Introduction
Liver cancer is the third leading cause of
cancer-related mortality, particularly in developing Asian
countries, where the incidence has increased in recent years
(1). Globally, approximately 630,000
new cases of liver cancer occur annually, and more than half of
these new cases occur in China (2).
Currently, systemic chemotherapy serves an important role in the
treatment of advanced liver cancer and in therapy for liver cancer
patients with extensive disease or with poor liver function
(3,4). Cisplatin (CDDP) is one of the most
commonly used chemotherapeutic agents for advanced liver cancer and
in postoperative patients when combined with intra-arterial
chemotherapy (5). Unfortunately, the
curative efficacy of CDDP may be limited due to intrinsic or
acquired chemoresistance (6).
DNA double-strand break (DSB) repair is an important
mechanism closely related to the development of chemoresistance
(7). DNA-dependent protein kinase
catalytic subunit (DNA-PKcs) is involved in nonhomologous end
joining (NHEJ) to repair DSBs (8).
DNA-PKcs mediates a variety of cellular responses via
phosphorylation of various downstream targets in the DNA-PKcs
signaling pathway (9). DNA-PKcs
could promote activation of the mitogen-activated protein kinase
(MAPK)/IκB kinase 2 (IKK-2)/NF-κB pathway, resulting in increased
cell survival in murine embryonic fibroblasts treated with
anthracycline doxorubicin chemotherapeutic agent (9). Elliott et al (10) reported that downregulation of
DNA-PKcs sensitized chronic lymphocytic leukemia cells to
chemotherapy treatment, while upregulation of DNA-PKcs was
associated with chemoresistance and shorter survival time.
Chemosensitivity was enhanced in multidrug-resistant human leukemia
CEM cells in response to inhibition of DNA-PKcs through combined
treatment with the DNA-PKcs inhibitor wortmannin (11). These results suggest that the
DNA-PKcs signaling pathway is closely associated with development
of chemoresistance.
MicroRNAs (miRNAs/miRs) are an important class of
endogenous small noncoding RNAs that are typically approximately 22
nucleotides in length and bind to the 3′-untranslated region (UTR)
of target mRNAs, leading to the degradation of the mRNA or blocking
translation (12). Dysregulation of
miRNAs is also involved in the development of chemoresistance and
radioresistance to therapy. MicroRNA-101 (miR-101) has been shown
to efficiently sensitize tumor cells to radiation in vitro
and in vivo through degradation of DNA-PKcs via binding to
the 3′-UTR of DNA-PKcs in tumor cells (13). It has been reported that miR-101
selectively targets and downregulates DNA-PKcs, mediating
sensitivity of pancreatic cancer cells to gemcitabine (14). In addition, Chang et al
(15) found that miR-101 sensitized
osteosarcoma U2OS cells in vitro to doxorubicin treatment
via inhibition of autophagy. In liver cancer cells (HepG2), miR-101
regulates the chemosensitivity of CDDP through inhibiting autophagy
and inducing apoptosis (16).
However, the regulatory mechanism of miR-101 and its function on
chemoresistance in liver cancer remains unclear.
The aim of the present study was to examine the
effects of miR-101 on the chemotherapeutic efficacy of CDDP in
liver cancer cells. miR-101 mimic and miR-101 inhibitors were
transfected into HepG2 cells to alter the expression levels of
miR-101. Next, HepG2 cells were exposed to different concentrations
of CDDP and cytotoxicity was assessed. Furthermore, the influence
of miR-101 on expression and activity of the DNA-PKcs/protein
kinase B(Akt) pathway were examined to explore the underlying
mechanism. This study may provide new insight into the potential
role of miR-101 as a novel therapeutic target for liver cancer
treatment.
Materials and methods
Cell culture and treatments
Human liver cancer cells (HepG2) were obtained from
American Type Culture Collection (ATCC) and maintained in
Dulbecco's-modified Eagle's medium (DMEM; Invitrogen) supplemented
with 10% fetal bovine serum (FBS; Invitrogen), 0.33% sodium
bicarbonate and antibiotics (100 U/ml penicillin and 0.1 mg/ml
streptomycin) (Sigma-Aldrich) at 37°C in an atmosphere with 5%
CO2. For cisplatin (CDDP; Selleck Chemicals) treated,
HepG2 cells were incubated with different concentrations of CDDP
(0, 2 and 5 µM) for 24 h.
Transfection of miRNA
The miR-101-mimic, control-mimic, inhibitor and
inhibitor-control were designed and synthesized by Shanghai
GenePharma Co., Ltd. miRNA (~50 nM) was transfected using
Lipofectamine™ 2000 (Invitrogen) as recommended by the
manufacturer. After 48 h of transfection, total RNA was extracted
and the efficiency of transfection was verified by reverse
transcription-quantitative PCR (RT-qPCR). For in vivo
chemosensitivity assays, miR-101 mimic, control-mimic, miR-101
inhibitor and inhibitor-control sequences were embedded into the
lentiviral pGIPZ plasmid (Genechem, Inc.). HepG2 cells were
subsequently infected with the lentiviruses and inoculated into
mice to produce tumor xenografts after 24 h of transfection. miRNA
sequences were as follows: miR-101 mimic sense,
5′-UACAGUACUGUGAUAACUGAA-3′ and antisense,
5′-CAGUUAUCACAGUACUGUAUU-3′; control-mimic sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′; miR-101 inhibitor,
5′-UUCAGUUAUCACAGUAUGUA-3′; and inhibitor-control,
5′-CAGUACUUUUGUGUAGUACAA-3′.
RT-qPCR analysis
Total RNA was prepared using TRIzol®
reagent (Invitrogen). For miRNA quantification, cDNA was
synthesized with specific miRNA reverse transcriptase primers using
the TaqMan microRNA Reverse Transcription kit (Applied Biosystems).
For mRNA quantification, cDNA was reversely transcribed at 37°C for
15 min, and 95°C for 10 min using the Moloney murine leukemia virus
(M-MLV) reverse transcriptase (Toyobo Life Science), according to
the manufacturer's protocol. RNA and cDNA samples were stored at
−80°C until use. The qPCR analysis was performed on an Exicyler 96
sequence detection system (Bioneer Corporation). The final 50 µl
reaction mixture contained 25 µl SYBR-Green PCR Master Mix
(Toyobo), 2 µl primer mix (5 µM) and 10 ng cDNA. Thermocycling was
carried out as follows: 95°C for 2 min for denaturation; followed
by 40 cycles of 94°C for 15 sec, 55°C for 30 sec for amplification;
and 72°C for 30 sec for termination. The standard curve was plotted
using Origin 5.0 software (Originlab Corporation). The relative
expression of miRNA was calculated using the 2−ΔΔCq
method (17) and normalized to
glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The following
primers for GAPDH were synthesized by Beijing Dingguo Changsheng
Biotechnology Co., Ltd.: Sense, 5′-CCATGGAGAAGGCTGGGG-3′ and
antisense, 5′-CAAAGTTGTCATGGATGACC-3′.
Cell viability assay
Cell viability was measured using the Cell Counting
Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc.)
according to the manufacturer's protocol. HepG2 cells were cultured
to 70–80% confluence and treated with various concentrations of
CDDP (0, 2 and 5 µM) for 24 h. Absorbance at 450 nm was recorded
using a Multiscan Mk3 plate reader (Thermo Fisher Scientific,
Inc.). All measurements were performed at least in triplicate. CDDP
was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich). Controls
were treated with vehicle only at a final concentration of 0.1%
w/w.
Apoptosis analysis
Apoptosis was assessed in HepG2 cells using the
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide(PI)
assay (Mbchem™, Shanghai, China) according to the manufacturer's
instructions. For flow cytometry analysis, HepG2 cells were treated
with CDDP and then collected and washed twice with cold PBS
solution. Cells were suspended with 400 µl binding buffer,
incubated with 5 µl Annexin V-FITC at 37°C for 15 min and 10 µl PI
at 37°C for 5 min in the dark. Stained cells were immediately
analyzed using a flow cytometer (Becton-Dickinson and Company) with
Cell Quest 4.0.2 software (Becton-Dickinson and Company).
Comet assay
DNA single-strand breaks (SSBs) were evaluated with
alkaline single-cell gel electrophoresis (comet assay) as described
in previous studies (18,19). After CDDP treatment, HepG2 cells were
immersed in cell lysis solution (2.5 M NaCl, 100 mM
Na2EDTA, 10 mM Tris, pH 10.0; 1% Triton X-100 and 10%
DMSO) at 4°C for 1 h, then placed in electrophoresis solution (300
mM NaOH, 1 mM Na2EDTA, pH >13) for 40 min at 4°C.
After electrophoresis (25 V, ~300 mA) and subsequent neutralization
with Tris-HCl (400 mM, pH 7.5), cells were stained with PI (5 mg/l;
Sigma-Aldrich). Finally, the comet images were analyzed under a
fluorescence microscope at ×40 magnification with CASP comet
analysis 1.2.2 software (Comet Assay Software Project Lab). The
Olive tail moment of DNA (referred as DNA percentage in the tail)
was used to evaluate the level of DNA damage.
Measurement of reactive oxygen species
(ROS)
Levels of intracellular ROS were detected using the
fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA)
(Sigma-Aldrich) according to the manufacturer's instructions. After
treatment with CDDP as aforementioned, HepG2 cells were harvested,
and cell solutions were incubated with 10 µM DCFH-DA at 37°C for 20
min. Serum-free culture medium and 50 µM tert-butyl hydroperoxide
(Sigma-Aldrich) were used as negative and positive controls,
respectively. Fluorescence intensity was observed under a
fluorescence microscope (Olympus BX-51) and analyzed using
Image-pro plus 6.0 software (Media Cybernetics, Inc.). All
experiments were performed at least three times with six replicates
for each sample.
Tumor xenograft mouse model
For in vivo chemosensitivity assays,
4-week-old male BALB/c nude mice (weight 10–12 g) from Shanghai
SLAC Laboratory Animal CO. Ltd. were housed and maintained in
specific pathogen-free laminar flow cabinets. Treatments of animals
were in accordance with the guidelines established by the
Institutional Animal Care and Ethics Committee. BALB/c nude mice
were randomly separated into five groups (20 mice in each group):
Control group, mimic control, miR-101 mimic, inhibitor control, and
miR-101 inhibitor group. Control HepG2 cells (~1×107
cells) and cells infected with lentivirus containing miR-101 mimic,
mimic control, miR-101 inhibitor, or inhibitor control were
subcutaneously inoculated into the right flank of nude mice. Ten
days after injection of HepG2 cells, with no obvious enlargement of
tumor volume, each group of mice was further divided into two
subgroups, CDDP (−) and CDDP (+). Mice in the CDDP (+) group
received intraperitoneal injection of CDDP at a dose of 5 mg/kg
every 5 days for 20 days. Tumor diameter and tumor volume were
examined every five days, and tumor volume (V) was calculated using
the equation V=AxB2/2 (mm3) where A is the
largest diameter and B is the perpendicular diameter. At different
time points, four tumor-bearing mice were sacrificed, and xenograft
tumors were separated and weighed. The average tumor weights of
each group were calculated. During the experimental procedure,
animals were maintained in a specific pathogen-free environment in
a laminar flow hood.
Luciferase assay
The putative miRNA binding sites from the 3′-UTR of
DNA-PKcs or a mutant version of this 3′-UTR were amplified by PCR
using DNA samples from HepG2 cells with a TaqMan®
Universal PCR Master Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and inserted into pGL3-control luciferase
reporter vectors (Promega) according to Yan et al (13). The sequences of primers were listed
as follows: Wild-type DNA-PKcs (sense,
5′-CTAGCTTTGCATTGAATTTGGGATAACTTCAA-3′, antisense,
5′-AGCTTTGAAGTTATCCCAAATTCAATGCAAAG-3′); mutant DNA-PKcs (sense,
5′-CTAGACATAAAAGTGCTTCAAAAATCCCATGG-3′, antisense,
5′-AGCTCCATGGGATTTTTGAAGCACTTTTATGT-3′). The thermocycling
conditions of PCR were as follows: 95°C for 2 min; then 42 cycles
of 95°C for 5 sec, 63°C for 20 sec and 72°C for 10 sec. Constructed
vectors were sequenced and denoted as pGL3-DNA-PKcs-WT and
pGL3-DNA-PKcs-MUT, respectively. HepG2 cells were cotransfected
with 0.05 mg constructed luciferase reporters, 0.01 mg control
pRL-TK vector containing Renilla luciferase and 100 nM miR-101 or
miRNA-control using Lipofectamine 2000 (Invitrogen). Cells were
harvested 48 h after transfection and measured with a Dual
Luciferase Assay kit (Promega) using the luminescence microplate
reader LUMIstar Galaxy (BMG Labtech GmbH) according to the
manufacturer's protocol. Firefly luciferase activity was normalized
to Renilla luciferase activity for each transfected well.
Western blotting
Total protein samples and nuclear protein samples
were prepared after CDDP treatments using Mammalian Protein
Extraction Reagent and the Nuclear and Cytoplasmic Extraction
Reagent Kit (Thermo Fisher Scientific, Inc.), respectively. Protein
concentration was determined using the bicinchoninic acid protein
assay (Thermo Fisher Scientific, Inc.). Protein samples (60 µg per
sample) were separated by 8% SDS-PAGE and transferred onto
nitrocellulose membranes (Merck KGaA). Membranes were then blocked
with 5% skim milk at 37°C for 1 h, followed by successive
incubation with primary antibodies for 12 h at 4°C and
corresponding secondary antibodies for 4 h at 4°C. The primary
antibodies were as follow: NF-κB (p65; 1:1,000; cat. no. 8242),
phospho-IKKα/β (1:1,000; cat. no. 2697), IKKα/β (1:1,000; cat. no.
2682/2684), p-IκBα (1:1,000; cat. no. 2859), IκBα (1:1,000; cat.
no. 4812), p-Akt (Ser473; 1:1,000; cat. no. 9271), H3 (1:1,000;
cat. no. 4499), Akt (1:1,000; cat. no. 9272), p53 (1:1,000; cat.
no. 9282), caspase-3 (1:1,000; cat. no. 9662) and mammalian target
of rapamycin (mTOR; 1:1,000; cat. no. 2972) antibodies were all
purchased from Cell Signaling Technology, Inc. The DNA-PKcs
antibody (1:1,000; cat. no. sc-135886) was purchased from Santa
Cruz Biotechnology, Inc. GAPDH (1:1,000; cat. no. 5465-040)
antibody was purchased from Multisciences (Lianke) Biotech Co.,
Ltd. Anti-Rabbit IgG (H+L)/HRP and anti-Mouse IgG (H+L)/HRP
secondary antibodies were purchased from Beijing Dingguo Changsheng
Biotechnology Co., Ltd. Finally, blots were visualized using
enhanced chemiluminescence (cat. no. P0018; Beyotime Institute of
Biotechnology) and quantified using the ChemiImager system (Alpha
Innotech). GAPDH was used as the internal reference, and H3 was
used as the nuclear protein loading control. Prior to quantifying
the ratio of phosphorylated to total protein, the intensities of
phosphorylated and total protein bands were normalized to GAPDH or
H3. Each experiment was repeated at least three times with three
replicates for each sample.
Caspase activity assay
Caspase 3 activity was measured with the Caspase 3
Assay Kit (Abcam) according to the manufacturer's protocol.
Following treatment, HepG2 cells were collected and resuspended in
cell lysis buffer. The supernatant (cytosolic extract) was
collected and incubated with reactive buffer at 37°C for 1–2 h, and
the absorbance was recorded at 405 nm using a Multiscan Mk3 plate
reader (Thermo Fisher Scientific, Inc.).
Statistical analyses
All experiments were performed in triplicate with at
least three replicates for each sample. Data are expressed as the
mean ± standard error of the mean (SEM). Two-way analysis of
variance (ANOVA) was performed to test the comparisons and
corrected by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
miR-101 affects cell survival and
apoptosis in HepG2 cells exposed to CDDP
To investigate the effects of miR-101 on the
therapeutic efficiency of CDDP treatment in HepG2 cells, miR-101
expression was altered through transfection with a miR-101 mimic or
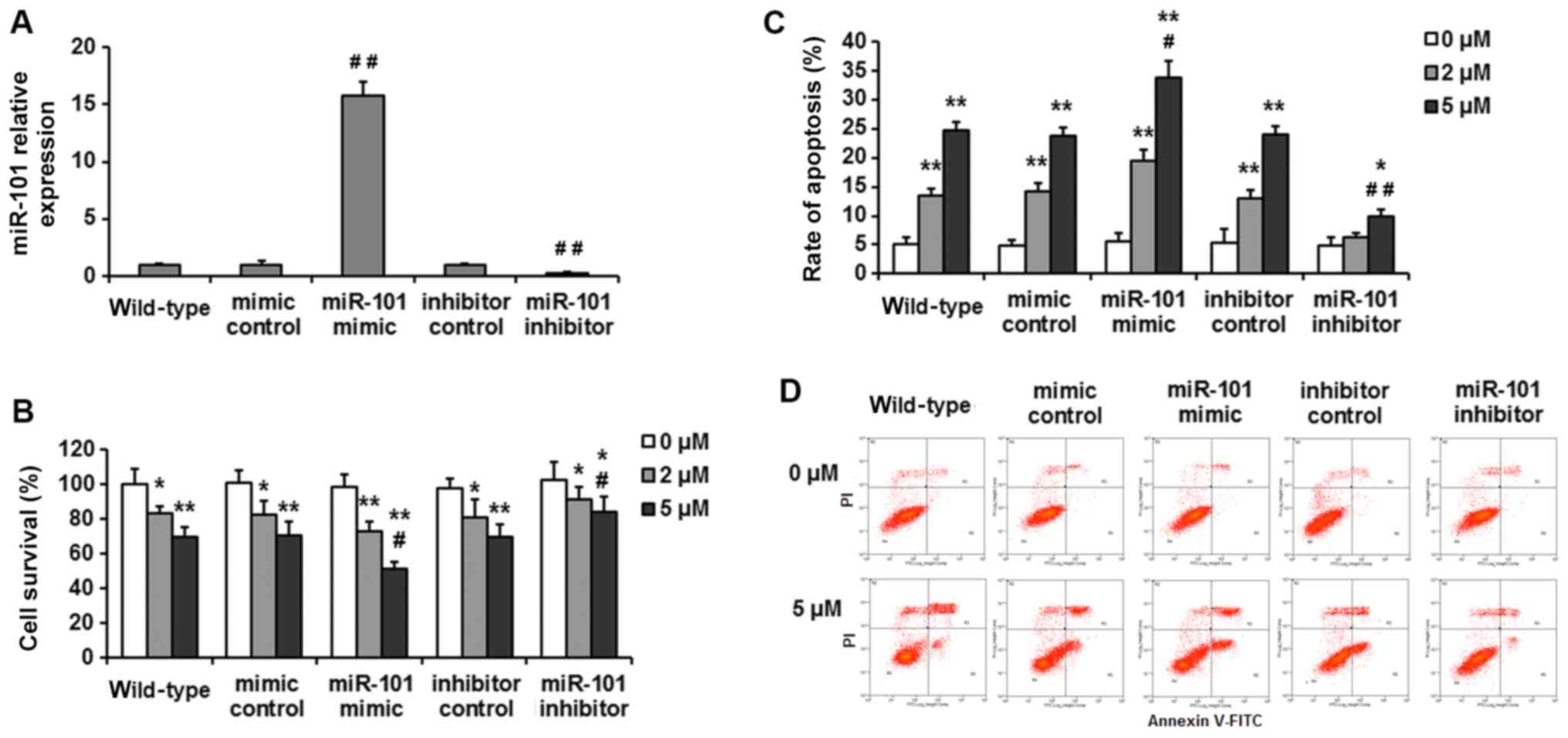
miR-101 inhibitor. Expression levels of miR-101 were confirmed by
RT-qPCR. miR-101 mimic-treated cells exhibited 15.8-fold higher
miR-101 expression levels compared with that of mimic-control cells
and wild-type HepG2 cells. In contrast, miR-101 inhibitor-treated
cells exhibited 70% reduced expression compared with
control-treated cells (Fig. 1A).
These HepG2 cells with differing expression of miR-101 were treated
with CDDP (0, 2 or 5 µM) in subsequent experiments.
Cell viability was assessed using the CCK-8 assay.
Transfection with mimic-control and inhibitor-control had no effect
on cell viability compared with wild-type HepG2 cells (Fig. 1B). Cell survival reduced by ~16.8±3.9
and 30.2±5.5% after 24-h treatment with 2 and 5 µM CDDP,
respectively in wild-type HepG2 cells. miR-101 overexpression
significantly enhanced the sensitivity of HepG2 cells to CDDP
treatment compared to the mimic control group, with cell viability
reduced by 25.6±5.1 and 47.4±4.0% for the two doses of CDDP. In
addition, miR-101 knockdown partially attenuated the cytotoxic
effects of CDDP treatment, with viability decreased by only ~10.5
and 17.3% for these two doses.
To investigate the role of miR-101 in apoptosis of
HepG2 cells, CDDP-induced apoptosis was assessed by flow cytometry.
The wild-type HepG2 cells had an apoptosis rate of 13.5±1.2 and
24.7±1.3% in response to 2 and 5 µM of CDDP treatment. The
miR-101-overexpressing cells exhibited an increased apoptosis rate
compared with the mimic control group (19.4±2.05 and 33.9±2.76%;
P<0.05), whereas miR-101-knockdown groups exhibited a reduced
apoptosis rate compared with the inhibitor control in response to
CDDP treatment (6.3±0.74 and 9.8±1.27%; P<0.05) (Fig. 1C and D). These findings indicate that
increased miR-101 expression may enhance cisplatin sensitivity in
HepG2 cells.
miR-101 expression impacts DNA damage
and ROS levels in HepG2 cells exposed to CDDP
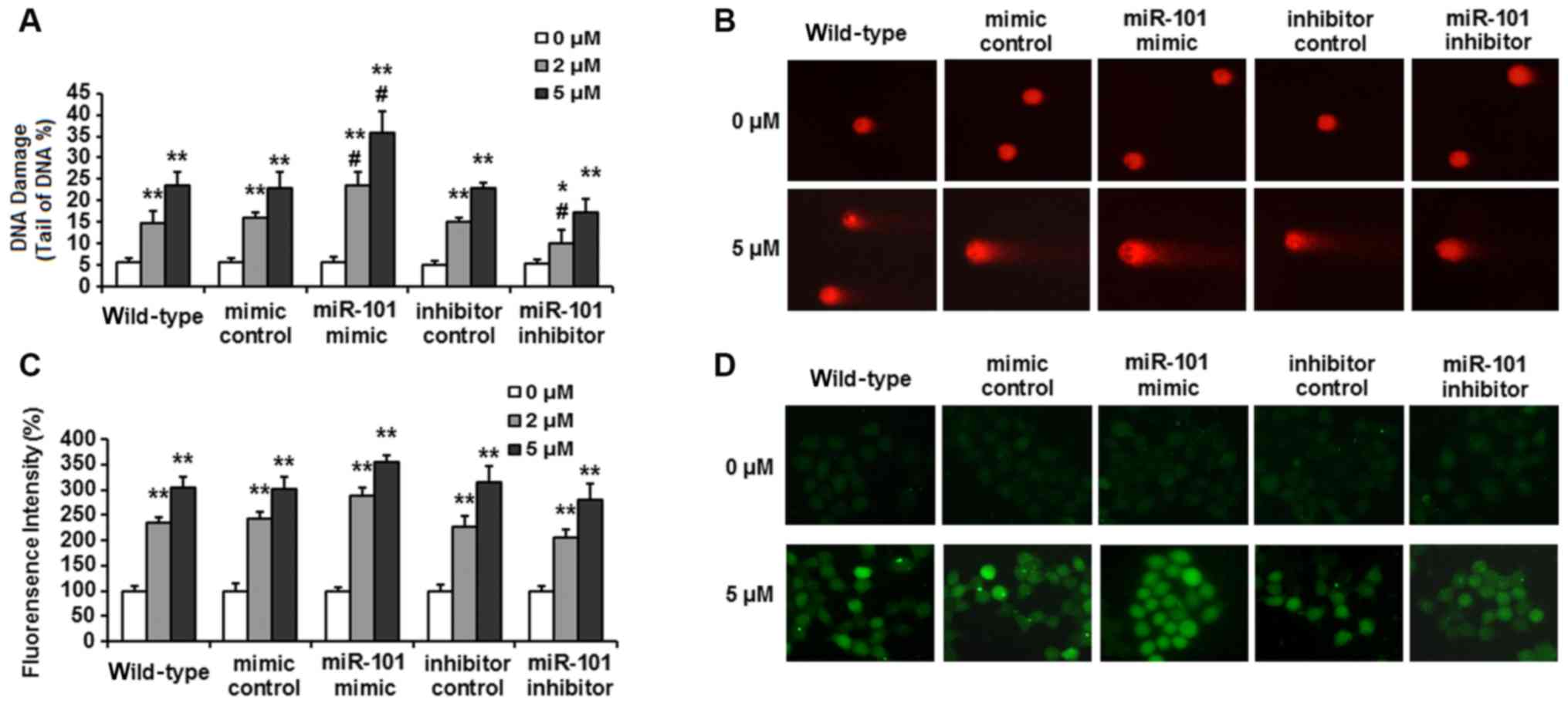
Increased repair or tolerance of DNA damage is one
of the potential mechanisms for cisplatin resistance (20,21). In
the present study, the results of a comet assay (Fig. 2A and B) showed that CDDP induced
significant DNA SSB damage in wild-type HepG2 cells, with the DNA
damage rate increasing from ~5.6±1.0% in untreated cells to
14.8±2.9 and 23.7±3.1% after treatment for 24 h with 2 and 5 µM of
CDDP, respectively. miR-101 overexpression significantly enhanced
DNA damage, with the DNA damage rate increasing to 23.6±3.2 and
35.9±4.9% (P<0.05; vs. mimic control group) for the two doses of
CDDP. miR-101 inhibition decreased CDDP-induced DNA damage to
10.1±3.2 and 17.3±3.0% (P<0.05; vs. inhibitor control).
In response to treatment with 2 and 5 µM CDDP for 24
h, intracellular ROS significantly increased to ~235 and 305% of
untreated cells in wild-type HepG2 cells (Fig. 2C). A similar degree of ROS increase
also occurred in HepG2 cells transfected with control-mimic and
inhibitor-control. With miR-101 overexpression, ROS levels of the
groups treated with 2 and 5 µM CDDP increased to 289±15.8 and
356±12.2% compared with the untreated group, respectively. In cells
with knockdown of miR-101, ROS levels increased to 205±17.2 and
280±32.5% of the untreated group (Fig.
2C and D). These results suggest that the chemosensitizing
effect of miR-101 in HepG2 cells may be associated with oxidative
stress.
miR-101 expression regulates tumor
growth and sensitivity to CDDP in BALB/c nude mice
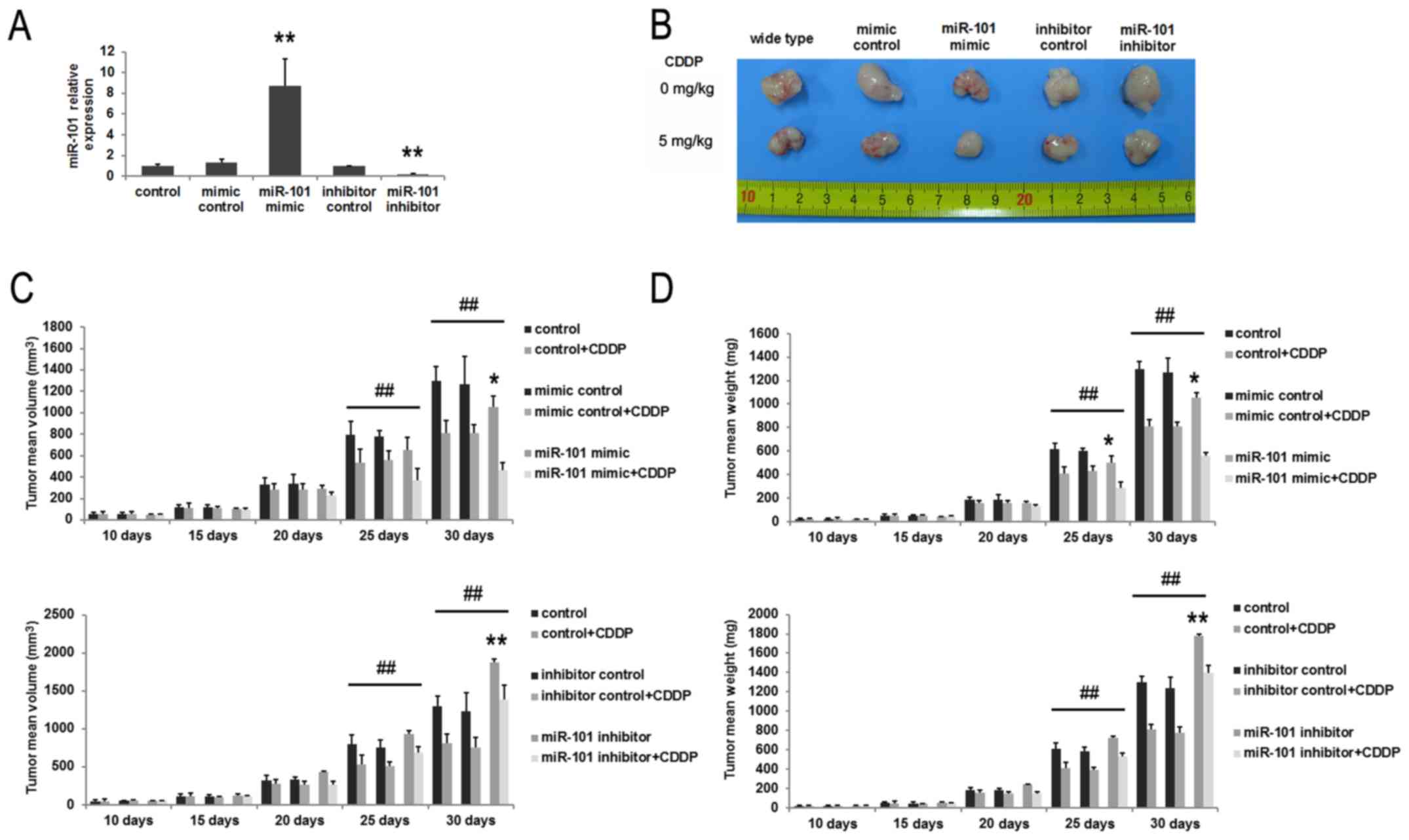
To assess tumor growth rates in vivo, HepG2
cells were infected with lentivirus containing miR-101 mimic,
control-mimic, miR-101 inhibitor, or inhibitor control. Expression
levels of miR-101 were confirmed by RT-qPCR. miR-101
mimic-transfected cells exhibited 8.7-fold higher expression
compared with that of mimic-control-transfected cells and control
HepG2 cells. By contrast, the expression level of miR-101 in cells
transfected with miR-101 inhibitor reduced by 81% compared with
that of control cells (Fig. 3A).
Next, infected HepG2 cells were subcutaneously inoculated into nude
mice (day 0). After 10 days, half the mice were treated with CDDP
at 5 mg/kg every 5 days for 20 days, and tumor volume and weight
were examined every 5 days. For non-CDDP groups, treatment of
miR-101 mimic control or inhibitor control had no effects on tumor
growth compared to controls. miR-101 overexpression, however,
inhibited tumor growth in vivo, and miR-101 suppression
promoted tumor growth (Fig. 3B, C and
D). CDDP treatment suppressed tumor growth (Fig. 3D), with a mean average inhibition
rate of 37.5% for tumor weight on day 25. Combined treatment of
CDDP plus miR-101 exerted the most marked inhibition of growth,
reducing tumor weight by 55.6% on day 25. By contrast, addition of
the miR-101 inhibitor attenuated the effects of CDDP to 25.8%
inhibition. Tumor growth in the groups treated with miR-101 mimic
control and CDDP and with miR-101 inhibitor control and CDDP was
roughly equivalent to that of the control plus CDDP groups
(Fig. 3C and D).
miR-101 expression regulates protein
expression of the DNA-PKcs/Akt pathway and alters expression of
apoptosis related proteins
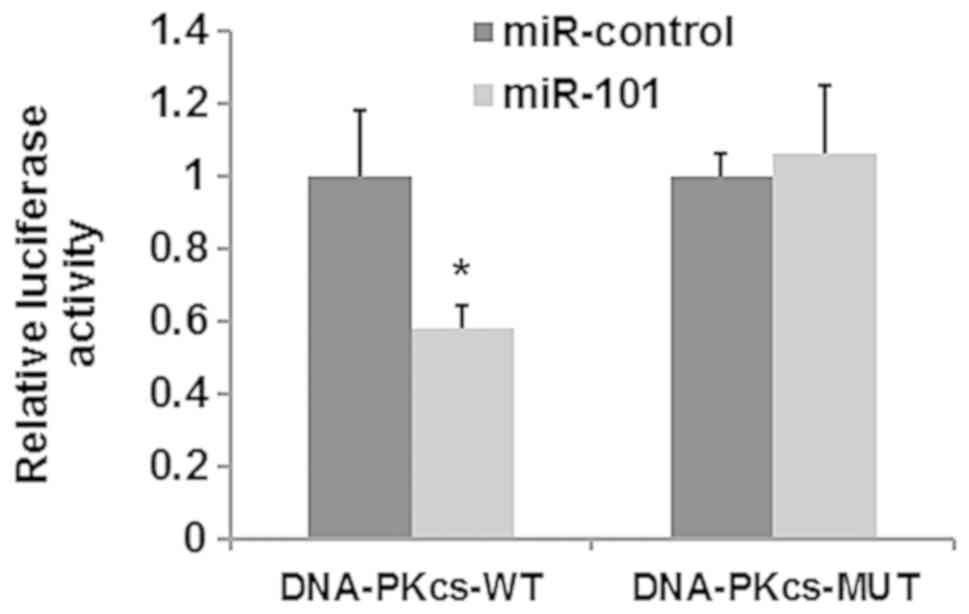
A luciferase reporter assay was performed to
identify whether DNA-PKcs is a target of miR-101. As shown in
Fig. 4, transfection with miR-101
mimic significantly suppressed the luciferase activity in cells
co-transfected with DNA-PKcs 3′-UTR-containing plasmid, while
having no effect on cells containing plasmids carrying a mutant
3′UTR sequence. These results suggested that miR-101 efficiently
targets DNA-PKcs via binding to the 3′-UTR of DNA-PKcs in HepG2
cells. To explore the underlying molecular mechanism of
miR-101-mediated chemosensitivity, expression of DNA-PKcs/Akt/NF-κB
pathway related proteins, including DNA-PKcs, Akt, phosphorylated
Akt, mTOR, caspase-3, p53, phospho-IKK-α/β and NF-κB were assessed.
Western blot results revealed that miR-101 mimic decreased Akt
phosphorylation in HepG2 cells compared to the control group, and
no change was observed in total levels of Akt protein (P<0.05;
Fig. 5A and C). In addition,
phosphorylation level of the downstream target protein mTOR was
significantly downregulated (Fig. 5A and
C). miR-101 inhibitor increased expression of DNA-PKcs, p-Akt
and p-mTOR compared with control group (Fig. 5A and C), indicating that miR-101 may
downregulate the DNA-PKcs/Akt pathway. In addition, CDDP treatment
(2 µM) for 24 h reduced the expression of DNA-PKcs, p-Akt and
p-mTOR compared with the CDDP treatment alone, while the
pro-apoptotic factors p53 and caspase-3 were elevated in response
to CDDP treatment (Fig. 5A-C).
Caspase 3 activity was also significantly increased in cells
treated with CDDP and miR-101 mimic compared to that in the control
or control + CDDP groups (Fig. 5D),
consistent with the effect of miR-101 mimic and CDDP on apoptosis
in vitro (Fig. 1C).
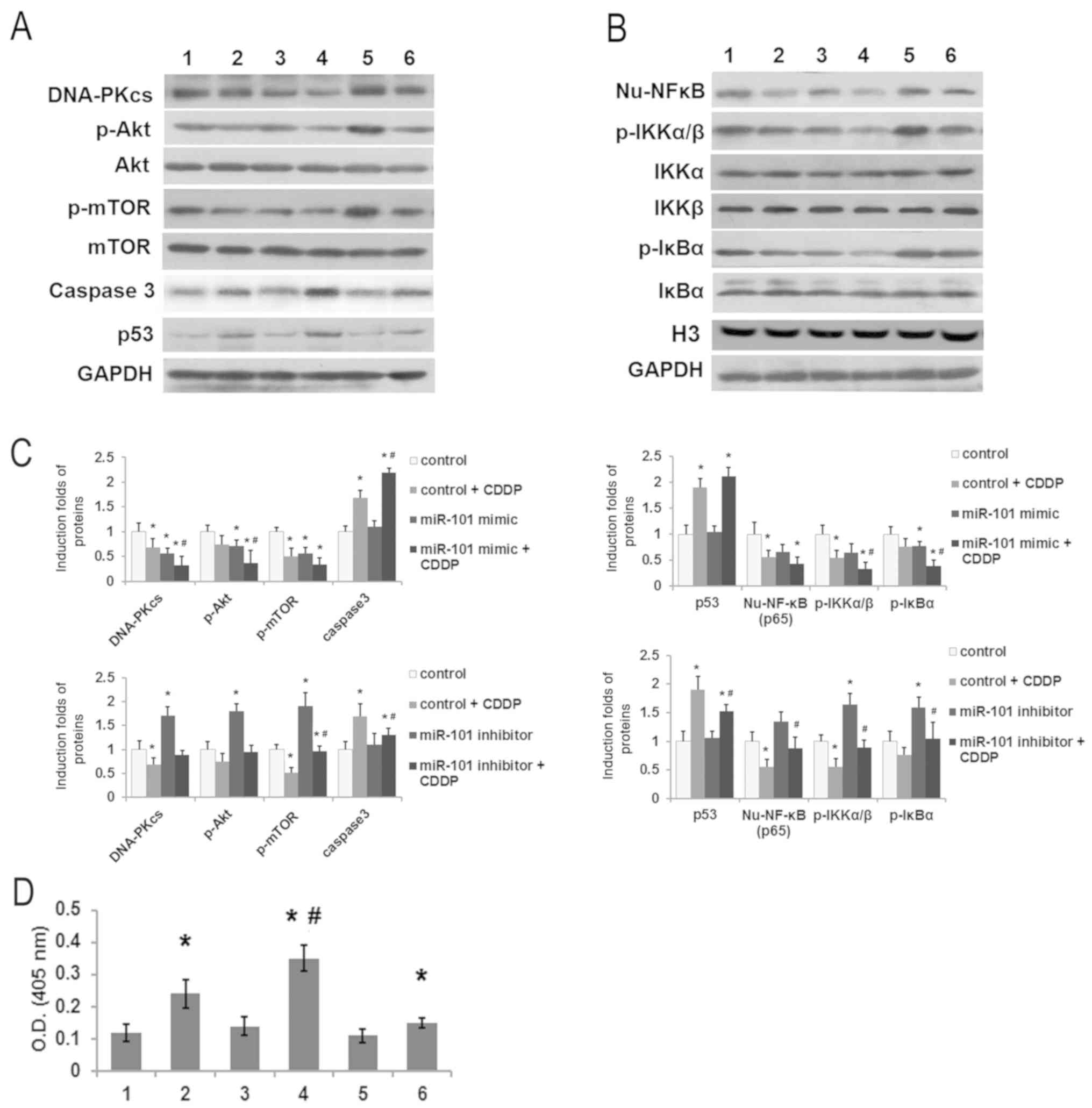 | Figure 5.miR-101 affects protein expression of
the DNA-PKcs/Akt/NF-κB pathway. HepG2 cells were transfected with
miR-101 mimic or miR-101 inhibitor and treated with 0 or 5 µM CDDP.
(A) Representative images of western blot analysis of DNA-PKcs,
p-Akt, Akt, p-mTOR, mTOR, caspase-3, p53 and GAPDH; (B)
representative images of western blot analysis of nuclear NF-κB
(Nu-NF-κB, P65), p-IKKα/β, IKKα, IKKβ, p-IκBα, IκBα, H3 and GAPDH.
(C) Quantified results of western blot analysis with three
replicates for each sample. Intensity of bands for DNA-PKcs,
caspase-3 and p53 were normalized using GAPDH as the internal
reference. Bands for p-Akt, p-mTOR, p-IKKα/β and p-IκBα were
normalized to GAPDH/H3 and are presented as a ratio of
phosphorylated to total protein for each. Expression of NF-κB was
quantified using H3 as the internal reference. (D) Activity of
caspase 3. 1, control; 2, control + CDDP; 3, miR-101 mimic; 4,
miR-101 mimic + CDDP; 5, miR-101 inhibitor; and 6, miR-101
inhibitor + CDDP. *P<0.05 compared to control group.
#P<0.05 compared to control + CDDP group. miR-101,
microRNA-101; DNA-PKcs, DNA-dependent protein kinase catalytic
subunit; CDDP, cisplatin; Akt, protein kinase B; p-Akt,
phosphorylated Akt; mTOR, mammalian target of rapamycin; p-mTOR,
phosphorylated mTOR; p53, tumor protein P53; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; NF-κB, nuclear factor κB;
IKKα/β, inhibitor of κB kinase α/β; p-IKKα/β, phosphorylated
IKKα/β; IκBα, inhibitor of NF-κB α; p-IκBα, phosphorylated IκBα;
H3, histone 3. |
The NF-κB pathway serves crucial roles in anticancer
strategies. Consistent with the results for the DNA-PKcs/Akt
pathway, miR-101 mimic also inhibited the NF-κB pathway, as
evidenced by the downregulation of nuclear NF-κB (Nu-NF-κB, p65)
and decreased phosphorylation of IKKα/β and IκBα. Additionally,
miR-101 inhibitor activated the NF-κB pathway, resulting in
enhanced protein expression and phosphorylation (Fig. 5B and C). This study also showed that
expression of Nu-NF-κB and phosphorylation of IKKα/β and IκBα were
reduced in HepG2 cells treated with CDDP alone for 24 h (Fig. 5B and C).
Discussion
CDDP is an effective cytotoxic platinum agent used
in chemotherapy, particularly in patients with unresectable liver
cancer (22). However,
chemoresistance to CDDP has become a barrier to its clinical
effectiveness, with low response rates varying from 22–29% in liver
cancer treatment (23). Although
several factors contributing to CDDP resistance have been reported
(24,25), the underlying mechanism of acquired
chemoresistance remains poorly understood. Drug resistance is an
important issue that remains to be resolved in liver cancer
chemotherapy.
Numerous studies have reported that miRNAs regulate
the response to carcinogenesis and chemosensitivity in multiple
types of cancer, including liver cancer (26,27).
miR-142-3p targets autophagy protein 5 and autophagy-related
protein 16-1 to inactivate autophagy and sensitizes liver cancer to
sorafenib (28). miR-205-5p
regulates the chemotherapeutic resistance of liver cancer to
5-fluorouracil by targeting the PTEN/JNK/ANXA3 pathway (29). miR-101 was initially identified as a
tumor suppressor miRNA in a human cancer by Xu et al
(16). It was demonstrated that
miR-101 serves an important role in cisplatin-induced apoptosis in
liver cancer cells through inhibition of autophagy via modulating
the expression of Ras-related protein RAB5A, stathmin and cysteine
protease ATG4D. He et al (30) reported that miR-101 sensitizes liver
cancer cell lines to doxorubicin-induced apoptosis by targeting
apoptosis regulator MCL1. In vitro testing demonstrated that
miR-101 binds to the 3′-UTR of DNA-PKcs to regulate protein levels
of DNA-PKcs and mediates the sensitivity of lung cancer cells to
radiation (13). However, the
molecular mechanism of how miR-101 regulates resistance of liver
cancer cells to CDDP treatment remains unclear.
CDDP induces DNA-adducts and DNA damage by DNA
crosslinking via displacement of the chloride ligand, which could
consequently induce cell proliferation inhibition and cell death
(24). As shown in Fig. 2, miR-101 overexpression enhanced
CDDP-induced DNA damage, and miR-101 knockdown partially relieved
DNA single-strand damage. This may partially explain the findings
in this study that overexpression of miR-101 significantly
sensitized HepG2 cells to CDDP treatment, while downregulation of
miR-101 reduced chemosensitivity (Fig.
1). Overexpression of miR-101 significantly elevated ROS
production, while miR-101 downregulation reduced ROS levels induced
by CDDP treatment in HepG2 cells, indicating that CDDP cytotoxicity
was associated with intracellular redox status. A xenograft mouse
model further confirmed that miR-101 overexpression inhibited tumor
growth, and miR-101 knockdown could promote tumor growth (Fig. 3). These results suggest that miR-101
may increase cisplatin sensitivity in HepG2 cells through
modulation of DNA damage and intracellular ROS levels.
The PI3K/Akt pathway is involved in stimulation of
tumor cell survival, invasive behavior and chemosensitivity in
numerous types of malignancy (31,32). As
one of the PI3K family members, DNA-PK plays an important role in
NHEJ to repair DSBs, which are a major mechanism of CDDP
cytotoxicity (23). A previous study
demonstrated that suppression of DNA-PKcs results in sensitization
of HepG2 cells to CDDP treatment (33). In the present study, the western blot
results revealed that miR-101 overexpression markedly reduced the
expression of DNA-PKcs, as well as phosphorylation of Akt and mTOR.
By contrast, miR-101 knockdown increased the expression of DNA-PKcs
and further elevated the activity of Akt/mTOR signaling. These
results confirm a regulatory role for DNA-PKcs in chemoresistance
mediated by miR-101. Moreover, combining treatment of miR-101 mimic
and CDDP significantly increased the expression of the apoptotic
protein caspase 3 compared with CDDP treatment alone, and miR-101
knockdown partially reversed caspase-3 expression induced by CDDP
treatment, consistent with results of the apoptosis assay.
Two serine threonine IκB kinases, IKKα and IKKβ, are
responsible for phosphorylation, ubiquitination and subsequent
degradation of IκB molecules, which facilitate the release of NF-κB
from its binding with IκB (34).
This allows free NF-κB to translocate from the cytoplasmic into the
nucleus, where it exerts transcriptional activity (35). Physiological processes such as cell
proliferation, invasion and cell death are regulated by NF-κB
release (36). CDDP is a potent
inhibitor of NF-κB pathway activation that inhibits tumor growth
(37). Western blot results in the
present study revealed that that CDDP treatment led to reduced
nuclear translocation of NF-κB/p65 protein, as well as reduced
phosphorylation levels of IKKα/β and IκB. Moreover, suppression of
NF-κB by CDDP was further aggravated by miR-101 mimic, which may
make HepG2 cells more susceptible to chemotherapeutic agents. These
data suggest that miR-101 attenuates chemoresistance to CDDP in
HepG2 cells through the DNA-PKcs/Akt/NF-κB pathway.
In conclusion, miR-101 overexpression augmented the
cytotoxicity of CDDP and reduced chemoresistance in HepG2 cells,
and these phenomena were associated with negative regulation of the
DNA-PKcs/Akt/NF-κB signaling pathway. The present study provides
new insight into a potential alternative therapeutic strategy for
liver cancer treatment using miR-101 as antitumor miRNA.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Funds of China (grant nos. 81272393 and 81702335).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZC, XY, FL and JC conducted the experiments. SC
designed the research. JS, JXS and CL acquired and analyzed the
data. ZC and SC drafted and revised the manuscript.
Ethics approval and consent to
participate
Animal experiments were approved by the Experimental
Animal Ethics Committee of Second Military Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Argyrou C, Moris D and Vernadakis S:
Hepatocellular carcinoma development in non-alcoholic fatty liver
disease and non-alcoholic steatohepatitis. Is it going to be the
‘Plague’ of the 21st century? A literature review focusing on
pathogenesis, prevention and treatment. J BUON. 22:6–20.
2017.PubMed/NCBI
|
|
2
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin DY, Lin SM and Liaw YF: Non-surgical
treatment of hepatocellular carcinoma. J Gastroenterol Hepatol.
12:S319–S328. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ueno H, Okada S, Okusaka T, Ikeda M and
Kuriyama H: Phase I and pharmacokinetic study of 5-fluorouracil
administered by 5-day continuous infusion in patients with
hepatocellular carcinoma. Cancer Chemother Pharmacol. 49:155–160.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Falkson G, Ryan LM, Johnson LA, Simson IW,
Coetzer BJ, Carbone PP, Creech RH and Schutt AJ: A random phase II
study of mitoxantrone and cisplatin in patients with hepatocellular
carcinoma. An ECOG study. Cancer. 60:2141–2145. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ghosh S: Cisplatin: The first metal based
anticancer drug. Bioorg Chem. 88:1029252019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roos WP, Frohnapfel L, Quiros S, Ringel F
and Kaina B: XRCC3 contributes to temozolomide resistance of
glioblastoma cells by promoting DNA double-strand break repair.
Cancer Lett. 424:119–126. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sirbu BM and Cortez D: DNA damage
response: Three levels of DNA repair regulation. Cold Spring Harb
Perspect Biol. 5:a0127242013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Panta GR, Kaur S, Cavin LG, Cortés ML,
Mercurio F, Lothstein L, Sweatman TW, Israel M and Arsura M: ATM
and the catalytic subunit of DNA-dependent protein kinase activate
NF-kappaB through a common MEK/extracellular signal-regulated
kinase/p90(rsk) signaling pathway in response to distinct forms of
DNA damage. Mol Cell Biol. 24:1823–1835. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Elliott SL, Crawford C, Mulligan E,
Summerfield G, Newton P, Wallis J, Mainou-Fowler T, Evans P,
Bedwell C, Durkacz BW and Willmore E: Mitoxantrone in combination
with an inhibitor of DNA-dependent protein kinase: A potential
therapy for high risk B-cell chronic lymphocytic leukaemia. Br J
Haematol. 152:61–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim SH, Um JH, Dong-Won B, Kwon BH, Kim
DW, Chung BS and Kang CD: Potentiation of chemosensitivity in
multidrug-resistant human leukemia CEM cells by inhibition of
DNA-dependent protein kinase using wortmannin. Leuk Res.
24:917–925. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan D, Ng WL, Zhang X, Wang P, Zhang Z, Mo
YY, Mao H, Hao C, Olson JJ, Curran WJ and Wang Y: Targeting
DNA-PKcs and ATM with miR-101 sensitizes tumors to radiation. PLoS
One. 5:e113972010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu H, He Y, Wang Y, Chen W, Hu B and Gu Y:
micorRNA-101 silences DNA-PKcs and sensitizes pancreatic cancer
cells to gemcitabine. Biochem Biophys Res Commun. 483:725–731.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang Z, Huo L, Li K, Wu Y and Hu Z:
Blocked autophagy by miR-101 enhances osteosarcoma cell
chemosensitivity in vitro. ScientificWorldJournal. 2014:7947562014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu Y, An Y, Wang Y, Zhang C, Zhang H,
Huang C, Jiang H, Wang X and Li X: miR-101 inhibits autophagy and
enhances cisplatin-induced apoptosis in hepatocellular carcinoma
cells. Oncol Rep. 29:2019–2024. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tice RR, Agurell E, Anderson D, Burlinson
B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC and Sasaki
YF: Single cell gel/comet assay: Guidelines for in vitro and in
vivo genetic toxicology testing. Environ Mol Mutagen. 35:206–221.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
An J, Wang X, Guo P, Zhong Y, Zhang X and
Yu Z: Hexabromocyclododecane and polychlorinated biphenyls increase
resistance of hepatocellular carcinoma cells to cisplatin through
the phosphatidylinositol 3-kinase/protein kinase B pathway. Toxicol
Lett. 229:265–272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mueller T, Voigt W, Simon H, Fruehauf A,
Bulankin A, Grothey A and Schmoll HJ: Failure of activation of
caspase-9 induces a higher threshold for apoptosis and cisplatin
resistance in testicular cancer. Cancer Res. 63:513–521.
2003.PubMed/NCBI
|
|
21
|
Okouoyo S, Herzer K, Ucur E, Mattern J,
Krammer PH, Debatin KM and Herr I: Rescue of death receptor and
mitochondrial apoptosis signaling in resistant human NSCLC in vivo.
Int J Cancer. 108:580–587. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamaguchi T, Nakajima N, Nakamura I,
Mashiba H, Kawashiro T, Ebara K, Ichimura E, Nishimura C, Okamoto
K, Ichikawa Y and Ichida T: Preclinical anticancer effects and
toxicologic assessment of hepatic artery infusion of fine-powder
cisplatin with lipiodol in vivo. Drug Discov Ther. 7:201–208.
2013.PubMed/NCBI
|
|
23
|
Ellis PA, Norman A, Hill A, O'Brien ME,
Nicolson M, Hickish T and Cunningham D: Epirubicin, cisplatin and
infusional 5-fluorouracil (5-FU) (ECF) in hepatobiliary tumours.
Eur J Cancer 31A. 1594–1598. 1995. View Article : Google Scholar
|
|
24
|
Pruefer FG, Lizarraga F, Maldonado V and
Melendez-Zajgla J: Participation of Omi Htra2 serine-protease
activity in the apoptosis induced by cisplatin on SW480 colon
cancer cells. J Chemother. 20:348–354. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stordal B and Davey M: Understanding
cisplatin resistance using cellular models. IUBMB Life. 59:696–699.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Callegari E, Gramantieri L, Domenicali M,
D'Abundo L, Sabbioni S and Negrini M: MicroRNAs in liver cancer: A
model for investigating pathogenesis and novel therapeutic
approaches. Cell Death Differ. 22:46–57. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lan H, Lu H, Wang X and Jin H: MicroRNAs
as potential biomarkers in cancer: Opportunities and challenges.
Biomed Res Int. 2015:1250942015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang K, Chen J, Zhou H, Chen Y, Zhi Y,
Zhang B, Chen L, Chu X, Wang R and Zhang C: PU.1/microRNA-142-3p
targets ATG5/ATG16L1 to inactivate autophagy and sensitize
hepatocellular carcinoma cells to sorafenib. Cell Death Dis.
9:3122018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shao P, Qu WK, Wang CY, Tian Y, Ye ML, Sun
DG, Sui JD, Wang LM, Fan R and Gao ZM: MicroRNA-205-5p regulates
the chemotherapeutic resistance of hepatocellular carcinoma cells
by targeting PTEN/JNK/ANXA3 pathway. Am J Transl Res. 9:4300–4307.
2017.PubMed/NCBI
|
|
30
|
He H, Tian W, Chen H and Deng Y:
MicroRNA-101 sensitizes hepatocellular carcinoma cells to
doxorubicin-induced apoptosis via targeting Mcl-1. Mol Med Rep.
13:1923–1929. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grandage VL, Gale RE, Linch DC and Khwaja
A: PI3-kinase/Akt is constitutively active in primary acute myeloid
leukaemia cells and regulates survival and chemoresistance via
NF-kappaB, Mapkinase and p53 pathways. Leukemia. 19:586–594. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pellegrino R, Calvisi DF, Neumann O,
Kolluru V, Wesely J, Chen X, Wang C, Wuestefeld T, Ladu S, Elgohary
N, et al: EEF1A2 inactivates p53 by way of PI3K/AKT/mTOR-dependent
stabilization of MDM4 in hepatocellular carcinoma. Hepatology.
59:1886–1899. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fang Y, Chai Z, Wang D, Kuang T, Wu W and
Lou W: DNA-PKcs deficiency sensitizes the human hepatoma HepG2
cells to cisplatin and 5-fluorouracil through suppression of the
PI3K/Akt/NF-κB pathway. Mol Cell Biochem. 399:269–278. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Birbach A, Gold P, Binder BR, Hofer E, de
Martin R and Schmid JA: Signaling molecules of the NF-kappa B
pathway shuttle constitutively between cytoplasm and nucleus. J
Biol Chem. 277:10842–10851. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Erstad DJ and Cusack JC Jr: Targeting the
NF-κB pathway in cancer therapy. Surg Oncol Clin N Am. 22:705–746.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Q, Jin M, Yang F, Zhu J, Xiao Q and
Zhang L: Matrix metalloproteinases: Inflammatory regulators of cell
behaviors in vascular formation and remodeling. Mediators Inflamm.
2013:9283152013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ito Y, Kikuchi E, Tanaka N, Kosaka T,
Suzuki E, Mizuno R, Shinojima T, Miyajima A, Umezawa K and Oya M:
Down-regulation of NF kappa B activation is an effective
therapeutic modality in acquired platinum-resistant bladder cancer.
BMC Cancer. 15:3242015. View Article : Google Scholar : PubMed/NCBI
|