Introduction
Cancer is a significant global health problem; in
2017 it was predicted that 600,920 cancer-associated mortalities
would occur in the USA and 26% of those cases would be associated
with lung cancer (1). Non-small cell
lung cancer (NSCLC) accounts for >80% of primary lung cancer
cases (2). The identification of
specific molecular targets against NSCLC has promoted a shift
towards personalized treatment strategies in clinics (3,4).
Abnormal activation of the epidermal growth factor
receptor (EGFR) signaling pathway has been reported in NSCLC, which
leads to the activation of subsequent intracellular signaling
pathways, including the phosphoinositide 3-kinase (PI3K)/AKT and
mitogen-activated protein kinase 1 (MAPK) signaling pathways, which
serve important roles in the proliferation, differentiation,
migration and apoptosis of tumor cells (5,6). To
attenuate the effects of EGFR-mediated proliferation of cancer
cells, EGFR tyrosine kinase inhibitors (EGFR-TKIs) that
specifically bind to the tyrosine kinase domain of EGFR and inhibit
its activity have been widely administered clinically (7).
Erlotinib is a first-generation EGFR-TKI for
patients with EGFR mutation-positive lung adenocarcinoma. Erlotinib
elicits effective treatment responses, however, these responses are
lost after a long period of time due to acquired resistance
(7,8). The most common mechanism of acquired
resistance is a secondary T790 mutation in EGFR termed EGFR T790M.
Other mechanisms include stimulation of alternative pathways either
by activation of other kinases, including hepatocyte growth factor
receptor (MET) and human epidermal growth factor receptor 2, or
alterations of key components in the EGFR pathway, including
activation of phosphatidylinositol-4,5-bisphosphate 3-kinase or
loss of phosphatase and tensin homolog (PTEN), which eliminate the
requirement for EGFR-mediated tumor cell activation (9–14). To
overcome EGFR-TKI resistance in lung cancer, numerous combinatorial
strategies have been reported that demonstrate effective results
and provide promising strategies to prevent resistance and
potentially reduce the toxicity of both agents (15–24).
The receptor tyrosine kinase-like orphan receptor 1
(ROR1) is a type 1 transmembrane protein expressed on the plasma
membrane (25). Previous studies
have demonstrated that ROR1 is an oncogene that is highly expressed
in numerous types of hematologic malignancy and several types of
solid tumor, including lung cancer (26,27).
ROR1 acts as a partner for the oncogenic tyrosine kinase MET and
sustains the MET-driven transformed phenotype (28). ROR1 is also required to sustain the
association between EGFR and receptor tyrosine-protein kinase
erbB-3 (ERBB3), the activation of ERBB3, consequentially, making
ROR1 an ideal target for therapies against EGFR-TKI resistance in
lung adenocarcinoma (29).
Our previous investigation of patients with lung
adenocarcinoma revealed that >60% of tumor tissues expressed
ROR1, and inhibition of ROR1 significantly downregulated the
proliferation of NSCLC cells and induced cell apoptosis (27). The current study analyzed the effect
of ROR1 inhibition combined with erlotinib on the induction of
apoptosis and the inhibition of proliferation via the AKT/mTOR
signaling pathway. In summary, the present study provided a novel
therapeutic strategy to increase the sensitivity of
ROR1+ lung adenocarcinoma to erlotinib treatment.
Materials and methods
Cell lines and cell culture
The NSCLC cell line NCI-H1975 was kindly provided by
the Stem Cell Bank, Chinese Academy of Sciences. The human lung
cancer cell line XLA-07 was a gift from Professor Yong Duan (First
Affiliated Hospital of Kunming Medical University, Kunming, China)
(30) and the PC-9 cell line was a
gift from Dr Jun Zhang (Shanghai Pulmonary Hospital, Shanghai,
China) (31). The cells were
cultured at 37°C in a 5% CO2 incubator (Panasonic
Healthcare,) in RPMI-1640 (HyClone; GE Healthcare Life Sciences)
supplemented with 10% fetal bovine serum (Beijing Transgen Biotech
Co., Ltd.) and 1% penicillin/streptomycin (HyClone; GE Healthcare
Life Sciences).
Establishment of an acquired
erlotinib-resistant cell line termed PC-9erlo
The acquired erlotinib-resistant cell line termed
PC-9erlo was established from the parental cell line PC-9. Briefly,
2×106 cells were seeded in a 10 cm2 dish and
then exposed to 10 µM erlotinib (Cayman Chemical Company).
Following incubation at 37°C for 48 h the cells were washed with 1X
PBS and then cultured in complete medium without erlotinib. To
acquire and maintain the erlotinib resistance of PC-9erlo cells,
the cultured cells were collected and gradually exposed to
increasing concentrations of erlotinib (0.1 µM for 2 months, 0.5 µM
for 2 months, 1.25 µM for 2 months and 2.5 µM for 2 months).
Following the 2 months of exposure to 2.5 µM erlotinib, the
half-maximal inhibitory concentration (IC50) value of erlotinib in
PC-9erlo was 2.62±0.82 µM.
Silencing of human ROR1
ROR1 small interfering RNA (siRNA), termed siROR1,
was obtained from Ambion (Thermo Fisher Scientific, Inc.). The
sequence of siROR1 was sense 5′-GUACUGCGAUGAAACUUCATT-3′. The
method of silencing ROR1 with siRNA was as previously described
(27) with modifications. Briefly,
the cells were seeded in 96- or 6-well plates, and incubated in a
CO2 incubator for 12 or 20 h. The cells were then
transfected with siROR1 or a non-targeting control siRNA (siNC;
Invitrogen; Thermo Fisher Scientific, Inc.) at a concentration of
20 or 25 nM, followed by culture in serum-free medium for 6 h.
Transfections were performed in Opti-MEM reduced serum medium
(Thermo Fisher Scientific, Inc.) using Lipofectamine RNA iMAX
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol.
ROR1 expression analysis
Expression of ROR1 in different cell lines was
examined by flow cytometry. Briefly, the cells were collected 72 h
after ROR1 silencing with 25 nM siROR1 (cat. no. 4457298; Ambion;
Thermo Fisher Scientific, Inc.) using Lipofectamine RNAiMAX (Thermo
Fisher Scientific, Inc.) for 6 h at 37°C and washed twice with
ice-cold PBS. R12 is a chimeric rabbit/human anti-ROR1 monoclonal
antibody with a hemagglutinin (HA) tag that was developed in
Christoph Rader's laboratory by the corresponding author (Jia-Hui
Yang, School of Basic Medicine, Chengdu University of TCM, Chengdu,
China) (32). R12 (5 µg/ml) or
normal human IgG antibodies (5 µg/ml; cat. no. 009-000-003; Jackson
ImmunoResearch Laboratories, Inc.) were added to the cells and
incubated at 4°C for 30 min. Following washing, 5 µl PE-conjugated
anti-HA monoclonal antibody (cat. no. 130-098-806; Miltenyi Biotec,
Inc.) was added and incubated at 4°C for 30 min. Finally, the cells
were suspended in 500 µl flow cytometry buffer and analyzed using
an Accuri C6 flow cytometer (BD Biosciences). Data were analyzed
using FlowJo v7.6.2 software (FlowJo LLC). The inhibition rate of
ROR1 expression level in different cell lines was calculated using
the formula: [value (siNC)-value (background)]-[value
(siROR1)-value (background)]/[value (siNC)-value (background)]
×100% (value, value of mean fluorescence intensity of cells in
different treated groups).
MTS cytotoxicity assay
Cells were seeded in 96-well plates at
4–6×103 per well and transfected with 20 nM siROR1 or
siNC, then cultured for 48 h prior to treatment with a range of
concentrations (0, 1.25, 2.5, 5, 10, 20 µM) of erlotinib (Cayman
Chemical Company). Cell cytotoxicity was examined using the
CellTiter 96® AQueous one solution reagent (Promega
Corporation) with the following steps: 20 µl of the reagent was
added to each well and cells were incubated in the dark at 37°C for
1 h. Cell viability was examined by measuring absorbance at 490 nm
using a microplate reader. Experiments were performed in
triplicate. Cell growth ratio values were calculated using the
formula: 100× [A490 (sample, T)-A490 (sample, T0)]/[A490 (control,
T)-A490 (control, T0)] (T, value of absorbance at 490 nm of wells
with different treatment cells; T0, value of absorbance at 490 nm
of wells without cells). Cell images in different treated groups
were observed by inverted optical microscope (magnification ×100;
XD-30; Sunny Optical Technology Co., Ltd.).
Apoptosis assay
Cell apoptosis was analyzed using flow cytometry.
Cells were seeded in 6-well plates at 1.2–1.8×105
cells/well and appropriate concentrations of erlotinib (NCI-H1975,
2.5 µM; PC-9erlo, 2.5 µM and XLA-07, 10 µM) were added to the wells
48 h after 20 nM siRNA transfection. The plates were then incubated
at 37°C for 2–5 days. Cells were collected and washed, then
incubated with 5 µl FITC-conjugated Annexin V and propidium iodide
(BD Biosciences) in the dark for 15 min at room temperature. Cell
apoptosis was measured using a FACSCalibur flow cytometer
(FACSCalibur; BD Biosciences).
Bio-Plex pro assays
Multiple proteins and the AKT signaling pathway were
evaluated using a Bio-Plex signaling AKT 8-plex panel (cat. no.
LQ00006JK0K0RR) and a Bio-Plex pro signaling reagent kit (cat. no.
171304006M) both from Bio-Rad Laboratories, Inc., according to the
manufacturer's protocol. Briefly, 24 h after transfection with 25
nM siRNA, NCI-H1975 cells were treated with 2.5 µM erlotinib at
37°C for 24 h. The cells were lysed 48 h later with RIPA buffer
(Beyotime Institute of Biotechnology) supplemented with 10%
phosphatase inhibitor (Roche Diagnostics) and 1% protease inhibitor
(EMD Millipore) at 4°C. Enhanced BCA Protein Assay kit (Beyotime
Institute of Biotechnology) was used to analyze the protein
concentration of each cell lysate. Suspended beads were added to a
96-well flat bottom plate at 50 µl per well and the plate was
washed with washing buffer using a Bio-Plex Pro II Wash station
(Bio-Rad Laboratories, Inc.). Subsequently, 10 µg cell lysate was
added to each well. Following incubation of the plate overnight at
room temperature with shaking (450 RPM), biotin conjugated
detecting antibody cocktail from the kit (Bio-Plex signaling AKT
8-plex panel, cat. no. LQ00006JK0K0RR; Bio-Plex pro signaling
reagent kit, cat. no. 171304006M; dilution 1:20; Bio-Rad
Laboratories, Inc.) and reagent Streptavidin-PE (Bio-Rad
Laboratories, Inc.) were added and measurements were obtained with
the Bio-Plex 200 system (Bio-Rad Laboratories, Inc.) at a
wavelength of 575 nm. Results were recorded as relative
fluorescence units and data were analyzed using GraphPad Prism
version 6 (GraphPad Software Inc.).
Western blot analysis
Cell treatment, protein extraction and
quantification were performed as aforementioned. Normalized amounts
of protein (approximately 30 µg each lane) were added to 12%
SDS-PAGE gels, then electrically separated and transferred onto
PVDF membranes (EMD Millipore). Membranes were blocked with 5%
skimmed milk for 1 h at 37°C and stained with rabbit
anti-phospho(p)-p70S6K (cat. no. 9205S; dilution 1:400), mouse
anti-AKT (cat. no. 2920S; dilution 1:500), mouse anti-p-AKT (cat.
no. 4051S; dilution 1:400) (all from Cell Signaling Technology,
Inc.), mouse anti-p70S6K (cat. no. 611260; dilution 1:800) and
mouse anti-Bcl-2 (cat. no. 51-6511GR; dilution 1:500) (all from BD
Biosciences). Mouse anti-β-actin antibody (cat. no. HC201; dilution
1:1000; Beijing Transgen Biotech Co., Ltd.) was used as the loading
control. Horseradish peroxidase-conjugated anti-rabbit antibody
(cat. no. HS101-01) and anti-mouse antibody (cat. no. HS201-01)
(both dilution 1:5,000; Beijing Transgen Biotech Co., Ltd.) were
used as secondary antibodies. Specific proteins were detected using
an enhanced Pierce ECL western blotting substrate (Thermo Fisher
Scientific, Inc.). Grayscale values were measured using ImageJ
v1.51s software (National Institutes of Health).
Statistical analysis
Data are presented as the mean ± standard error of
the mean of at least three independent experiments. Statistical
significance of data was determined by analysis of variance with
LSD post hoc test, using GraphPad Prism version 6 (GraphPad
Software Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Silencing ROR1 with siRNA enhances the
cytotoxicity of erlotinib in erlotinib-resistant cell lines
The present study first examined ROR1 expression
levels by flow cytometry following ROR1 silencing with siRNA in
NCI-H1975, PC-9erlo and XLA-07 cell lines. The results indicated
that the ROR1 expression level was reduced by siROR1 with all
inhibition rates >75% (Fig. 1).
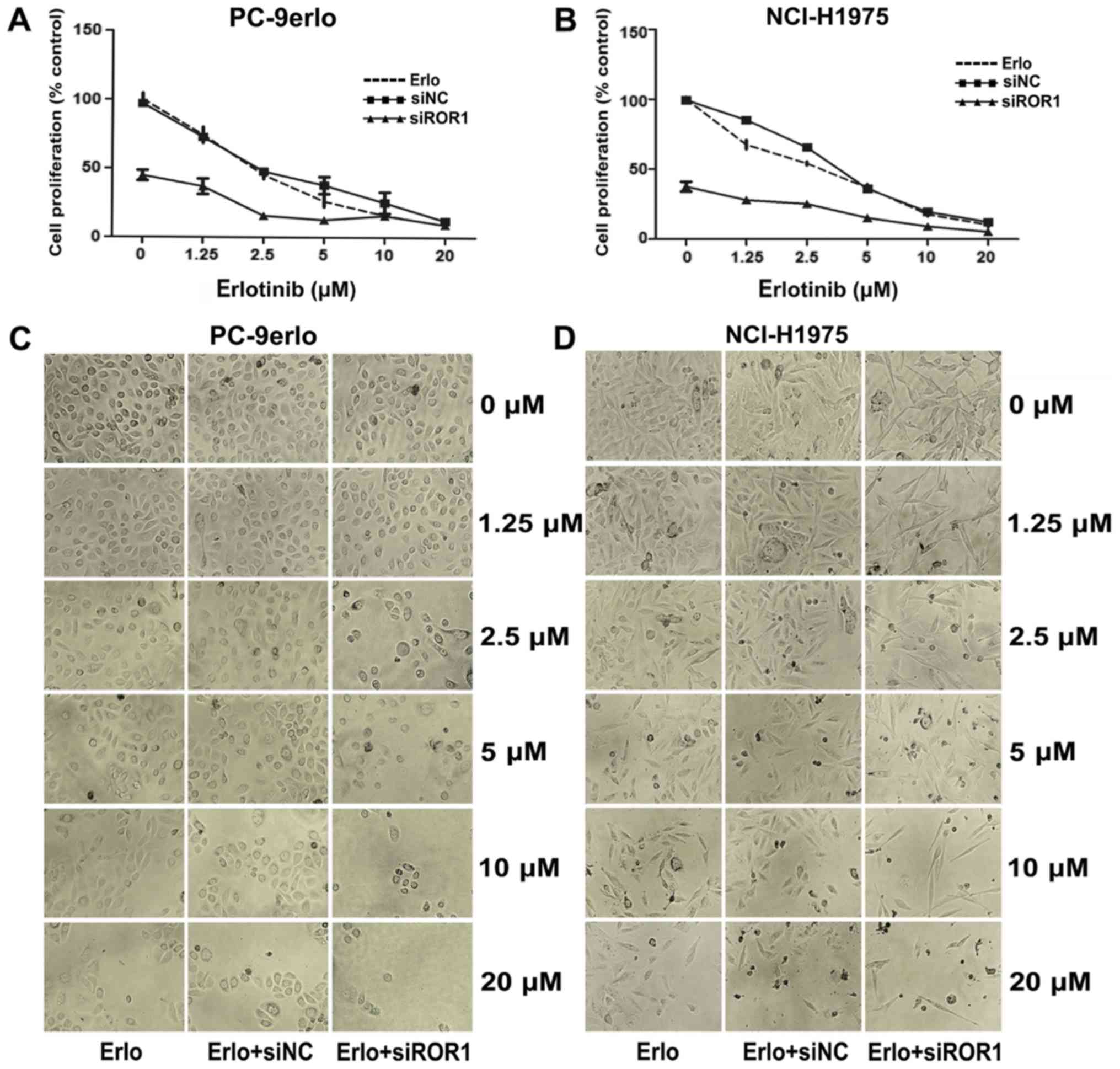
The growth inhibitory efficacy of erlotinib following ROR1
silencing was evaluated using the MTS assay. Compared with
erlotinib alone, the specific cell proliferation rates of blocking
ROR1 together with erlotinib at concentrations of 1.25, 2.5, and 5
µM in PC-9erlo cells were 36.96 vs. 74.31%, 15.67 vs. 45.27%, and
12.67 vs. 26.13%, respectively. In addition, compared with
erlotinib alone the cell proliferation rates of blocking ROR1
together with erlotinib at the aforementioned concentrations in
NCI-H1975 cells were, 27.98 vs. 67.51%, 25.18 vs. 54.01% and 15.17
vs. 36.72%, respectively (Fig. 2A and
B).
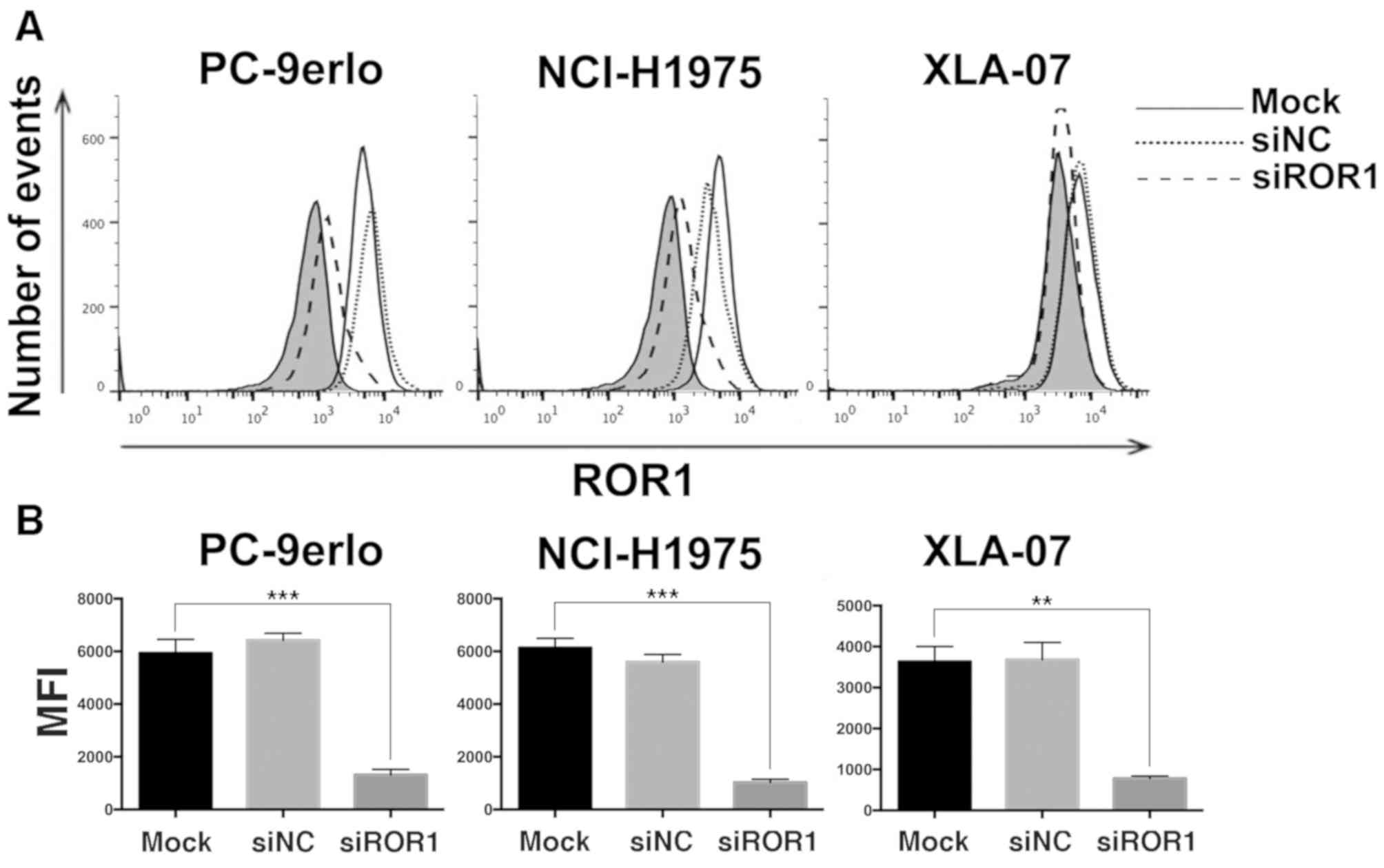 | Figure 1.Silencing ROR1 with siRNA
significantly reduces the expression of ROR1 in non-small cell lung
cancer cell lines. (A) NCI-H1975, PC-9erlo and XLA-07 cell lines
were treated with Mock, 25 nM siROR1 or siNC for 72 h, and ROR1
expression levels were examined using flow cytometry with R12, a
chimeric rabbit/human anti-ROR1 monoclonal antibody, or normal
human IgG. The background signal stained with human IgG is
presented in gray. (B) MFI value of ROR1 expression in NCI-H1975,
PC-9erlo and XLA-07 cell lines (**P<0.01, ***P<0.001, mock
vs. siROR1). Experiments were performed 3 times (n=3). The y-axis
represents the number of cells acquired by flow cytometry. ROR1,
receptor tyrosine kinase-like orphan receptor 1; si, small
interfering; NC, negative control; Mock, complete medium; MFI, Mean
fluorescence intensity. |
Blocking ROR1 enhances the
apoptosis-inducing role of erlotinib in erlotinib-resistant cell
lines
To gain further insight into the additive roles of
blocking ROR1 combined with erlotinib in erlotinib-resistant cells,
NCI-H1975, PC-9erlo and XLA-07 cell lines were treated with
complete medium (mock group), siNC alone, siROR1 alone, erlotinib
alone (Erlo), siNC plus erlotinib (Erlo+siNC) and siROR1 plus
erlotinib (Erlo+siROR1). The apoptosis rates of the cells were then
analyzed using flow cytometry (Fig.
3). All three erlotinib-resistant cell lines demonstrated a
limited response to erlotinib alone; however significantly
different apoptosis rates were revealed when treated with
Erlo+siROR1 compared with that in the Erlo group (NCI-H1975, 16.5
vs. 1.51%; PC-9erlo, 28.2 vs. 2.15%; XLA-07, 30.4 vs. 18.0%). The
expression level of Bcl-2 was then further analyzed as Bcl-2 is
considered an important antiapoptotic protein. Using western blot
analysis it was revealed that the expression level of Bcl-2 was
markedly decreased in the Erlo+siROR1 group compared with that in
the Erlo group (Fig. 4C and D),
which indicated that the activity of Bcl-2 was downregulated.
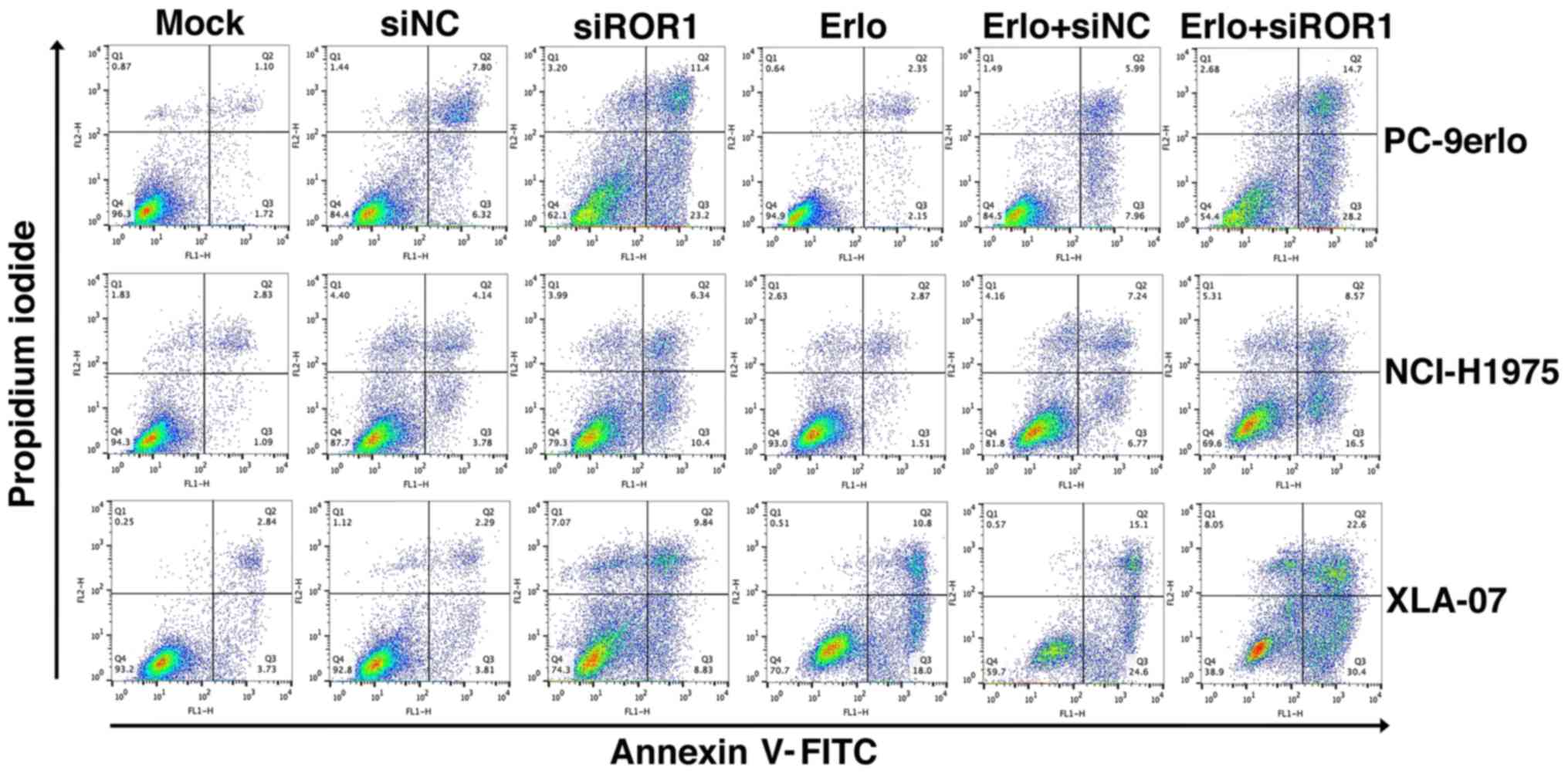 | Figure 3.Blocking ROR1 with siRNA enhances the
apoptosis-inducing role of erlotinib in non-small cell lung cancer
cell lines. NCI-H1975, PC-9erlo and XLA-07 cell lines were treated
with Mock, 20 nM siNC, 20 nM siROR1, erlotinib alone, erlotinib +
20 nM siNC or erlotinib + 20 nM siROR1. The concentration of
erlotinib was 2.5 µM for PC-9erlo and NCI-H1975 cells, and 10 µM
for XLA-07 cells. Following treatment, apoptosis of the cells was
analyzed by Annexin V/propidium iodide staining. Experiments were
performed 3 times (n=3). ROR1, receptor tyrosine kinase-like orphan
receptor 1; si, small interfering; NC, negative control; Mock,
complete medium. |
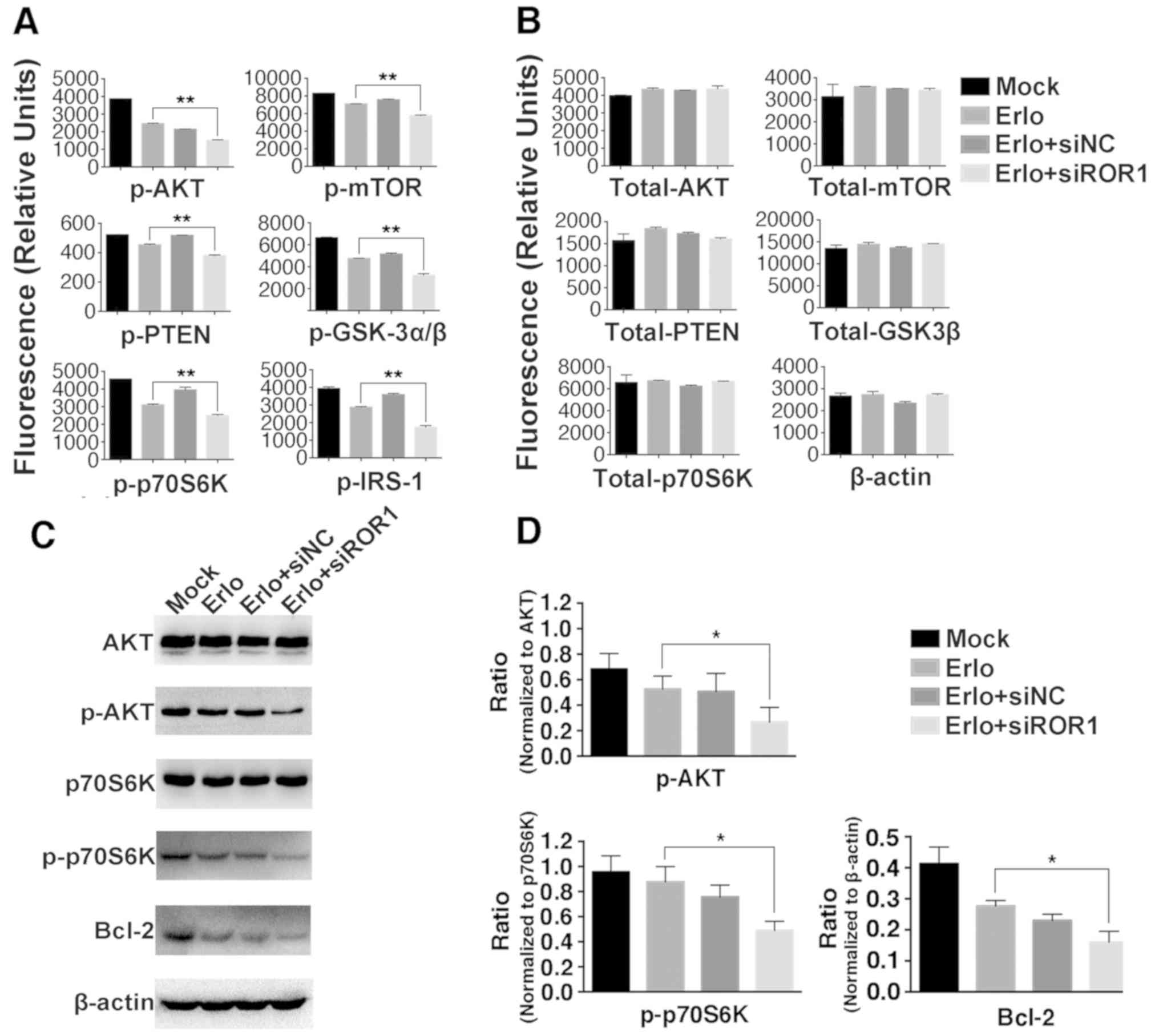 | Figure 4.Inhibition of ROR1 has an additive
role with erlotinib in the NCI-H1975 cell line via the AKT/mTOR
signaling pathway. NCI-H1975 cells were treated with Mock, 2.5 µM
erlotinib alone, 2.5 µM erlotinib + 25 nM siNC or 2.5 µM erlotinib
+ 25 nM siROR1. The (A) phosphorylated and (B) total protein levels
were analyzed using the Bio-Plex signaling AKT 8-plex panel and
Bio-Plex pro signaling reagent kit, because the kit did not contain
antibody detecting total protein of IRS-1, β-actin was used as
control instead. Values are presented as relative fluorescence
units. Data are presented as the mean of three independent
experiments. (C) The phosphorylated and total protein expression
levels of AKT, p70S6K, Bcl-2 and β-actin were determined using
western blot analysis. (D) The integrated density analysis
demonstrated the changes in the expression levels of p-AKT,
p-p70S6K and Bcl-2, and difference in phosphorylated protein levels
was analyzed using ratios between the phosphorylated/total protein.
Experiments were performed 3 times (n=3). Statistical analysis was
performed using analysis of variance. *P<0.05 and **P<0.01,
erlotinib treated group vs. erlotinib+siROR1 treated group. ROR1,
receptor tyrosine kinase-like orphan receptor 1; si, small
interfering; NC, negative control; Mock, complete medium; mTOR,
mammalian target of rapamycin; IRS-1, insulin receptor substrate 1;
GSK-3α/β, glycogen synthase kinase-3α/β; p, phosphorylated. |
Silencing ROR1 with siRNA prevents
erlotinib resistance via the AKT/mTOR signaling pathway in
NCI-H1975 cells
To investigate the molecular mechanisms of
ROR1-silencing-enhanced cytotoxicity and the apoptosis-inducing
roles of erlotinib, key molecules in the AKT/mTOR signaling pathway
were analyzed in the erlotinib-resistant NCI-H1975 cell line using
the Bio-Plex Pro assay (Fig. 4A and
B), and since the kit did not contain an antibody detecting
total protein of IRS-1, β-actin was used as control instead. It was
identified that the phosphorylation levels of IRS-1, GSK-3α/β, AKT,
p70S6K, PTEN and mTOR were significantly lower in the
Erlo+siROR1-treated group compared with that in the
erlotinib-treated group. To confirm these findings, the
phosphorylation of AKT and p70S6K was further analyzed using the
western blot assay (Fig. 4C and D).
This revealed that the activity of AKT and p70S6K was significantly
downregulated in the Erlo+siROR1-treated group compared with the
Erlo treatment group alone, which was consistent with the data from
the Bio-Plex assay.
Discussion
Treatment with TKIs provides significant benefits
for patients with EGFR mutations, particularly for those with lung
cancer. However, the majority of patients with NSCLC will acquire
resistance to first-generation EGFR-TKIs, including gefitinib and
erlotinib, following 9–14 months of treatment (7). There are two central mechanisms that
are involved in this process: EGFR secondary mutations and
alternative signaling activation (5,6). In
addition, in an EGFR-independent manner, dysregulation of other
receptor tyrosine kinases (RTKs) or abnormal activation of
downstream compounds have compensatory functions against the
inhibition of EGFR by altering the PI3K/AKT and MAPK signaling
axis. Certain studies have revealed that the proline-rich region of
the intracellular domain of ROR1 is directly activated by MET and
the pseudokinase domain is phosphorylated by Src (26,28).
Yamaguchi et al (29) also
demonstrated that a cysteine-rich domain of the extracellular
domain of ROR1 is associated with EGFR and sustains EGFR-ERBB3-PI3K
signaling. It may be beneficial to clarify whether ROR1 silencing
has an additive role with erlotinib in lung adenocarcinoma, which
could provide a potential new therapeutic strategy for patients
with lung cancer, and TKI insensitivity and resistance.
The present study selected an erlotinib-resistant
cell line NCI-H1975, which is known to be a T790M-mutant, and
another erlotinib-resistant cell line XLA-07, and an
erlotinib-acquired resistant cell line PC-9erlo, which was
developed from its parental cell line PC-9 and mimics the situation
that occurs in clinical treatment. The current results demonstrated
that ROR1 inhibition plus erlotinib have additional cytotoxic
effect in ROR1 positive lung adenocarcinoma cell lines. In
addition, it was identified that the expression level of Bcl-2, a
key regulator of antiapoptotic signaling (33), was significantly lower in
Erlo+siROR1-treated cells (Fig. 4D),
which was in accordance with the apoptosis-inducing role of ROR1
inhibition combined with erlotinib.
ROR1-mediated signaling pathways in lung cancer are
not fully understood. Our previous data suggested that the AKT/mTOR
signaling pathway is necessary for ROR1-mediated proliferation and
antiapoptosis in lung adenocarcinoma. The AKT/mTOR signaling
pathway is important for regulating cell proliferation, cancer
growth and longevity (6,34,35). The
present study investigated the association of the AKT/mTOR
signaling pathway with ROR1 silencing against erlotinib resistance
in lung cancer. Compared with erlotinib alone, phosphorylation of
key molecules in the AKT/mTOR signaling pathway, including insulin
receptor substrate 1 (IRS-1), glycogen synthase kinase-3α/β
(GSK-3α/β), PTEN, AKT, mTOR and p70S6K, was significantly lower
when ROR1 was silenced in combination with erlotinib. This supports
the hypothesis that inhibiting the ROR1-mediated signaling pathway
could partially overcome erlotinib-resistance via upregulation of
the activity of IRS-1, AKT, mTOR and p70S6K, and downregulation of
the activity of GSK-3α/β and PTEN, which are negative regulators of
PI3K/AKT (Fig. 5). Inhibiting ROR1
with small molecules and monoclonal antibodies, or inhibiting the
key regulators involved in AKT/mTOR signaling could effectively
increase the sensitivity of tumor cells to erlotinib.
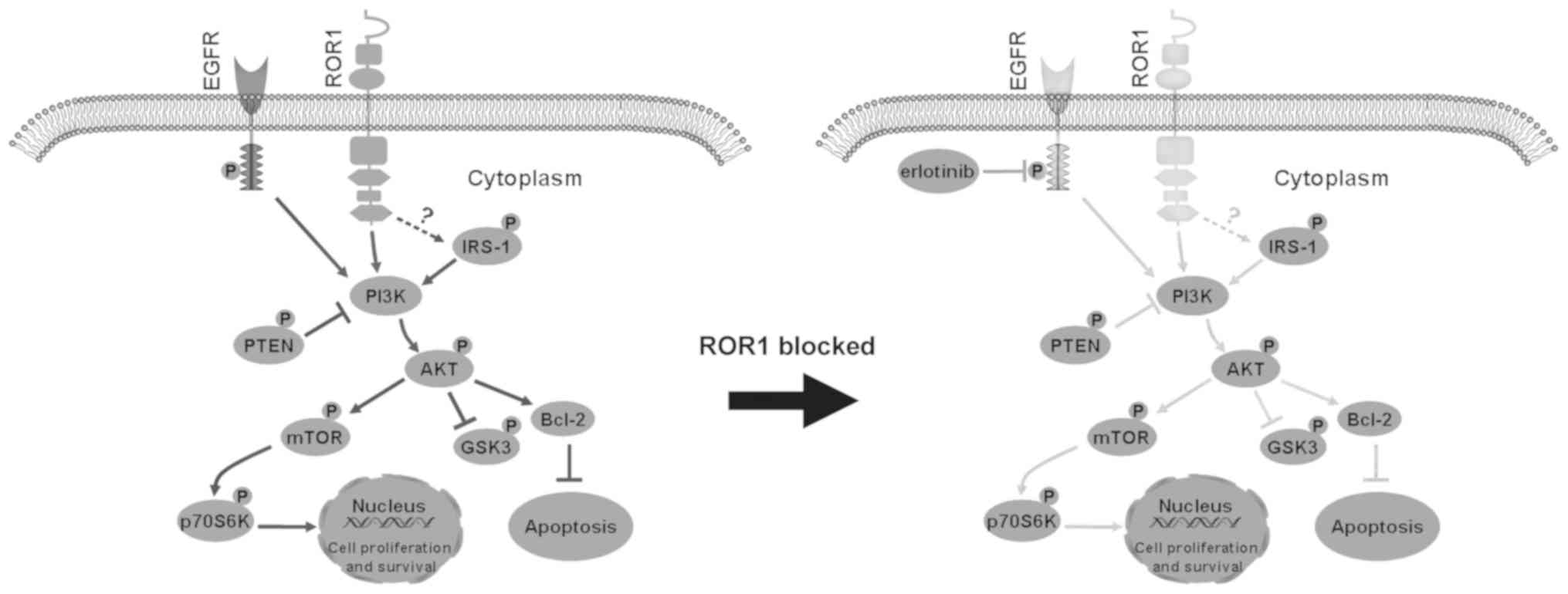 | Figure 5.Proposed model of the combined effect
of ROR1 inhibition and erlotinib treatment in non-small cell lung
cancer cell lines via inhibition of the AKT/mTOR signaling pathway.
Inhibition of ROR1 significantly decreased the activity of IRS-1,
AKT, mTOR and p70S6K, and activated GSK-3α/β and PTEN, which are
two negative regulators of PI3K/AKT signaling. This enhances cell
apoptosis, and reduces cell proliferation and survival. ROR1,
receptor tyrosine kinase-like orphan receptor 1; IRS-1, insulin
receptor substrate 1; GSK-3α/β, glycogen synthase kinase-3α/β; p,
phosphorylated; EGFR, epidermal growth factor receptor. |
The present study revealed that the protein
expression level of IRS-1, which is involved in cell proliferation,
was also significantly reduced through the inhibition of ROR1 in
combination with erlotinib. The underlying association between ROR1
and IRS-1 is unclear; however, targeting IRS-1 in NSCLC has been
reported to exhibit an antitumor effect in a number of studies
(36–38). It remains to be determined whether
interactions between ROR1 and IRS-1 directly activate IRS-1
following binding to its ligand, or whether an indirect
stabilization occurs through the association of IRS-1 with
insulin-like growth factor-1 receptors, thus transmitting signals
to the intracellular AKT/mTOR pathway.
In conclusion, the present study identified that
ROR1 is a potential target for preventing erlotinib resistance in
lung adenocarcinomas via the AKT/mTOR signaling pathway. Targeting
ROR1 with small molecules or immunological procedures may increase
the sensitizing of tumor cells, particularly erlotinib-resistant
cells, to erlotinib. Further studies should investigate the
functions of ROR1 in lung adenocarcinoma to promote the development
of ROR1-targeting therapies in the future.
Acknowledgements
The authors would like to thank Dr Christoph Rader
(Department of Immunology and Microbiology, the Scripps Research
Institute, FL, USA) and Dr Rose Mage (Laboratory of Immune System
Biology, NIAID, NIH, MD, USA) for their critical reading of this
manuscript. The authors would also like to thank Professor Yong
Duan (First Affiliated Hospital of Kunming Medical University,
Kunming, China) and Dr Jun Zhang (Shanghai Pulmonary Hospital,
Shanghai, China) for providing the NSCLC cell lines.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81401904; http://www.nsfc.gov.cn), Science and Technology
Department of Sichuan Province, China (grant no. 2018HH0005;
http://www.scst.gov.cn) and Xinglin Scholar
Discipline Talents Scientific Research Promotion Program (grant no.
XSGG2018004).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JHY and CZ conceived the study. HLW, YCL and MPL
performed the experiments. HLW, YCL, MPL and JHY analyzed the data.
HLW, YCL, CZ and JHY drafted the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ROR1
|
receptor tyrosine kinase-like orphan
receptor 1
|
|
EGFR
|
epidermal growth factor receptor
|
|
TKI
|
tyrosine kinase inhibitor
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
mTOR
|
mammalian target of rapamycin
|
|
IRS-1
|
insulin receptor substrate 1
|
|
GSK-3
|
glycogen synthase kinase 3
|
|
ERBB3
|
receptor tyrosine-protein kinase
erbB-3
|
|
MAPK
|
mitogen-activated protein kinase 1
|
|
p70S6K
|
ribosomal protein S6 kinase β-1
|
|
PTEN
|
phosphatase and tensin homolog
|
|
MET
|
hepatocyte growth factor receptor
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Osmani L, Askin F, Gabrielson E and Li QK:
Current WHO guidelines and the critical role of immunohistochemical
markers in the subclassification of non-small cell lung carcinoma
(NSCLC): Moving from targeted therapy to immunotherapy. Semin
Cancer Biol. 52:103–109. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reck M, Popat S, Reinmuth N, De Ruysscher
D, Kerr K and Peters S; ESMO Guidelines Working Group, : Metastatic
non-small-cell lung cancer (NSCLC): ESMO clinical practice
guidelines for diagnosis, treatment and follow-up. Ann Oncol. 25
(Suppl 3):iii27–iii39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nascimento A, Bousbaa H, Ferreira D and
Sarmento B: Non-small cell lung carcinoma: An overview on targeted
therapy. Curr Drug Targets. 16:1448–1463. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mitsudomi T and Yatabe Y: Epidermal growth
factor receptor in relation to tumor development: EGFR gene and
cancer. FEBS J. 277:301–308. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gadgeel SM and Wozniak A: Preclinical
rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for
epidermal growth factor receptor inhibitor-resistant non-small-cell
lung cancer. Clin Lung Cancer. 14:322–332. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tan CS, Kumarakulasinghe NB, Huang YQ, Ang
YL, Choo JR, Goh BC and Soo RA: Third generation EGFR TKIs: Current
data and future directions. Mol Cancer. 17:292018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu SG and Shih JY: Management of acquired
resistance to EGFR TKI-targeted therapy in advanced non-small cell
lung cancer. Mol Cancer. 17:382018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin Y, Wang X and Jin H: EGFR-TKI
resistance in NSCLC patients: Mechanisms and strategies. Am J
Cancer Res. 4:411–435. 2014.PubMed/NCBI
|
|
10
|
Morgillo F, Della Corte CM, Fasano M and
Ciardiello F: Mechanisms of resistance to EGFR-targeted drugs: Lung
cancer. ESMO Open. 1:e0000602016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martinez-Marti A, Felip E, Matito J, Mereu
E, Navarro A, Cedrés S, Pardo N, Martinez de Castro A, Remon J,
Miquel JM, et al: Dual MET and ERBB inhibition overcomes intratumor
plasticity in osimertinib-resistant-advanced non-small-cell lung
cancer (NSCLC). Ann Oncol. 28:2451–2457. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Heydt C, Michels S, Thress KS, Bergner S,
Wolf J and Buettner R: Novel approaches against epidermal growth
factor receptor tyrosine kinase inhibitor resistance. Oncotarget.
9:15418–15434. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Q, Yu S, Zhao W, Qin S, Chu Q and Wu
K: EGFR-TKIs resistance via EGFR-independent signaling pathways.
Mol Cancer. 17:532018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Westover D, Zugazagoitia J, Cho BC, Lovly
CM and Paz-Ares L: Mechanisms of acquired resistance to first- and
second-generation EGFR tyrosine kinase inhibitors. Ann Oncol.
29:i10–i19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Laurila N and Koivunen JP: EGFR inhibitor
and chemotherapy combinations for acquired TKI resistance in
EGFR-mutant NSCLC models. Med Oncol. 32:2052015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tricker EM, Xu C, Uddin S, Capelletti M,
Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK, et al:
Combined EGFR/MEK inhibition prevents the emergence of resistance
in EGFR-mutant lung cancer. Cancer Discov. 5:960–971. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cavazzoni A, La Monica S, Alfieri R,
Ravelli A, Van Der Steen N, Sciarrillo R, Madeddu D, Lagrasta CAM,
Quaini F, Bonelli M, et al: Enhanced efficacy of AKT and FAK kinase
combined inhibition in squamous cell lung carcinomas with stable
reduction in PTEN. Oncotarget. 8:53068–53083. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu X, Shi S, Wang H, Yu X, Wang Q, Jiang
S, Ju D, Ye L and Feng M: Blocking autophagy improves the
anti-tumor activity of afatinib in lung adenocarcinoma with
activating EGFR mutations in vitro and in vivo. Sci Rep.
7:45592017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang X, Yan L, Zhu L, Jiao D, Chen J and
Chen Q: Salvianolic acid A reverses cisplatin resistance in lung
cancer A549 cells by targeting c-met and attenuating Akt-mTOR
pathway. J Pharmacol Sci. 135:1–7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ye M, Wang S, Wan T, Jiang R, Qiu Y, Pei
L, Pang N, Huang Y, Huang Y, Zhang Z and Yang L: Combined
inhibitions of glycolysis and AKT/autophagy can overcome resistance
to EGFR-targeted therapy of lung cancer. J Cancer. 8:3774–3784.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fu Y, Li C, Luo Y, Li L, Liu J and Gui R:
Silencing of long non-coding RNA MIAT sensitizes lung cancer cells
to gefitinib by epigenetically regulating miR-34a. Front Pharmacol.
9:822018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ni J, Zhou LL, Ding L, Zhang XQ, Zhao X,
Li H, Cao H, Liu S, Wang Z, Ma R, et al: Efatutazone and T0901317
exert synergistically therapeutic effects in acquired
gefitinib-resistant lung adenocarcinoma cells. Cancer Med.
7:1955–1966. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang L, Dong X, Ren Y, Luo J, Liu P, Su D
and Yang X: Targeting EHMT2 reverses EGFR-TKI resistance in NSCLC
by epigenetically regulating the PTEN/AKT signaling pathway. Cell
Death Dis. 9:1292018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang YC, Wu DW, Wu TC, Wang L, Chen CY and
Lee H: Dioscin overcome TKI resistance in EGFR-mutated lung
adenocarcinoma cells via down-regulation of tyrosine phosphatase
SHP2 expression. Int J Biol Sci. 14:47–56. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Masiakowski P and Carroll RD: A novel
family of cell surface receptor with tyrosine kinase-like domain. J
Biol Chem. 267:26181–26190. 1992.PubMed/NCBI
|
|
26
|
Borcherding N, Kusner D, Liu GH and Zhang
W: ROR1, an embryonic protein with an emerging role in cancer
biology. Protein Cell. 5:496–502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Y, Yang H, Chen T, Luo Y, Xu Z, Li Y
and Yang J: Silencing of receptor tyrosine kinase ROR1 inhibits
tumor-cell proliferation via PI3K/AKT/mTOR signaling pathway in
lung adenocarcinoma. PLoS One. 10:e01270922015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gentile A, Lazzari L, Benvenuti S,
Trusolino L and Comoglio P: The ROR1 pseudokinase diversifies
signaling outputs in MET-addicted cancer cells. Int J Cancer.
135:2305–2316. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamaguchi T, Yanagisawa K, Sugiyama R,
Hosono Y, Shimada Y, Arima C, Kato S, Tomida S, Suzuki M, Osada H
and Takahashi T: NKX2-1/TITF1/TTF-1-Induced ROR1 is required to
sustain EGFR survival signaling in lung adenocarcinoma. Cancer
Cell. 21:348–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma LJ, Wang HZ, Bian L, Shao WP, Tang RZ,
Wang QQ and Jin KW: Establishment and characterization of lung
adenocarcinoma cell line XLA-07. Zhonghua Bing Li Xue Za Zhi.
41:335–339. 2012.(In Chinese). PubMed/NCBI
|
|
31
|
Zhao YM, Su B, Yang XJ, Shi JY, Tang L,
Zhang J, Li JY and Chen J: Small molecule inhibitor SB203580
enhances the antitumor effect of gefitinib in PC-9 and A549 lung
cancer cell lines. Zhonghua Zhong Liu Za Zhi. 35:103–108. 2013.(In
Chinese). PubMed/NCBI
|
|
32
|
Yang J, Baskar S, Kwong KY, Kennedy MG,
Wiestner A and Rader C: Therapeutic potential and challenges of
targeting receptor tyrosine kinase ROR1 with monoclonal antibodies
in B-cell malignancies. PLoS One. 6:e210182011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang T, Zhang Y, Li Y, Hao Y, Zhou M, Dong
N and Duan X: High amounts of fluoride induce apoptosis/cell death
in matured ameloblast-like LS8 cells by downregulating Bcl-2. Arch
Oral Biol. 58:1165–1173. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bonelli MA, Digiacomo G, Fumarola C,
Alfieri R, Quaini F, Falco A, Madeddu D, La Monica S, Cretella D,
Ravelli A, et al: Combined inhibition of CDK4/6 and PI3K/AKT/mTOR
pathways induces a synergistic anti-tumor effect in malignant
pleural mesothelioma cells. Neoplasia. 19:637–648. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu J, Xing Y and Rong L: miR-181
regulates cisplatin-resistant non-small cell lung cancer via
downregulation of autophagy through the PTEN/PI3K/AKT pathway.
Oncol Rep. 39:1631–1639. 2018.PubMed/NCBI
|
|
36
|
Goetsch L, Gonzalez A, Leger O, Beck A,
Pauwels PJ, Haeuw JF and Corvaia N: A recombinant humanized
anti-insulin-like growth factor receptor type I antibody (h7C10)
enhances the antitumor activity of vinorelbine and anti-epidermal
growth factor receptor therapy against human cancer xenografts. Int
J Cancer. 113:316–328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cosaceanu D, Carapancea M, Alexandru O,
Budiu R, Martinsson HS, Starborg M, Vrabete M, Kanter L, Lewensohn
R and Dricu A: Comparison of three approaches for inhibiting
insulin-like growth factor I receptor and their effects on NSCLC
cell lines in vitro. Growth Factors. 25:1–8. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu F, Zhang YJ, Li L, Zhang YJ, Han JC, Yu
YY and Ma CN: MicroRNA-214 inhibits the proliferation of non-small
cell lung cancer via the suppression of IRS1. Int J Clin Exp
Pathol. 9:22–29. 2016.
|