be significantly associated with OS
(P<0.001). These genes were subsequently subjected to stepwise
multivariate Cox regression analysis. As illustrated in Table II, 7 independent genes were selected
using step-wise multivariate Cox regression analysis, and a
gene-based prognostic model was established to estimate the
survival risk of patients using the following equation:
| Table II.Seven prognostic genes significantly
associated with overall survival in patients with breast
cancer. |
Table II.
Seven prognostic genes significantly
associated with overall survival in patients with breast
cancer.
| Gene name | Coefficient | Hazard ratio | 95% confidence
interval |
P-valuea |
|---|
| TMEM190 | −0.1735 | 0.8407 | 0.6531–1.0300 | 0.05765 |
| lncRNA
AL049749.1 | −0.1510 | 0.8598 | 0.5017–1.2051 | 0.08750 |
| lncRNA
AC123595.1 | −0.2924 | 0.7465 | 0.6183–0.9259 |
0.04453b |
| TUBA3D | −0.1024 | 0.9027 | 0.6526–1.7124 | 0.13771 |
| LYVE1 | 0.1990 | 1.2202 | 1.0347–1.5063 |
0.04394b |
| LILRB5 | 0.4676 | 1.5962 | 1.3441–1.9163 |
0.00084c |
| CD209 | 0.1744 | 1.1906 | 1.0275–1.4055 |
0.04598b |
Risk score=(−0.1735 × TMEM190) + (−0.1510 ×
AL049749.1) + (−0.2924 × AC123595.1) + (−0.1024 × TUBA3D) + (0.1990
× LYVE1) + (0.4676 × LILRB5) + (0.1744 × CD209).
Training dataset: Risk score
performance
Final calculations indicated a median and mean risk
score of 0.95 and 1.32, respectively. The minimum and maximum
values were 0.01 and 6.62, respectively. To confirm the performance
of the risk score in predicting the survival rates of patients with
BC, the prognostic, 7-gene signature-based model was used to
allocate a risk score for each patient. Patients were categorized
as high-risk (n=198) or low-risk (n=167), where the median risk
score was used as the cut-off value. Kaplan-Meier analysis revealed
that the OS curves of these 2 groups were significantly different
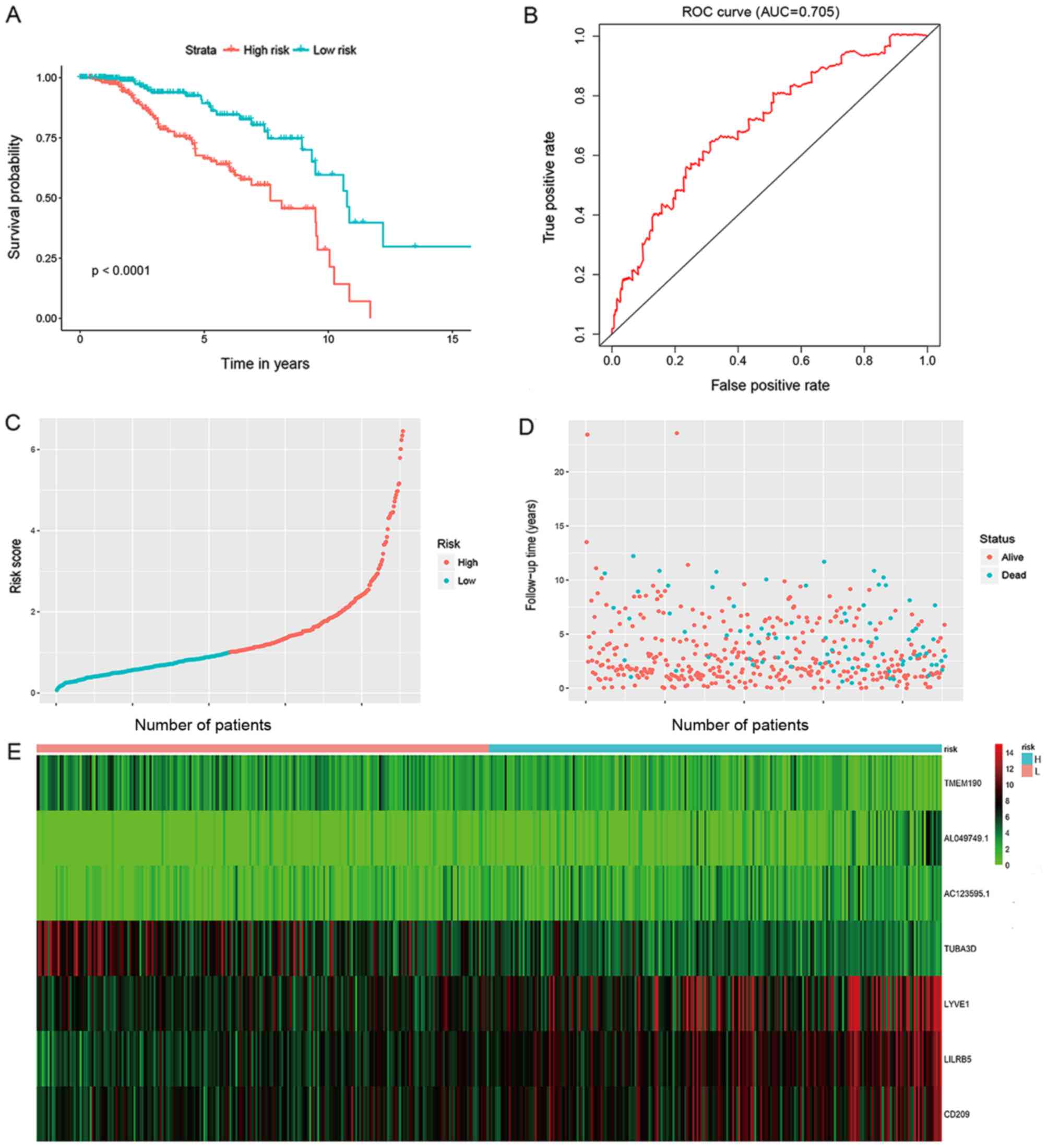
(P<0.001; Fig. 1A). ROC curve
analysis of the 10-year survival rate was used to evaluate the
projection potential of the 7 genes. Moreover, the area under curve
(AUC) for the 7-gene signature-based prognostic model was 0.705 at
120 months OS (Fig. 1B). The
scattering of the risk score (Fig.
1C), survival status (Fig. 1D)
and gene expression levels of the 7 genes (Fig. 1E) from each patient were also
analyzed.
Validation of the performance of risk
score in test datasets
To assess the strength of the prognostic model in
patients with BC, the risk scores in the test dataset (n=179) and
the entire dataset (n=544) were determined. In the test dataset,
the patients were categorized into high-risk (n=81) and low-risk
(n=98) groups per the risk-score model, and cut-off points were
defined using the training dataset. Kaplan-Meier survival curves of
the high- and low-risk groups were considerably dissimilar in the
test dataset. Compared with patients from the high-risk group,
those from the low-risk group had significantly longer survival
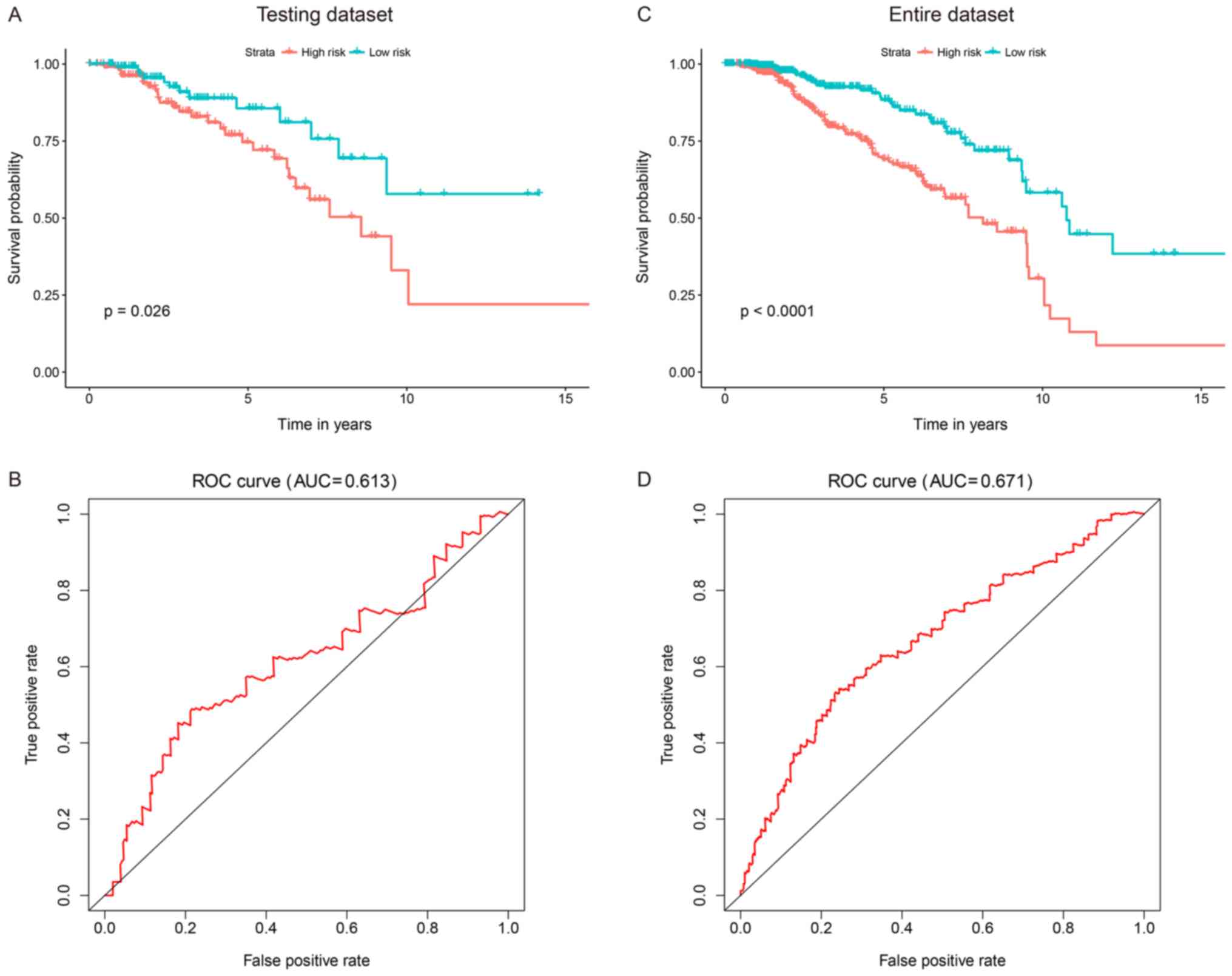
times (P=0.026; Fig. 2A). The AUC of
the time-dependent ROC curves for the 7-gene signature in the test
dataset was 0.613 at 10 years (Fig.
2B). When this signature was applied to the entire dataset, a
conclusion was reached. Moreover, the 7-gene signature was used to
classify patients in the entire TCGA dataset into high-risk (n=279)
and low-risk (n=265) groups. The patients in the high-risk group
exhibited shorter OS times compared with those in the low-risk
group (P<0.0001; Fig. 2C).
Authentication of the signature using all 544 patients generated a
ROC AUC of 0.671 at 10 years (Fig.
2D). These outcomes suggested that the risk score was a robust
predictor of clinical outcome for patients with BC. The
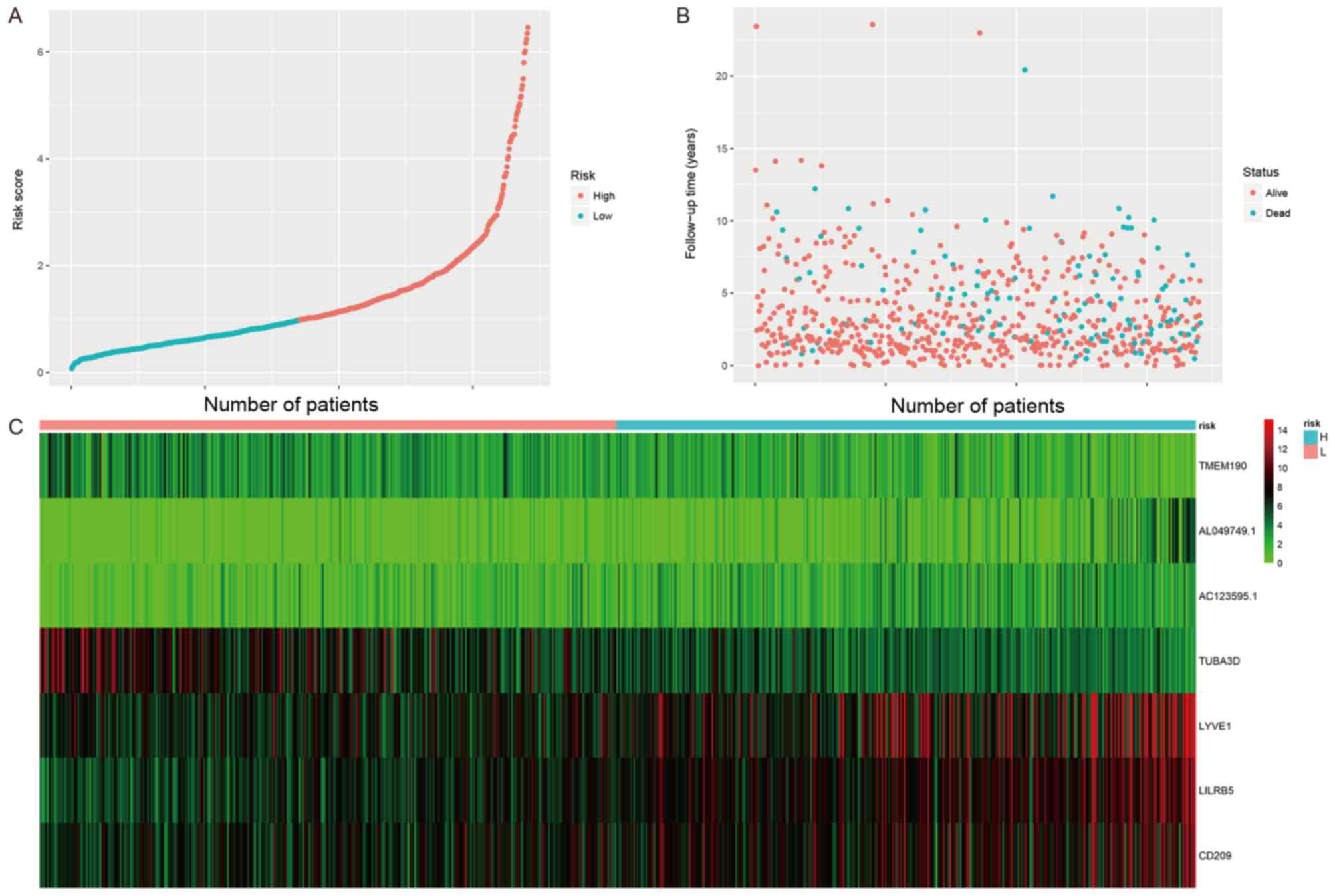
distribution of the risk score model, survival status and gene
expression patterns of patients in the test and entire datasets
were also analyzed (Figs. 3 and
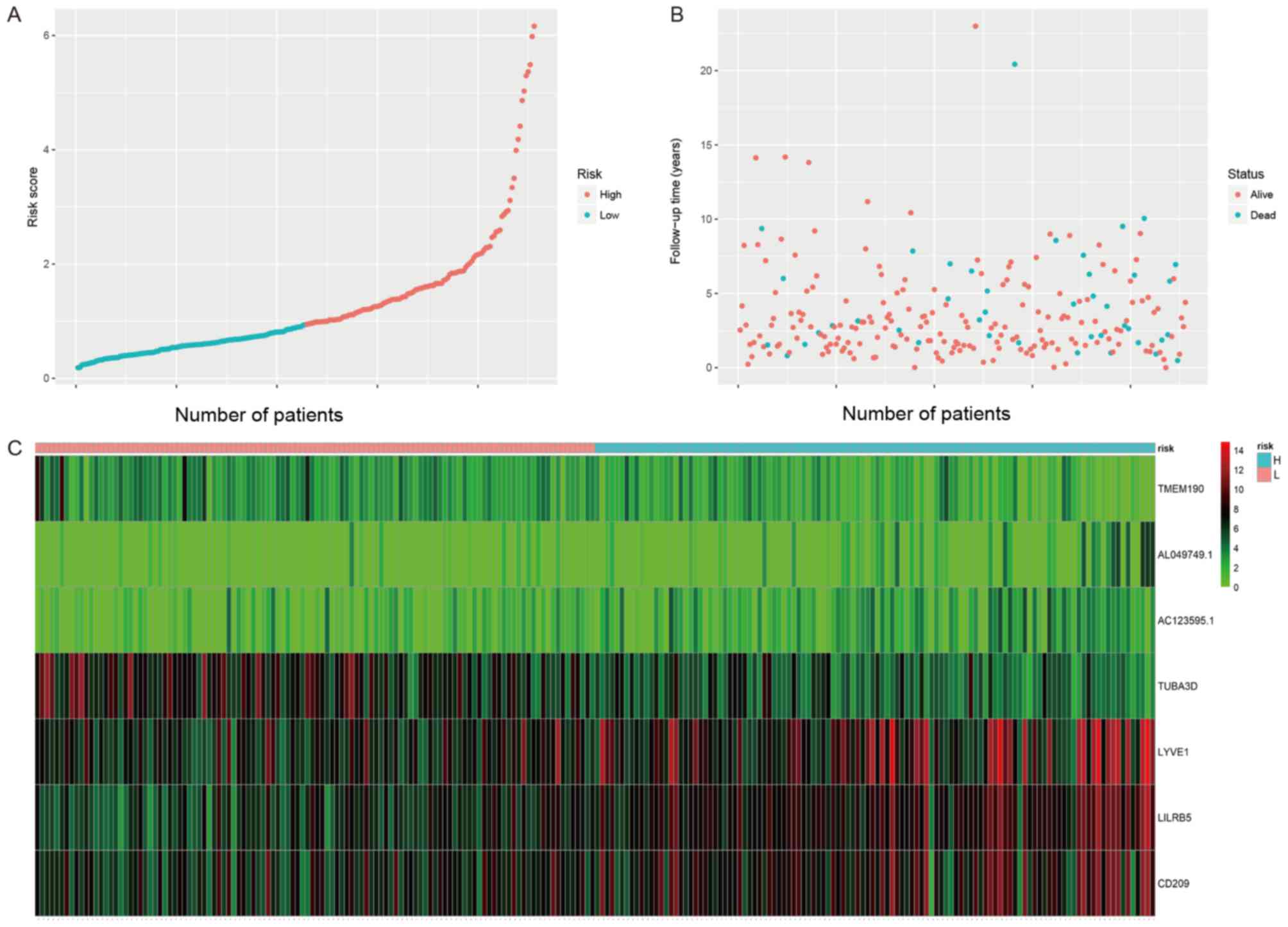
4). Fig.
3A shows that the cut-off value of the risk scores in the test
data set is 0.95. The distributions of survival status in the test
data set is shown in Fig. 3B.
Fig. 3C shows the expression
profiles of patients in the test data set. LYYE1, LILRB5, CD209 was
highly expressed in tumor tissues and TMEM190, AC123595.1,
AL049749.1 and TUBA3D expression was low in tumor tissues. The same
conclusion was reached based on the data shown in Fig. 4.
Association between risk score and
other clinical factors in the entire dataset
To determine the potential association between the
risk score and clinical parameters [including age, sex, oestrogen
receptor (Er) status, progesterone receptor (Pr) status, tumor
stage and histological type], multiple Cox hazard analyses were
performed utilizing the entire dataset. As presented in Fig. 5, the risk score possessed a
predictive ability separate from that of the other clinical
parameters [hazard ratio (HR), 2.464; 95% confidence interval (CI),
1.546–3.929; P<0.0010] (Fig. 5).
This suggests that the prognostic capacity of the risk score was
also independent of these other clinical variables.
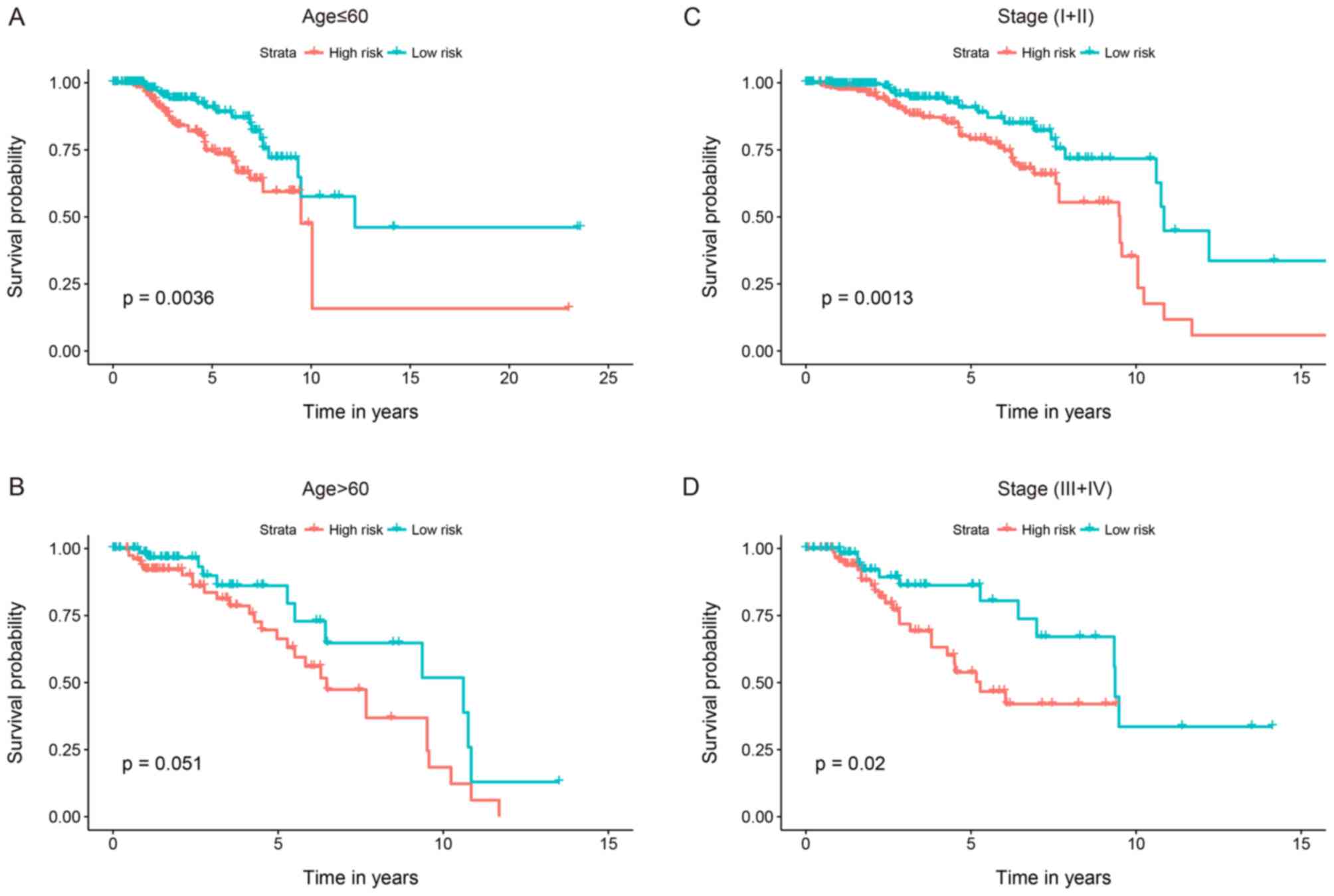
Stratified analyses were conducted to determine
whether the 7-gene signature held predictive importance. Patients
were categorized into younger (n=388) and elder (n=256) strata
depending on the median age (60 years); younger patients were
subsequently divided into high-risk (n=193) and low-risk (n=195)
groups. Patients in the low-risk group had significantly longer OS
times compared with those in the high-risk group (P=0.0036;
Fig. 6A). Likewise, patients in the
elder group were separated into low- and high-risk groups with
different OS times (P=0.051; Fig.
6B). The patients were concurrently categorized into
early-stage (n=399) and advanced-stage (n=45) groups depending on
the primary tumor stage. The early-stage patients were then divided
into a high-risk group (n=203) with shorter survival, and a
low-risk group (n=196) with an extended survival period (P=0.0013;
Fig. 6C). Similarly, the
advanced-stage patient group was divided into 2 risk subgroups with
significantly different survival times (P=0.02; Fig. 6D). The results of these analyses
suggested that the 7-gene signature may function as an autonomous
indicator of survival for patients with BC.
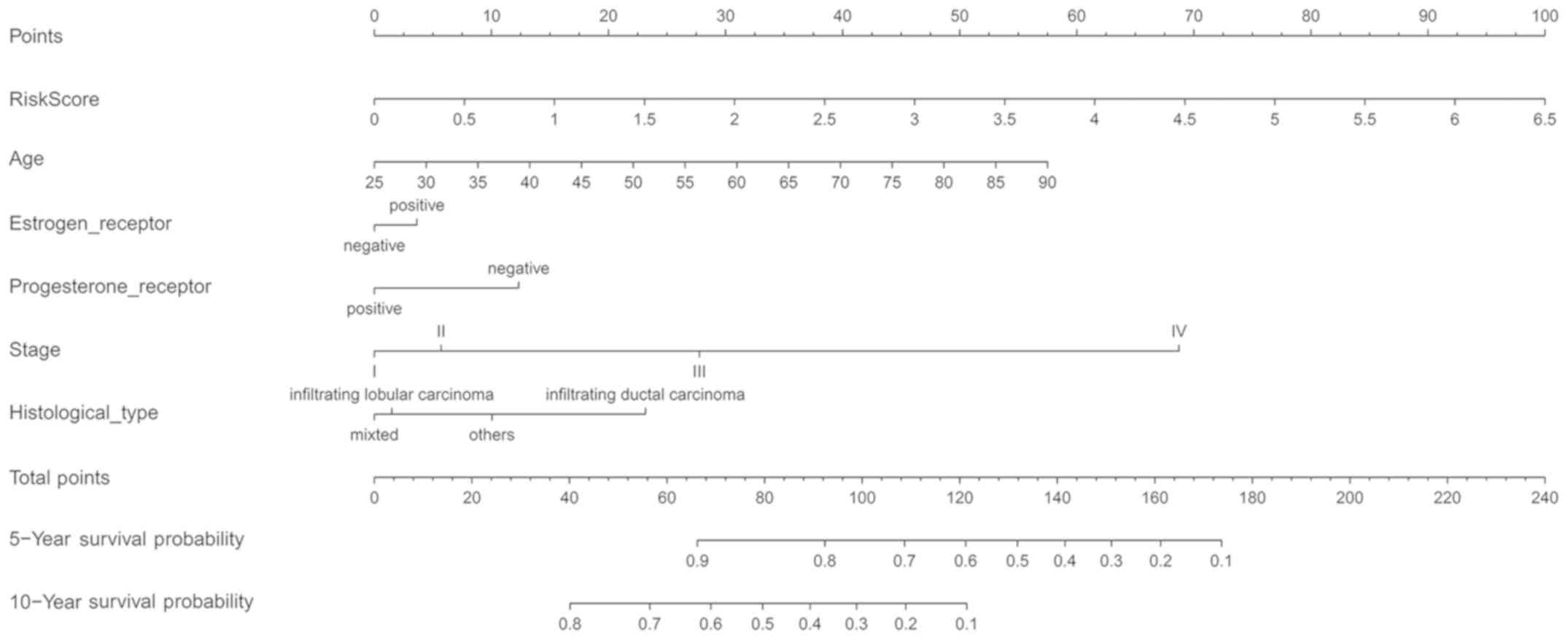
An innovative nomogram for the
prediction of patient outcome
To complement the predictive capacity of the risk
score, an innovative nomogram was developed to predict the
prognosis of patients with BC. The nomogram was based on 6
projecting factors and comprised risk score, age, sex, Er status,
Pr status, tumor stage and histological type. A high total score
indicated low 5- and 10-year survival rates, whilst a low total
score indicated improved survival rates. The C-index of the
nomogram for predicting OS was 0.755 (95% CI, 0.719–0.791) in the
main cohort. This suggested that in medical practice, the model was
appropriate for predicting the outcomes of patients with BC
(Fig. 7).
Discussion
A number of previously published studies have
reported numerous individual prognostic biomarkers associated with
BC. Using reverse transcription-quantitative PCR and western
blotting, Zhao et al (21)
analyzed the expression of inosine monophosphate dehydrogenase
2(IMPDH2) in 40 matched normal and BC tissues, the results of which
indicated that a high level of IMPDH2 expression was associated
with poor patient outcome. Another study revealed that the
expression level of lncRNA-AK058003 was upregulated in BC tissues
compared with healthy adjacent tissue, and that this was also
indicative of poor prognosis and associated with tumor cell
invasion and metastasis (22) In
addition, Guo et al (23)
demonstrate that the upregulation of miR-1915-3p and downregulation
of miR-455-3p in the serum of patients with BC promoted lymph node
metastasis in patients with in situ carcinomas, compared
with patients without lymph node metastasis. However, the
aforementioned studies were based on the assessment of single gene
biomarkers only, which when used as a prognostic standard,
inevitably lead to clinical bias (16). Therefore, it is necessary to develop
novel multi-gene models to predict the survival of patients with
BC, and to establish personalized treatment programs. The use of
multi-gene biomarkers increases the sensitivity and specificity of
the predictive model, ultimately improving overall credibility
(15).
In the present study, Cox multiple regression
analysis of BC RNA-Sequencing data downloaded from TCGA was
performed in order to identify genes associated with patient OS; 7
genes were found to meet the selection criteria. Survival analysis
indicated that patients with high-risk scores possessed
considerably shorter OS times than patients with low risk scores
(P<0.001). The AUC of this model was 0.705 at 120 months OS,
indicating that the predictive value of the 7-gene signature may be
utilized for survival prediction. Compared with other specific
medical parameters (including age, sex, tumor stage and
histological type) risk scores were better predictors of patient
survival, indicating that the 7-gene signature may be of value in
further research. Additionally, in order to better adapt the
multi-gene risk score model to current clinical requirements, other
clinical factors were combined to develop a novel nomogram that
could accurately and conveniently predict the 5- and 10-year
survival rates of patients. The nomogram may be able to more
accurately determine the correct course of treatment for patients
with a low survival rate, in comparison to the traditional
Tumor-Node-Metastasis classification systems or nomograms
containing clinical features alone or which utilized only a single
biomarker.
Among the 7 genes identified in the present study,
lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1),
leukocyte immunoglobulin-like receptor B5 (LILRB5) and CD209
molecule (CD209) were risk-associated, demonstrating that the
expression levels of these genes negatively correlated with BC
survival time; conversely, the expression levels of AC123595.1,
AL049749.1, transmembrane protein 190 and tubulin α-3D chain were
positively associated with survival. LYVE1 and CD209 are reportedly
associated with cancer as discussed further below. LYVE1 is a type
I integral membrane glycoprotein (24) which acts as a receptor, binding to
both soluble and immobilized hyaluronan (HA), and may also be
involved in lymphatic HA transport and lymph angiogenesis (25,26). In
1999, Banerji et al (24)
were the first to reveal that LYVE1 is a lymph-specific HA receptor
and a unique marker for lymph vessels. Subsequently, Bono et
al (27) demonstrated that a
high LYVE1-positive lymphatic vessel number was associated with
poor outcome in patients with ductal BC (28). The present study supports this
conclusion, defining LYVE1 as a risk factor, and with upregulated
expression increasing the risk score and the likelihood of poor
prognosis. It was concluded that LYVE1 was an essential protein in
the lymph angiogenesis and tumor metastasis of BC, and that it may
be a favorable candidate for targeted treatment. Furthermore, CD209
is a C-type lectin receptor expressed on the surface of macrophages
and dendritic cells (DCs) (27). In
the present study, CD209 expression was identified as a protective
biomarker in the prognosis of BC. This conclusion was supported by
van Gisbergen et al (29),
who found revealed that the binding of SKBR3 cells to immature DCs
was inhibited by CD209-resistant antibodies, thereby inhibiting the
maturation of DCs and promoting tumor cell immune escape.
Nevertheless, the functions of the other 5 genes are not currently
known, and thus there is a requirement for further investigation.
Using Cox regression analysis, a risk score model merging the
aforementioned genes was established and may aid to predict the
survival of patients with BC.
Although the 7-gene signature effectively predicted
the outcome of patients with BC, there are certain limitations to
the present study. The risk score model was developed based on TCGA
datasets and future studies require its validation in additional
patient cohorts. However, due to a lack of data from patients in
this age range, a reliable model of the younger subgroup could not
be established. Furthermore, the treatment method serves an
important role in disease prognosis, and the inclusion of treatment
data in these analyses would increase the value of the subsequent
results. However, there was insufficient data on the patients'
treatment programs in the datasets, which was a major limitation
and will be addressed in the collection of subsequent clinical
data.
In conclusion, the 7-gene signature established in
the present study was effective and stable in BC samples acquired
from TCGA. Additional analysis indicated that the 7-gene signature
functioned as an autonomous element for the prognosis of patients
with BC. The results may potentially contribute to the development
of more effective prognostic tools, and ultimately improve patient
outcome.
Acknowledgements
Not applicable.
Funding
The authors acknowledge the support received from
the National Natural Science Foundation of China (grant no.
81560526), the Natural Science Foundation of Guang Xi (grant no.
2014GXNSFCA118011) and the College Young and Middle-Aged Teachers'
Basic Ability Improvement Project of Guang Xi (grant no.
2017KY0484).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WP and HL designed the experiment, provided
financial support, revised the manuscript and gave final approval
of the version to be published. LL and ZC performed the statistical
analysis and wrote the paper. WS made substantive contributions to
the work, including data collecting and manuscript revising.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gewefel H and Salhia B: Breast cancer in
adolescent and young adult women. Clin Breast Cancer. 14:390–395.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Clough KB, Kaufman GJ, Nos C, Buccimazza I
and Sarfati IM: Reply to comments on: Improving breast cancer
surgery: A classification and quadrant per quadrant atlas for
oncoplastic surgery. Ann Surg Oncol. 18:259–260. 2011. View Article : Google Scholar
|
|
3
|
Graham PJ, Brar MS, Foster T, McCall M,
Bouchard-Fortier A, Temple W and Quan ML: Neoadjuvant chemotherapy
for breast cancer, is practice changing? A population-based review
of current surgical trends. Ann Surg Oncol. 22:3376–3382. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dai D, Jin H and Wang X: Nomogram for
predicting survival in triple-negative breast cancer patients with
histology of infiltrating duct carcinoma: A population-based study.
Am J Cancer Res. 8:1576–1585. 2018.PubMed/NCBI
|
|
5
|
Lee SK, Yang JH, Woo SY, Lee JE and Nam
SJ: Nomogram for predicting invasion in patients with a
preoperative diagnosis of ductal carcinoma in situ of the breast.
Br J Surg. 100:1756–1763. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Balachandran VP, Gonen M, Smith JJ and
Dematteo RP: Nomograms in oncology: More than meets the eye. Lancet
Oncol. 16:e173–e180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mortazavi A, Williams BA, McCue K,
Schaeffer L and Wold B: Mapping and quantifying mammalian
transcriptomes by RNA-Seq. Nat Methods. 5:621–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Han Y, Xu GX, Lu H, Yu DH, Ren Y, Wang L,
Huang XH, Hou WJ, Wei ZH, Chen YP, et al: Dysregulation of miRNA-21
and their potential as biomarkers for the diagnosis of cervical
cancer. Int J Clin Exp Pathol. 8:7131–7139. 2015.PubMed/NCBI
|
|
9
|
Lee E, Collazolorduy A, Castillomartin M,
Gong Y, Wang L, Oh WK, Galsky MD, Cordon-Cardo C and Zhu J:
Identification of microR-106b as a prognostic biomarker of p53-like
bladder cancers by ActMiR. Oncogene. 37:5858–5872. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yue X, Lan F, Hu M, Pan Q, Wang Q and Wang
J: Downregulation of serum microRNA-205 as a potential diagnostic
and prognostic biomarker for human glioma. J Neurosurg.
124:122–128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lelo A, Prip F, Harris BT, Solomon D,
Berry DL, Chaldekas K, Kumar A, Simko J, Jensen JB, Bhattacharyya
P, et al: STAG2 is a biomarker for prediction of recurrence and
progression in papillary non-muscle-invasive bladder cancer. Clin
Cancer Res. 24:4145–4153. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feng A, Tu Z and Yin B: The effect of
HMGB1 on the clinicopathological and prognostic features of
non-small cell lung cancer. Oncotarget. 7:20507–20519. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peng F, Shi X, Meng Y, Dong B, Xu G, Hou
T, Shi Y and Liu T: Long non-coding RNA HOTTIP is upregulated in
renal cell carcinoma and regulates cell growth and apoptosis by
epigenetically silencing of LATS2. Biomed Pharmacother.
105:1133–1140. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang L, Song X, Wang X, Xie Y, Wang Z, Xu
Y, You X, Liang Z and Cao H: Circulating DNA of HOTAIR in serum is
a novel biomarker for breast cancer. Breast Cancer Res Treat.
152:199–208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Salomaa V, Havulinna A, Saarela O, Zeller
T, Jousilahti P, Jula A, Muenzel T, Aromaa A, Evans A, Kuulasmaa K
and Blankenberg S: Thirty-one novel biomarkers as predictors for
clinically incident diabetes. PLoS One. 5:e101002010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van de Vijver MJ, He YD, van't Veer LJ,
Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C,
Marton MJ, et al: A gene-expression signature as a predictor of
survival in breast cancer. N Engl J Med. 347:1999–2009. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paik S, Tang G, Shak S, Kim C, Baker J,
Kim W, Cronin M, Baehner FL, Watson D, Bryant J, et al: Gene
expression and benefit of chemotherapy in women with node-negative,
estrogen receptor-positive breast cancer. J Clin Oncol.
24:3726–3734. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang M, Klevebring D, Lindberg J, Czene K,
Grönberg H and Rantalainen M: Determining breast cancer
histological grade from RNA-sequencing data. Breast Cancer Res.
18:482016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Heagerty PJ, Lumley T and Pepe MS:
Time-dependent ROC curves for censored survival data and a
diagnostic marker. Biometrics. 56:337–344. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Harrell FE, Califf RM, Pryor DB, Lee KL
and Rosati RA: Evaluating the yield of medical tests. JAMA.
247:2543–2546. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao Y, Yang Y, Dai J, Xing D and Dong Y:
IMPDH2 is highly expressed in breast cancer and predicts
unfavourable prognosis. Biomarkers. Jul 2–2018.(Epub ahead of
print). doi: 10.1080/1354750X.2018.1496360. View Article : Google Scholar
|
|
22
|
He K and Wang P: Unregulated long
non-coding RNA-AK058003 promotes the proliferation, invasion and
metastasis of breast cancer by regulating the expression levels of
the γ-synuclein gene. Exp Ther Med. 9:1727–1732. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo J, Liu C, Wang W, Liu Y, He H, Chen C,
Xiang R and Luo Y: Identification of serum miR-1915-3p and
miR-455-3p as biomarkers for breast cancer. PLoS One.
13:e02007162018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Banerji S, Ni J, Wang SX, Clasper S, Su J,
Tammi R, Jones M and Jackson DG: LYVE-1, a new homologue of the
CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J
Cell Biol. 144:789–801. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mattila MM, Ruohola JK, Karpanen T,
Jackson DG, Alitalo K and Härkönen PL: VEGF-C induced
lymphangiogenesis is associated with lymph node metastasis in
orthotopic MCF-7 tumors. Int J Cancer. 98:946–951. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Skobe M, Hawighorst T, Jackson DG, Prevo
R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K and Detmar
M: Induction of tumor lymphangiogenesis by VEGF-C promotes breast
cancer metastasis. Nat Med. 7:192–198. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bono P, Wasenius VM, Heikkilä P, Lundin J,
Jackson DG and Joensuu H: High LYVE-1-positive lymphatic vessel
numbers are associated with poor outcome in breast cancer. Clin
Cancer Res. 10:7144–7149. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mcgreal EP, Miller JL and Gordon S: Ligand
recognition by antigen-presenting cell C-type lectin receptors.
Curr Opin Immunol. 17:18–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
van Gisbergen KP, Aarnoudse CA, Meijer GA,
Geijtenbeek TB and Van Kooyk Y: Dendritic cells recognize
tumor-specific glycosylation of carcinoembryonic antigen on
colorectal cancer cells through dendritic cell-specific
intercellular adhesion molecule-3-grabbing nonintegrin. Cancer Res.
65:5935–5944. 2005. View Article : Google Scholar : PubMed/NCBI
|