Introduction
Non-small-cell lung carcinoma (NSCLC) accounts for
~85% of all cases of lung cancer worldwide, and the most common
histological subtypes of NSCLC are lung adenocarcinoma (LA) and
lung squamous cell carcinoma (LSCC) (1). LA and LSCC cells originate from lung
epithelial cells and differentiate into glandular and squamous
phenotypes, lining the larger airways and the peripheral small
airways (2,3). LSCC exhibits many similarities with LA
in terms of somatic copy number alterations (4), which raises the possibility of the
presence of common genetic features between these two diseases
(5,6). Clinical, genetic and biochemical
evidence also suggest that different types of lung cancer may share
similar molecular pathways (6).
However, clinical or pathological phenotypes alone may be
insufficient to understand the underlying mechanisms of lung cancer
(5,6).
Investigation of disease-associated genes can
improve the understanding of disease etiology and development,
thereby facilitating design and development of novel preventive and
treatment strategies (7,8). Cross disease-gene studies and further
pathway analyses provide an opportunity to resolve overlapping
associations into discrete pathways and investigate possible shared
etiologies (9,10).
The aim of the present study was to identify shared
risk genes and to improve the understanding of shared pathways and
biological mechanisms involved in LA and LSCC using a mega-analysis
of gene expression data. Considering that the range of genetic
alterations in LSCC is less understood compared with LA, the
present study investigated genes that were involved in LA but not
with LSCC using LSCC gene expression datasets.
Materials and methods
Study design
First, a large-scale literature-based analysis of
disease-associated genes was performed to identify genes involved
in LSCC and LA. Subsequently, for each of the LA-associated genes
identified, a mega-analysis was performed using LSCC gene
expression data. Pathway analysis was then performed to identify
possible functional pathways associated with LSCC-specific genes.
Finally, a co-expression-based protein-protein interaction (PPI)
analysis was performed using LSCC expression data to evaluate the
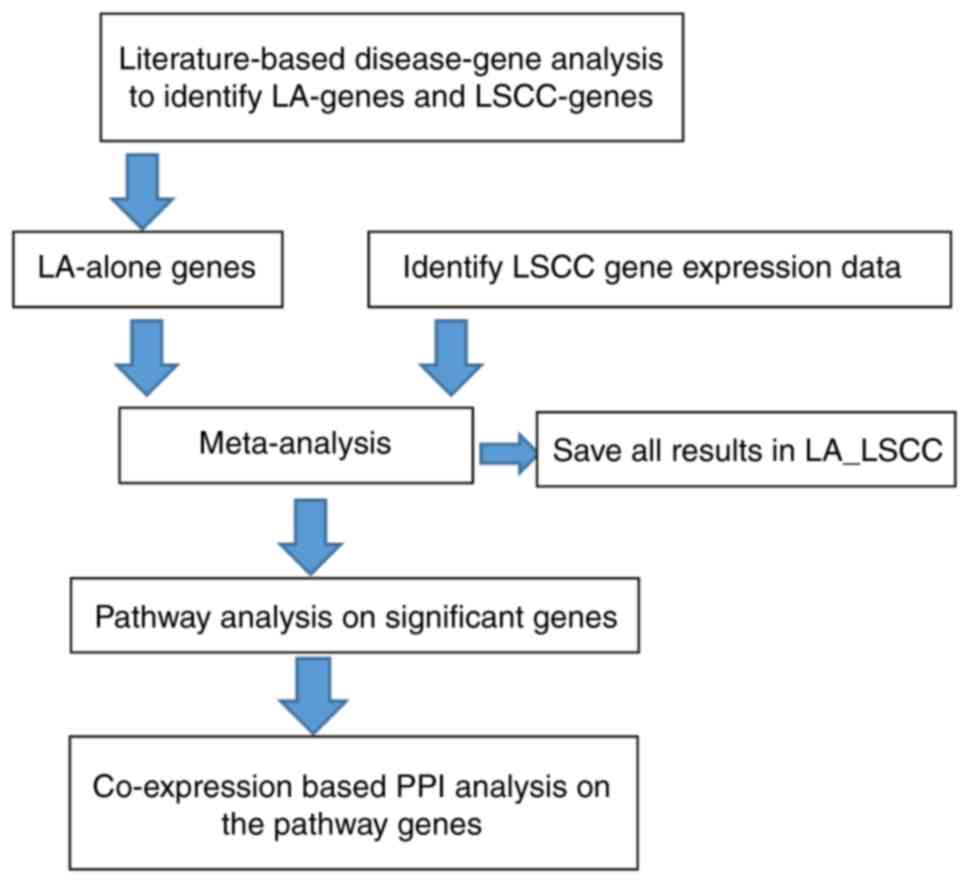
pathways identified. The workflow diagram is presented in Fig. 1.
LA-and LSCC-associated gene data
LA-and LSCC-associated gene data were acquired from
the Pathway Studio (version 12.1.0.9; www.pathwaystudio.com) (11) mammalian database, which is a group of
real-time updated literature knowledge databases, including curated
signaling pathways, cellular processes, megabolic pathways,
ontologies, annotations, molecular interactions and functional
associations (http://pathwaystudio.gousinfo.com/ResNetDatabase.html).
Association data were extracted from >41,000,000 references,
including PubMed (https://www.ncbi.nlm.nih.gov/pubmed) abstracts and
full-text articles. The Pathway database employs an automated
natural language processing-based information extraction system,
MedScan, with a precision >91% (12). Association data within the database
are supported with one or more reference. The Pathway Studio ResNet
Database is the largest literature database (13). These data were organized into a
genetic dataset termed ‘LA_LSCC’, which is available at the
Bioinformatics Database (http://database.gousinfo.com). The downloadable excel
spreadsheet containing the dataset is available at http://gousinfo.com/database/Data_Genetic/LA_LSCC.xlsx.
The full lists of genes associated with LA and/or LSCC are
presented in the groups ‘LA_alone genes’, ‘LSCC_alone genes’ and
‘Common genes’. In addition, the references for every disease-gene
association are presented in the groups ‘Ref for LA_alone genes’
for LA-specific genes, ‘Ref for LSCC_alone genes’ for LSCC-specific
genes and ‘Ref for common genes’ for shared genes. Information
regarding the titles of the references and the sentences where the
disease-gene associations were identified are presented in the
‘LA_LSCC’ dataset.
Gene expression data selected for
mega-analysis
Following the initial search with ‘lung squamous
cell carcinoma’, 158 microarray expression datasets were identified
on gene expression omnibus (https://www.ncbi.nlm.nih.gov/geo/) (14,15).
Subsequently, the following criteria were applied: i) The organism
used in the study was Homo sapiens; ii) the data type was
microarray expression profiling; and iii) the studies were limited
to comparison between LSCC and healthy controls. A total of 12
datasets satisfied the inclusion criteria for the mega-analysis.
However, one dataset (GSE27489 (16); www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE27489)
was excluded from further investigation as each gene in this
dataset demonstrated a small variation in expression level, which
may lead to biased results in the mega-analysis. The 11 included
datasets are listed in Table I
(17–27).
 | Table I.Datasets used for lung squamous cell
carcinoma-gene association mega-analysis. |
Table I.
Datasets used for lung squamous cell
carcinoma-gene association mega-analysis.
| Study name | Dataset GEO ID | Control (n) | Case (n) | Study age
(years) | Country | (Refs.) |
|---|
| Nazarov et
al, 2017 | GSE84784 | 9 | 9 | 2 | Luxembourg | (17) |
| Tong et al,
2016 | GSE67061 | 8 | 69 | 3 | China | – |
| Mascaux et
al, 2014 | GSE33479 | 27 | 14 | 5 | USA | – |
| Rousseaux et
al, 2014 | GSE30219 | 14 | 61 | 5 | France | (18) |
| Girard et
al, 2012 | GSE32036 | 59 | 12 | 7 | USA | (19,20) |
| Philipsen et
al, 2010 | GSE19188 | 65 | 27 | 9 | Netherlands | (21) |
| Boelens et
al, 2009 | GSE12472 | 28 | 35 | 10 | Netherlands | (22) |
| Ishikawa et
al, 2009 | GSE2088 | 30 | 48 | 10 | Japan | (23) |
| Boelens et
al, 2008 | GSE12428 | 28 | 34 | 11 | Netherlands | (24) |
| Rosskopf et
al, 2006 | GSE6044 | 5 | 14 | 13 | Germany | (25) |
| Takeuchi et
al, 2009 | GSE11969 | 5 | 35 | 10 | Japan | (26,27) |
Mega-analysis models
The log2 fold-change (LFC) of the gene expression
level was used to indicate the effect size. Both fixed-effect and
random-effects models were employed to investigate and compare the
effect size (28). The heterogeneity
of the mega-analysis was analyzed to study the variance within and
between different studies. In the case that the total variance (Q)
was equal to or smaller than the expected between-study variance
(df), the within-study variance percentage (ISq) =100% × (Q-df)/Q
was set at 0 and a fixed-effect model was selected for the
mega-analysis. Otherwise, a random-effects model was selected. Q-p
represents the probability that the total variance was only due to
within-study variance. Significantly associated genes from this
mega-analysis were identified using the following criteria: i)
P<1×10−7; and ii) |LFC| >1. When a gene exhibited
a |LFC| >1 in the mega-analysis, the change in the expression
level of the gene was >2-fold or <0.5-fold. The current study
presented all the mega-analysis results identified in the
‘Mega-analysis’ group in the ‘LA_LSCC’ dataset; however, only genes
with a |LFC| >1 were further discussed. All analyses were
performed using Matlab (version R2017a; http://www.mathworks.com/products/matlab.html).
Multiple linear regression
analysis
A multiple linear regression (MLR) model was
employed to investigate the possible influence of sample size,
country of origin and study date on the gene expression in LSCC.
P-values and 95% CIs were reported for each of these factors.
Pathway analysis
To test the functional profile of the common genes
associated with LA and LSCC, a Gene Set Enrichment Analysis (GSEA)
was conducted using Pathway Studio (version 12.1.0.9; www.pathwaystudio.com) against Gene Ontology (GO;
http://geneontology.org) and Pathway Studio
Ontology (version 12.1.0.9; www.pathwaystudio.com). In addition, functional
pathway analysis was performed to investigate potential biological
associations between the identified risk genes and LSCC. The
analysis was performed using the ‘Shortest Path’ module of Pathway
Studio in order to identify various ‘entities’, including
complexes, proteins and functional classes, that were associated to
both the genes and LSCC. The reference information included the
types of associations, the number of underlying supporting
references and the sentences where these associations had been
identified and described.
Co-expression analysis
For each pair of the genes and proteins identified
in the aforementioned pathway analysis, another mega-analysis was
performed to investigate their co-expression using the 11 LSCC
expression datasets. The Fisher's Z-value (FisherZ) of Pearson's
correlation was used to determine the effect size, and the
following equation was used to calculate it: FisherZ=0.5 × log [(1+
Correlation)-(1- Correlation)]. The purpose of this analysis was to
validate the associations identified in the pathway analysis. The
present study used the following criteria for the selection of a
non-random meaningful association: i) An absolute value of FisherZ
>0.3; and ii) P<0.05. The detailed FisherZ values and
P-values are presented in the ‘Co-expression’ analysis.
Results
LA and LSCC genes
LA-and LSCC-associated gene analyses revealed 1,178
genes associated with LA, supported by 7,355 references, and 334
genes associated with LSCC, supported by 838 references. The full
list of these genes and the associated references are presented in
the ‘LA_LSCC’ dataset. A significant overlap of 187 genes, which
are presented in the ‘Common genes’ group, was identified for both
LA and LSCC (right tail Fisher's Exact test;
P=1.02×10−161). This accounted for 55.99% of all the
LSCC-associated genes and 15.87% of all the LA-associated
genes.
To test the functional profile of the 187 common
genes associated with both LA and LSCC, a GSEA was conducted using
Pathway Studio against the GO and Pathway Studio Ontology. In
total, nine pathways/gene sets (73 unique genes) associated with
protein kinase, three pathways/gene sets (71 unique genes)
associated with cell growth proliferation, two pathways/gene sets
(nine unique genes) associated with cell apoptosis and one
pathway/gene set (ten unique genes) associated with transcription
factors were significantly enriched. The full list of the 39
pathways/gene sets enriched with P<1.7×10−5 (with 144
out of 187 unique genes) are presented in the ‘Common pathways’
group contained in the ‘LA_LSCC’ dataset. The majority of these
pathways were involved in LA and LSCC, indicating a shared genetic
basis for these diseases.
Three novel common genes in LA and
LSCC
Although an overlap was identified between LA-and
LSCC-associated genes, the majority of the LA-specific genes (991
genes, 84.13%) were not implicated in LSCC. A systematic
mega-analysis was performed to collectively assess differential
expressed mRNAs and determine whether previously investigated
LA-associated genes were also linked to LSCC. Notably, certain
datasets do not contain the three genes and therefore will not be
included in the current study. However, the LFCs of the genes were
estimated from the majority of the 11 studies (>9 studies). The
associations between the LA-specific genes with 11 LSCC gene
expression datasets (Table I) were
evaluated. A total of three genes, including solute carrier family
2 member 1 (SLC2A1), endothelial PAS domain protein 1 (EPAS1) and
cyclin-dependent kinase 4 (CDK4), passed the significant criteria
(P<1×10−7 and |LFC| >1) and are presented in
Table II. The detailed results are
presented in the ‘Mega-analysis’ group in the ‘LA_LSCC’
dataset.
| Table II.Statistically significant genes
identified from the mega-analysis of lung squamous cell
carcinoma. |
Table II.
Statistically significant genes
identified from the mega-analysis of lung squamous cell
carcinoma.
|
|
|
| MLR analysis
results (P-values) | Mega-analysis
results |
|---|
|
|
|
|
|
|
|---|
| Gene name | Random effects
model | Datasets included
(n) | LFC | SD of LFC | P-value | Sample size | Population
region | Study age |
|---|
| SLC2A1 | 0 | 11 |
1.63 | 0.25 |
4.31×10−11 | 0.59 |
4×10−3 | 0.69 |
| EPAS1 | 1 | 9 | −1.50 | 0.26 |
1.27×10−8 | 0.49 |
2×10−5 | 0.09 |
| CDK4 | 1 | 11 |
1.02 | 0.18 |
1.60×10−8 | 0.87 |
6×10−5 | 0.98 |
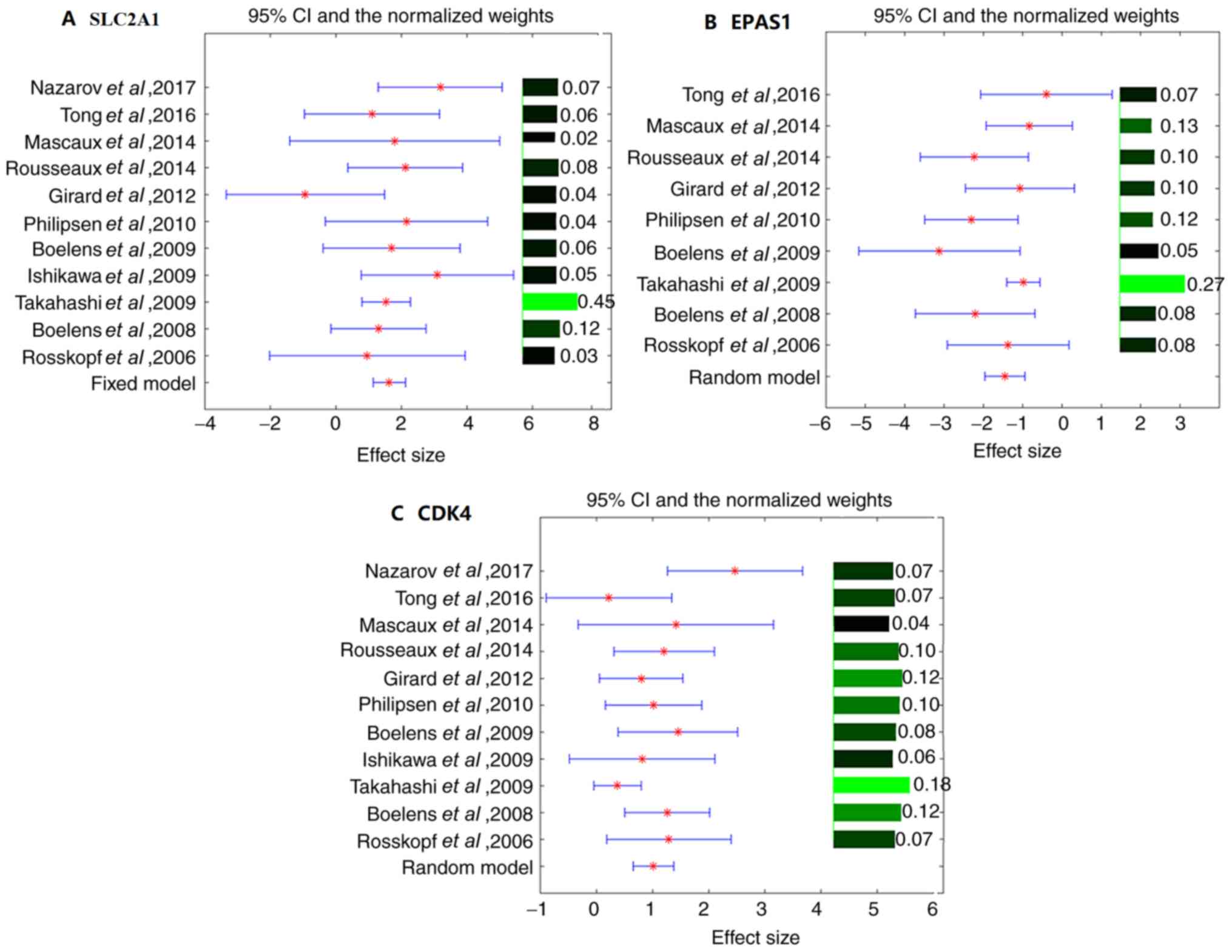
The effect sizes, 95% CIs and weights of different
studies for the three identified genes (SLC2A1, EPAS1 and CDK4) are
presented in Fig. 2. EPAS1 and CDK4
exhibited significant variances between studies (ISq >0% and
Q-test P<0.1). Therefore, the random-effects model was selected
for their mega-analysis. By contrast, no significant between-study
variance was observed for SLC2A1 (Q-test P>0.4), and the
fixed-effect model was selected for SLC2A1 (Fig. 2). Notably, multiple line regression
analyses demonstrated that the country of origin was a significant
factor that influenced the LFC of all three genes in the case of
LSCC (P<0.004; Table II).
Functional pathway analysis
According to the approach used to identify the genes
associated with LSCC, SLC2A1, EPAS1 and CDK4 exhibited no direct
link with LSCC. However, functional pathway analysis revealed
multiple potential pathways through which these three genes may
serve roles in the pathology of LSCC (Fig. 3). Each edge in Fig. 3 was supported by ≥1 references, and
details of these associations are presented in the
‘LSCC-3Genes_potential pathways’ group in the ‘LA_LSCC’
dataset.
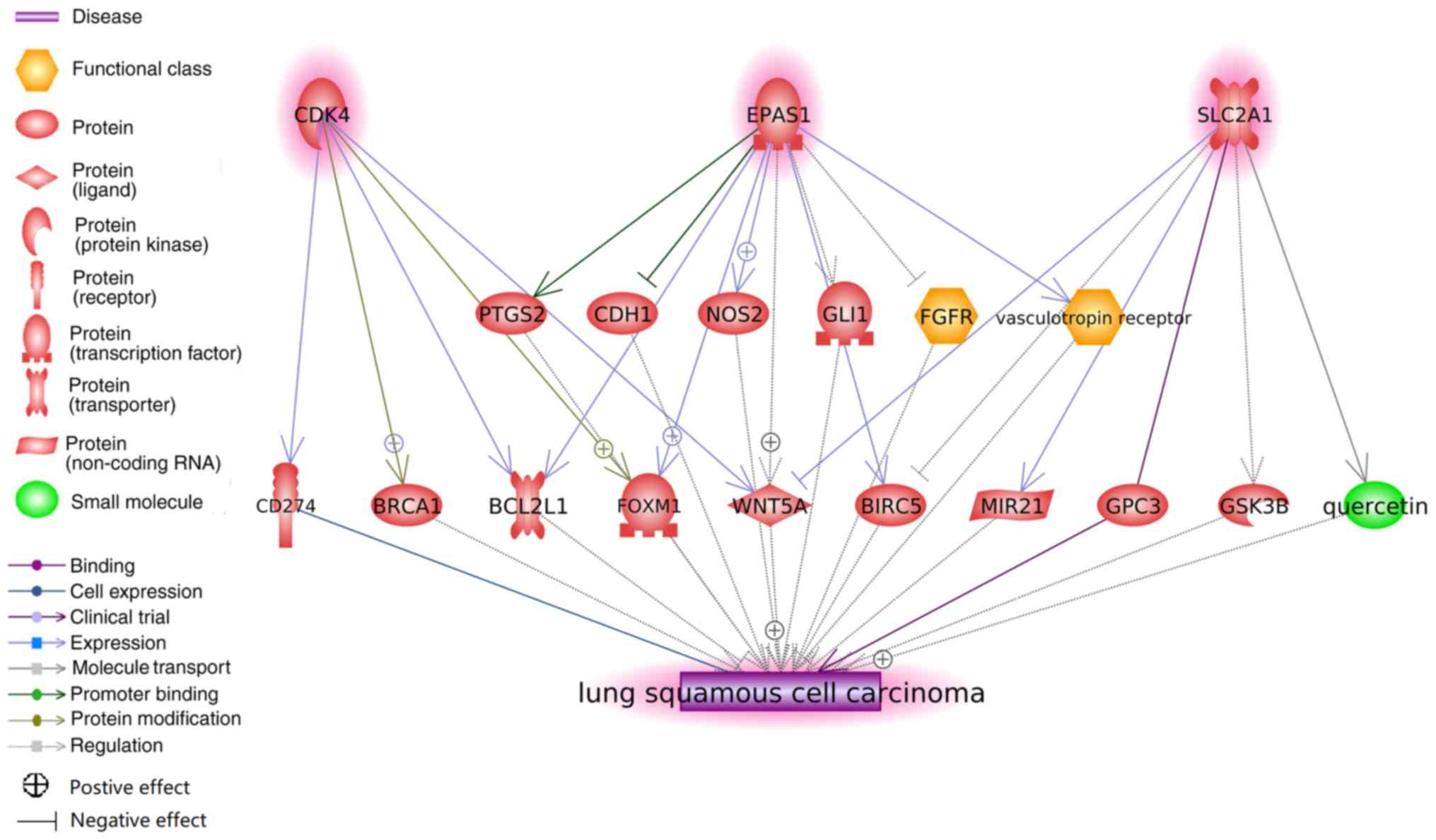 | Figure 3.Potential pathways associating
SLC2A1, EPAS1 and CDK4 to lung squamous cell carcinoma. Network was
generated using Pathway Studio. Each association (edge) has ≥1
supporting reference. SLC2A1, solute carrier family 2 member 1;
EPAS1, endothelial PAS domain protein 1; CDK4, cyclin-dependent
kinase 4; PTGS2, prostaglandin-endoperoxide synthase 2; CDH1,
cadherin 1; NOS2, nitric oxide synthase 2; GLI1, GLI family member
zinc finger 1; FGFR, fibroblast growth factor receptor; BCL2L1,
BCL2-like 1; FOXM1, forkhead box M1; WNT5A, Wnt family member 5A;
BIRC5, baculoviral IAP repeat containing 5; MIR21, microRNA-21;
GPC3, glypican 3; GSK3B, glycogen synthase kinase 3β. |
To confirm the associations presented in Fig. 3, a co-expression PPI analysis was
conducted with the purpose of validating the associations between
CD4K4, EPAS1 and SLC2A1, and the 13 other genes presented in
Fig. 3. The majority of the entities
presented in Fig. 3 also exhibited
significant associations in the co-expression analysis (Fig. 4), supporting the pathway analysis
results. Co-expression analysis results are presented in the
‘Co-expression’ group in the ‘LA_LSCC’ dataset.
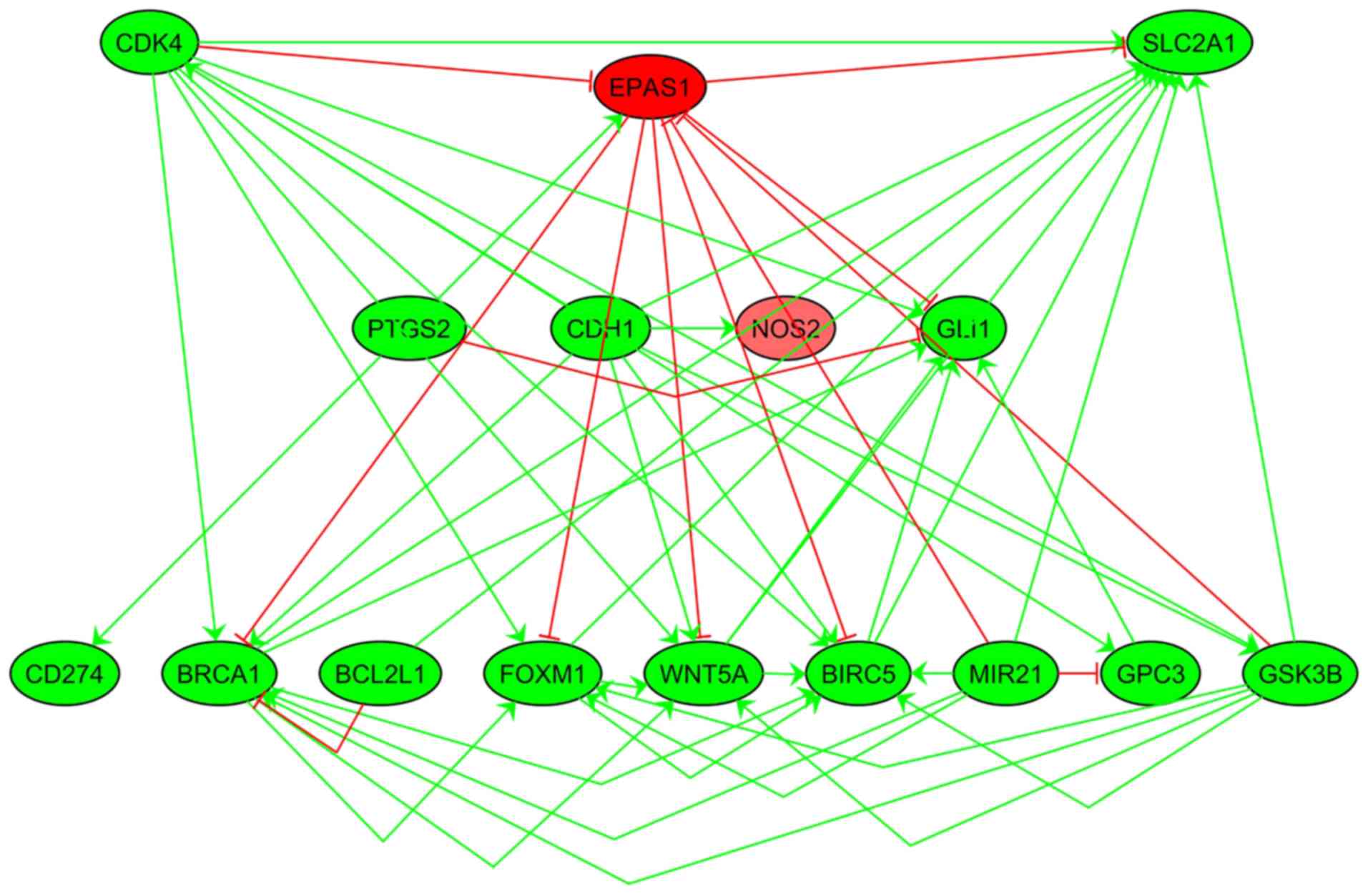 | Figure 4.Co-expression analysis. Each edge
represents a significant association between the corresponding two
entities (P<0.05). Positive associations are highlighted in
green and negative associations are highlighted in red. Nodes in
red indicate decreased expression and green nodes indicate
increased expression. SLC2A1, solute carrier family 2 member 1;
EPAS1, endothelial PAS domain protein 1; CDK4, cyclin-dependent
kinase 4; PTGS2, prostaglandin-endoperoxide synthase 2; CDH1,
cadherin 1; NOS2, nitric oxide synthase 2; GLI1, GLI family member
zinc finger 1; BCL2L1, BCL2-like 1; FOXM1, forkhead box M1; CD274,
cluster of differentiation 274; WNT5A, Wnt family member 5A; BIRC5,
baculoviral IAP repeat containing 5; MIR21, microRNA-21; GPC3,
glypican 3. |
Discussion
The cross-analysis of different lung cancer
phenotypes may facilitate the development of novel strategies and
approaches for the treatment of lung cancer. In the present study,
LA-specific genes were systematically mega-analyzed with LSCC
differential expression data and three genes, including SLC2A1,
EPAS1 CDK4, were identified as potential risk genes for LSCC.
Importantly, whether these associations between the genes and LSCC
indicate causality requires further investigation.
It is a major concern that a disease-gene
association derived from experiment-based literature is heavily
dependent on the quality and access of the text data, and the
efficiency of the mining algorithms. Candidate disease-gene
analysis is more appropriate for monogenic diseases because the
association between genotype and phenotype is clearer (11,12). In
lung cancer, a complex disease, the etiology can be attributed to
tobacco smoking, sex, ethnicity, age, diet, obesity, infections and
numerous genes that work in combination to elicit the disease
phenotype (29–31). It has also been observed that when
individually investigated, the genes potentially responsible for
the disease may not result in disease in certain patients (32–35).
In this context, cross-disease analysis based on
mega-analysis can overcome the limitations of sample size and
identify more reliable and robust common genes between LA and LSCC
through the quantitative combination and assessment of multiple
studies (36,37). In the present study, disease-gene
association data were retrieved from the Pathway Studio database
and mega-analysis was performed to detect their significance in
terms of gene expression levels. All of these analyses can provide
a more reliable and robust result.
The present study used MLR analysis to demonstrate
that lung cancer outcome varies among different populations and
ethnicities. In addition, the present study identified that the
country of origin may be associated with the expression levels of
SLC2A1, EPAS1 and CDK4 in the case of LSCC. It is therefore
necessary to assess the generalizability of the present results in
different ethnic groups. Socioeconomic and cultural differences
among different racial groups may account for some degree of the
current disparities and a personalized molecular approach may help
to resolve such problems (38–41).
The current literature-based functional pathway
analysis revealed several possible pathways that link the three
novel genes identified to LSCC. For example, CDK4, a member of the
serine/threonine protein kinase family, may contribute to the
development of LSCC via a CDK4-forkhead box M1 (FOXM1)-LSCC
pathway. It has been reported that CDK4 activity can increase the
transcriptional activity of FOXM1 without phosphorylating FOXM1
(42), while the expression of FOXM1
has been suggested to contribute to the development or progression
of LSCC (43). A previous study also
suggested that CDK4 can stimulate the BRCA1 promoter in an E2F
transcription factor 1-dependent manner, regulating cell cycle, DNA
replication and cell proliferation processes (44). BRCA1 serves an important role in LSCC
via cell cycle and DNA replication signaling pathways (44), which indicates a potential
CDK4-BRCA1-LSCC pathway.
EPAS1 can bind to and inhibit the expression of the
calcium-dependent cell adhesion molecule cadherin 1 (CDH1)
(45,46). CDH1 has been reported to serve a dual
role in the maintenance of the LSCC phenotype (47). These previous studies indicate that
the EPAS1-CDH1-LSCC pathway may serve a complex role in LSCC
development. EPAS1 regulates the production of
prostaglandin-endoperoxide synthase (PTGS) (48), which has been indicated to promote
the carcinogenesis of LSCC, suggesting a potential EPAS1-PTGS-LSCC
pathway.
SLC2A1 is a major glucose transporter responsible
for constitutive or basal glucose uptake, which can bind with
glypican 3 (GPC3) to decrease glucose transport activity (49) and transport quercetin to balance the
glucose efflux (50). Mechanisms
associated with glucose efflux are also identified in the
pathological process of LSCC (51,52),
suggesting the existence of SLC2A1-GPC3-LSCC and
SLC2A1-quercetin-LSCC pathways.
Co-expression analysis revealed that the majority of
the identified genes were associated with each other in terms of
expression. The majority of the literature-based pathway identified
was validated by the expression data-based associations found.
However, a certain number of the associations identified in the
present study may not be consistent with the present co-expression
analysis. For example, SLC2A1 inhibits the expression of
baculoviral IAP repeat containing 5 (BIRC5) in the pathway
analysis; however, SLC2A1 and BIRC5 exhibit positive co-expression
in the co-expression analysis, which indicates the presence of a
more complex genetic network including more regulators.
The present cross-disease analysis between LA and
LSCC suggested common genes may contribute to disease comorbidities
and trait manifestations. The novel common genes identified may
facilitate the development of novel strategies targeting shared
mechanisms across diseases. However, the conclusion of the current
study was only based on a statistical analysis of previous
experimental data and a literature-based pathway study. Therefore,
further biological experiments, including gene-knockout or
knockdown experiments, are required to validate the associations
between the three genes identified and LSCC.
In conclusion, cross-disease analysis could provide
a powerful tool to investigate new targets and reveal common
biological mechanisms. Genes associated with LA require further
analysis to identify their association with LSCC. SLC2A1, EPAS1 and
CDK4 genes identified in the present study may be novel common risk
genes associated with both LA and LSCC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the http://database.gousinfo.com repository, http://gousinfo.com/database/Data_Genetic/LA_LSCC.xlsx.
Authors' contributions
GZ and ZL developed the study design, supervised the
whole the study process and prepared the manuscript. WW and HC
contributed to data analysis and manuscript drafting and revision.
WH and XX contributed to data collection and manuscript drafting
and revision. All authors approve the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ansari J, Shackelford RE and El-Osta H:
Epigenetics in non-small cell lung cancer: From basics to
therapeutics. Transl Lung Cancer Res. 5:155–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Catacchio I, Scattone A, Silvestris N and
Mangia A: Immune prophets of lung cancer. The prognostic and
predictive landscape of cellular and molecular immune markers
Transl Oncol. 11:825–835. 2018.PubMed/NCBI
|
|
3
|
Chalela R, Curull V, Enríquez C, Pijuan L,
Bellosillo B and Gea J: Lung adenocarcinoma: From molecular basis
to genome-guided therapy and immunotherapy. J Thorac Dis.
9:2142–2158. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cancer Genome Atlas Research Network, .
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hirsch FR, Spreafico A, Novello S, Wood
MD, Simms L and Papotti M: The prognostic and predictive role of
histology in advanced non-small cell lung cancer: A literature
review. J Thorac Oncol. 3:1468–1481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pankratz VS, Sun Z, Aakre J, Li Y, Johnson
C, Garces YI, Aubry MC, Molina JR, Wigle DA and Yang P: Systematic
evaluation of genetic variants in three biological pathways on
patient survival in low stage non-small cell lung cancer. J Thorac
Oncol. 6:1488–1495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yandell M, Huff C, Hu H, Singleton M,
Moore B, Xing J, Jorde LB and Reese MG: A probabilistic
disease-gene finder for personal genomes. Genome Res. 21:1529–1542.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Shen F, Mojarad MR, Li D, Liu S,
Tao C, Yu Y and Liu H: Systematic identification of latent
disease-gene associations from PubMed articles. PLoS One.
13:e01915682018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ellinghaus D, Jostins L, Spain SL, Cortes
A, Bethune J, Han B, Park YR, Raychaudhuri S, Pouget JG, Hübenthal
M, et al: Analysis of five chronic inflammatory diseases identifies
27 new associations and highlights disease-specific patterns at
shared loci. Nature Genetics. 48:510–518. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen D, Che N, Le J and Pan QL: A
co-training based entity recognition approach for cross-disease
clinical documents. Con Comput Pract Exp. e45052018. View Article : Google Scholar
|
|
11
|
Nikitin A, Egorov S, Daraselia N and Mazo
I: Pathway studio-the analysis and navigation of molecular
networks. Bioinformatics. 19:2155–2157. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Daraselia N, Yuryev A, Egorov S,
Novichkova S, Nikitin A and Mazo I: Extracting human protein
interactions from MEDLINE using a full-sentence parser.
Bioinformatics. 20:604–611. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lorenzi PL, Claerhout S, Mills GB and
Weinstein JN: ‘A curated census of autophagy-modulating proteins
and small molecules: Candidate targets for cancer therapy’.
Autophagy. 10:1316–1326. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res 41 (Database Issue).
D991–D995. 2013.
|
|
16
|
Kahn N, Meister M, Eberhardt R, Muley T,
Schnabel PA, Bender C, Johannes M, Keitel D, Sültmann H, Herth FJ,
et al: Early detection of lung cancer by molecular markers in
endobronchial epithelial-lining fluid. J Thorac Oncol. 7:1001–1008.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nazarov PV, Muller A, Kaoma T, Nicot N,
Maximo C, Birembaut P, Tran NL, Dittmar G and Vallar L: RNA
sequencing and transcriptome arrays analyses show opposing results
for alternative splicing in patient derived samples. BMC Genomics.
18:4432017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rousseaux S, Debernardi A, Jacquiau B,
Vitte AL, Vesin A, Nagy-Mignotte H, Moro-Sibilot D, Brichon PY,
Lantuejoul S, Hainaut P, et al: Ectopic activation of germline and
placental genes identifies aggressive metastasis-prone lung
cancers. Sci Transl Med. 5:186ra662013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Byers LA, Diao L, Wang J, Saintigny P,
Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al: An
epithelial-mesenchymal transition gene signature predicts
resistance to EGFR and PI3K inhibitors and identifies Axl as a
therapeutic target for overcoming EGFR inhibitor resistance. Clin
Cancer Res. 19:279–290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schuster K, Venkateswaran N, Rabellino A,
Girard L, Peña-Llopis S and Scaglioni PP: Nullifying the CDKN2AB
locus promotes mutant K-ras lung tumorigenesis. Mol Cancer Res.
12:912–923. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hou J, Aerts J, den Hamer B, van Ijcken W,
den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens
JA, Hoogsteden HC, et al: Gene expression-based classification of
non-small cell lung carcinomas and survival prediction. PLoS One.
5:e103122010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boelens MC, Gustafson AM, Postma DS, Kok
K, van der Vries G, van der Vlies P, Spira A, Lenburg ME, Geerlings
M, Sietsma H, et al: A chronic obstructive pulmonary disease
related signature in squamous cell lung cancer. Lung Cancer.
72:177–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fujiwara T, Hiramatsu M, Isagawa T,
Ninomiya H, Inamura K, Ishikawa S, Ushijima M, Matsuura M, Jones
MH, Shimane M, et al: ASCL1-coexpression profiling but not single
gene expression profiling defines lung adenocarcinomas of
neuroendocrine nature with poor prognosis. Lung Cancer. 75:119–125.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boelens MC, van den Berg A, Fehrmann RS,
Geerlings M, de Jong WK, te Meerman GJ, Sietsma H, Timens W, Postma
DS and Groen HJ: Current smoking-specific gene expression signature
in normal bronchial epithelium is enhanced in squamous cell lung
cancer. J Pathol. 218:182–191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rohrbeck A, Neukirchen J, Rosskopf M,
Pardillos GG, Geddert H, Schwalen A, Gabbert HE, von Haeseler A,
Pitschke G, Schott M, et al: Gene expression profiling for
molecular distinction and characterization of laser captured
primary lung cancers. J Transl Med. 6:692008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takeuchi T, Tomida S, Yatabe Y, Kosaka T,
Osada H, Yanagisawa K, Mitsudomi T and Takahashi T: Expression
profile-defined classification of lung adenocarcinoma shows close
relationship with underlying major genetic changes and
clinicopathologic behaviors. J Clin Oncol. 24:1679–1688. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Matsuyama Y, Suzuki M, Arima C, Huang QM,
Tomida S, Takeuchi T, Sugiyama R, Itoh Y, Yatabe Y, Goto H and
Takahashi T: Proteasomal non-catalytic subunit PSMD2 as a potential
therapeutic target in association with various clinicopathologic
features in lung adenocarcinomas. Mol Carcinog. 50:301–309. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Borenstein M, Hedges LV, Higgins JP and
Rothstein HR: A basic introduction to fixed-effect and
random-effects models for mega-analysis. Res Synth Methods.
1:97–111. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Davila DG and Williams DE: The etiology of
lung cancer. Mayo Clin Proc. 68:170–182. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Williams MD and Sandler AB: The
epidemiology of lung cancer. Cancer Treat Res. 105:31–52. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lemjabbar-Alaoui H, Hassan OU, Yang YW and
Buchanan P: Lung cancer: Biology and treatment options. BBA-Reviews
on Cancer. 1856:189–210. 2015.PubMed/NCBI
|
|
32
|
Piro RM and Di Cunto F: Computational
approaches to disease-gene prediction: Rationale, classification
and successes. FEBS J. 279:678–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Opap K and Mulder N: Recent advances in
predicting gene-disease associations. F1000Res. 6:5782017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Doncheva NT, Kacprowski T and Albrecht M:
Recent approaches to the prioritization of candidate disease genes.
Wiley Interdiscip Rev Syst Biol Med. 4:429–442. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee TI and Young RA: Transcriptional
regulation and its misregulation in disease. Cell. 152:1237–1251.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Botling J, Edlund K, Lohr M, Hellwig B,
Holmberg L, Lambe M, Berglund A, Ekman S, Bergqvist M, Pontén F, et
al: Biomarker discovery in non-small cell lung cancer: Integrating
gene expression profiling, mega-analysis and tissue microarray
validation. Clin Cancer Res. 19:194–204. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ramasamy A, Mondry A, Holmes CC and Altman
DG: Key issues in conducting a mega-analysis of gene expression
microarray datasets. PLoS Med. 5:e1842008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
El-Telbany A and Ma PC: Cancer genes in
lung cancer: Racial disparities: Are there any? Genes Cancer.
3:467–480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: The impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. Ca Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schabath MB, Cress D and Munoz-Antonia T:
Racial and ethnic differences in the epidemiology and genomics of
lung cancer. Cancer Control. 23:338–346. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hardy D, Liu CC, Xia R, Cormier JN, Chan
W, White A, Burau K and Du XL: Racial disparities and treatment
trends in a large cohort of elderly black and white patients with
nonsmall cell lung cancer. Cancer. 115:2199–2211. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wierstra I: CyclinD1/Cdk4 increases the
transcriptional activity of FOXM1c without phosphorylating FOXM1c.
Biochem Biophys Res Commun. 431:753–759. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang DK, Son CH, Lee SK, Choi PJ, Lee KE
and Roh MS: Forkhead box M1 expression in pulmonary squamous cell
carcinoma: Correlation with clinicopathologic features and its
prognostic significance. Hum Pathol. 40:464–470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang F, Chen X, Wei K, Liu D, Xu X, Zhang
X and Shi H: Identification of key transcription factors associated
with lung squamous cell carcinoma. Med Sci Monit. 23:172–206. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Maru S, Ishigaki Y, Shinohara N, Takata T,
Tomosugi N and Nonomura K: Inhibition of mTORC2 but not mTORC1
up-regulates E-cadherin expression and inhibits cell motility by
blocking HIF-2α expression in human renal cell carcinoma. J Urol.
189:1921–1929. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Murugesan T, Rajajeyabalachandran G, Kumar
S, Nagaraju S and Jegatheesan SK: Targeting HIF-2α as therapy for
advanced cancers. Drug Discov Today. 23:1444–1451. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pallier K, Cazes A, El Khattabi L, Lecchi
C, Desroches M, Danel C, Riquet M, Fabre-Guillevin E, Laurent-Puig
P and Blons H: DeltaN TP63 reactivation, epithelial phenotype
maintenance, and survival in lung squamous cell carcinoma. Tumor
Biol. 33:41–51. 2012. View Article : Google Scholar
|
|
48
|
Xiong J, Zhu FF and Nie MF:
Hypoxia-inducible factor-2α (HIF-2α) mediates the effects of
hypoxia on the promotion of HeLa cell viability, colony formation,
and invasion capacity in vitro. Genet Mol Res. 14:3281–3292. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cho HS, Ahn JM, Han HJ and Cho JY:
Glypican 3 binds to GLUT1 and decreases glucose transport activity
in hepatocellular carcinoma cells. J Cell Biochem. 111:1252–1292.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cunningham P, Afzal-Ahmed I and Naftalin
RJ: Docking studies show that d-glucose and quercetin slide through
the transporter GLUT1. J Biol Chem. 281:5797–5803. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li K, Pan X, Bi Y, Xu W, Chen C, Gao H,
Shi B, Jiang H, Yang S, Jiang L and Li Z: Adoptive immunotherapy
using T lymphocytes redirected to glypican-3 for the treatment of
lung squamous cell carcinoma. Oncotarget. 7:2496–2507.
2016.PubMed/NCBI
|
|
52
|
Yang JH, Hsia TC, Kuo HM, Chao PD, Chou
CC, Wei YH and Chung JG: Inhibition of lung cancer cell growth by
quercetin glucuronides via G2/M arrest and induction of apoptosis.
Drug Metab Dispos. 34:296–304. 2006. View Article : Google Scholar : PubMed/NCBI
|