Introduction
Endometrial cancer has the highest mortality rate of
malignant tumors in women in the USA, and the incidence rate is
rising; 61,380 American women were diagnosed with endometrial
cancer in 2017, and 10,920 were projected to succumb to the disease
(1). Of all patients, ~80% are
diagnosed at an early stage with an overall favorable prognosis;
however, ~20% of patients will eventually succumb to the disease
(1,2). Despite progress in the fields of
integrated diagnosis and treatment, there are still some
limitations, including disease biology, morbidity and mortality; in
particular, the prognosis of endometrial carcinoma has not markedly
improved (3–5). At present, the predominant treatment
strategy for endometrial cancer is surgery combined with adjuvant
chemotherapy. Taxol® is widely used as the most
promising antitumor agent for women with endometrial cancer;
however, some patients exhibit Taxol resistance, which results in
cancer recurrence or metastasis (6).
MicroRNAs (miRNAs/miRs) are an abundant group of
small endogenous non-coding RNA molecules (~22 nucleotides), which
are single-stranded and bind to target mRNAs mainly at their
3′untranslated region (3′UTR) (7–9).
Previous studies have revealed that miRNAs are involved in numerous
types of cellular processes in various types of human cancer,
including endometrial cancer (7–10). A
study reported that the downregulation of miR-106b is associated
with chemoresistance in endometrial cancer (11). Another study revealed that miR-218 is
significantly downregulated in Taxol-resistant endometrial cancer
cells compared with in non-drug-resistant cell lines, and that
miR-218 may directly bind to the 3′UTR of the high mobility group
box 1 (HMGB1) gene, which mediates autophagy and contributes to
chemotherapy resistance in endometrial carcinoma in vitro
(12). The results of this previous
study revealed the effect of miR-218 on HMGB1-mediated cell
autophagy during chemotherapy resistance in endometrial carcinoma
cells (12). Additionally, it has
been suggested that miR-194 could inhibit the
epithelial-mesenchymal transition (EMT) of endometrial cancer cells
by targeting oncogene BMI1 proto-oncogene polycomb ring finger,
which regulates the expression levels of chemoresistance markers
(SOX-2, Krüppel like factor 4 and mobility related protein-1)
(13). Prior to the present study,
our group used microarray analysis to identify that miR-23a
expression was decreased and associated with chemoresistance in
endometrial cancer (data not shown). These results were similar to
the results of another study, which demonstrated that miR-23a
expression is downregulated in endometrial cancer and miR-23a could
directly downregulate human SMAD3 protein levels (14). Furthermore, it has been demonstrated
that the overexpression of miR-23a could inhibit EMT by targeting
SMAD3 in endometrial cancer (14).
However, another study identified that overexpression of miR-23a
could enhance the chemoresistance of colorectal cancer cells by
targeting ATP binding cassette subfamily F member 1, and that
miR-23a may promote cisplatin chemoresistance and protect against
cisplatin-induced apoptosis through Twist in tongue squamous cell
carcinoma cells (15). Thus far,
there is only one study on the role of miR-23a in cancer (14), and the mechanism of
miR-23a-inhibition in endometrial cancer remains unclear.
In the present study, the role of miR-23a in the
development of endometrial cancer was examined. The results
demonstrated that sine oculis homeobox homolog 1 (SIX1), a
biomarker for carcinogenesis in human endometrial cancer, was a
direct target of miR-23a. In the present study, miR-23a expression
in endometrial cancer tissues was detected and the function of
miR-23a was investigated in vitro, including a systematic
analysis of miR-23a and its role in endometrial cancer
development.
Materials and methods
Cell culture and reagents
Following approval by the Ethics Committee of The
Secondary Hospital of Tianjin Medical University (Tianjin, China),
16 paired tissue sections were obtained from 16 female patients
(age range, 35–50 years, mean age 38.7 years old) undergoing
surgical resection at Tianjin Medical University General Hospital
between July 2010 and May 2012. The patients or their families
provided written informed consent for the use of these tissues.
Human endometrial cancer cell lines, Ishikawa and HEC1B, were
obtained from the American Type Culture Collection (Manassas, VA,
USA). All cells were maintained in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.),
and 1% penicillin and 1% streptomycin at 37°C with 5%
CO2.
RNA analysis
TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to extract total RNA from the cells,
according to the manufacturer's protocol. A First Strand cDNA
Synthesis kit (Takara Biotechnology Co., Ltd., Dalian, China) was
used to synthesize cDNA. The following temperature protocol was
used: 30°C for 10 min, 42°C for 60 min and 95°C for 5 min. Mean
values were used for calculations and β-actin was used as a loading
control. The comparative detailed ΔCq method was utilized to
analyze the results. SYBR Green PCR Master mix (Applied Biosystems;
Thermo Fisher Scientific, Inc.) was used to perform reverse
transcription-quantitative (RT-q) PCR analysis (16): Pre-denature was performed at 94°C for
5 min; followed by 33 cycles of pre-denaturing at 94°C for 30 sec,
64°C for 30 sec, 72°C for 45 sec and 72°C for 10 min. The obtained
PCR products were routinely subjected to agarose gel
electrophoresis and scanned using an imaging system. The gray-scale
ratio of target genes and internal parameters was used to represent
the relative mRNA expression levels of each target gene. The levels
of miR-23a was normalized to the U6 snRNA. Primer sequences used
for RT-qPCR analysis were as follows: SIX1 forward,
5′AAGGAGAAGTCGAGGGGTGT-3′ and reverse 5′-TGCTTGTTGGAGGAGGAGTT-3′;
miR-23a forward, 5′-CCTACTGTCGTCCCAAGACCT-3′ and reverse,
5′-GGGGCTCGTGCAGAAGAAT-3′; and β-actin forward,
5′-CTCCATCATGAAGTGTGACGTT-3′ and reverse,
5′-ATCTCCTTCTGCATCCTGTCAG-3′. The experiment was repeated three
times, independently.
Reagents for the transient
transfection assays
miR-23a mimics (5′-AUCACAUUGCCAGGGAUUUCC-3′),
miR-23a mimic NC (5′-UUCUCCGAACGUGUCACGUTT-3′), miR-23a antisense
oligonucleotide (ASO; 5′-GUGGUAAUCCCUGGCAAUGUGAU-3′ and ASO-NC
(5′-CAGUACUUUUGUGUAGUACAA-3′) were obtained from Sangon Biotech
Co., Ltd. (Shanghai, China) SIX1 small interfering (si)RNA and a
pcDNA3.1 SIX1 overexpression plasmid were obtained from Guangzhou
Ribobio Co., Ltd. (Guangzhou, China) (17,18).
miR-23a mimics were transfected into Ishikawa cells, miR-23a ASO
into HEC1B cells, and SIX1 siRNA and plasmid into Ishikawa and
HEC1B cells. Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.) was used for transfection, according to the
manufacturer's protocol. The final concentration of ASO control
(con) and miR-23a ASO used was 100 nM. SIX1 plasmids were obtained
from OriGene Technologies, Inc. (Rockville, MD, USA).
The sequences were as follows: SIX1 siRNA forward
5′-GGAGCUCACAAGGCAAUAU-3′, and reverse 3′-CCUCGAGUGUUCCGUUAUA-5′;
and SIX1 control siRNA forward 5′-GGAGUUCUCAAGGGAGUAU-3′, and
reverse 3′-CCUCAAGAGUUCCCUCAUA-5′). Subsequent experimentation was
performed at 48 h after transfection.
Colony formation assay
Cells (~400 cells/well) were seeded into 60-mm
dishes and cultured for 10 days at 37°C with 5% CO2.
Following fixation in methanol for 15 min at room temperature, the
cells were stained with Giemsa for 10–30 min at room temperature.
The number of colonies were counted under a light microscope. The
experiment was performed in triplicate.
Western blotting
Whole cell extracts were prepared using cell lysis
reagent (cat. no. C2978; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). Total proteins were quantified using a bicinchoninic acid
assay (Pierce; Thermo Fisher Scientific, Inc.). A total of 50 µg
protein was separated by 10% SDS-PAGE and then transferred to
nitrocellulose membranes. The membranes were blocked with 10% goat
serum (dilution, 1:1,000; cat. no. ZLI-9022; OriGene Technologies,
Inc.) at room temperature for 60 min and incubated with primary
antibodies overnight at 4°C. The primary antibodies used included a
rabbit polyclonal SIX1 antibody (1:500 dilution; cat. no. ab211359;
Abcam, Cambridge, UK) and a mouse monoclonal β-actin antibody
(1:2,500 dilution; cat. no. sc-8432; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA). This was followed by incubation with a
horseradish peroxidas-conjugated secondary antibody (cat. no.
RI2341; polyclonal goat anti-rabbit/mouse; 1:5,000 dilution;
Rockland Immunochemicals Inc., Limerick, PA, USA) at 37°C for 1 h.
The visualization reagent used was Coomassie brilliant blue G-250
(cat. no. C8420; Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China). The gray values were analyzed using Odyssey
v3.0 software (Thermo Fisher Scientific, Inc.).
Scratch assay
A total of 1×105 cells/well were seeded
in 6-well plates overnight. Subsequently, a sterile pipette tip was
used to introduce a scratch in the middle of the well when the
confluency had reached 100%. The migration of cells towards the
center of the scratch was measured at the indicated time point (48
h). A light microscope was used to observe the cells and the
migration was measured using a caliper by testing the scratch
distance.
Transwell assay
Invasion assays were performed with an 8.0-µm pore
inserts in a 24-well Transwell chamber plate (Costar; Corning Inc.,
Corning, NY, USA). For this assay, 2×105 cells were
isolated and added to the upper chamber in serum-free RPMI-1640
medium, which was coated with Matrigel (BD Biosciences, San Jose,
CA, USA). RPMI-1640 (500 µl) with 10% FBS was added to the lower
chamber, followed by incubation for 24 h. Cells that had migrated
to the bottom of the filter were stained fixed in 4%
paraformaldehyde and stained with 0.1% crystal violet for 5 min at
room temperature. The cells left on the lower side of each membrane
were counted under a light microscope for five fields of view.
Bioinformatics analysis of miR-23a
target genes
Putative miR-23a targets were predicted using
several algorithms, including microRNA.org
(http://www.microrna.org/microrna/getGeneForm.do),
TargetScan (http://www.targetscan.org/) and miRanda (http://microrna.sanger.ac.uk/).
Dual-luciferase reporter assay
A Dual-Glo Luciferase Assay System (Promega
Corporation, Madison, WI, USA) was used to detect luciferase
activity, according to the manufacturer's protocol and as
previously described (19). A total
of 4×104 cells/well in 12-well plates were cultured
without antibiotics for 12 h. Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to transfect
cloned SIX1 wild-type 3′UTR and mutant 3′UTR target sequence.
Renilla luciferase plasmids (pRL-SV40; Promega Corporation)
were used as a control. After 48 h, a Dual Luciferase assay kit
(Promega Corporation) was used to detect luciferase intensity. All
data were normalized to Renilla luciferase expression. The
wild-type miR-23a target site in SIX1 3′UTR was AAGUGUA of the SIX1
3′UTR region. The mutant miR-23a target site was AACUCUU. The two
were designed and purchased from Shanghai GeneChem Co., Ltd.
(Shanghai, China).
Xenograft assays in vivo
The animal study protocols were approved by the
Animal Experimentation Ethics Committee of The Secondary Hospital
of Tianjin Medical University. All efforts were made to minimize
suffering and relieve pain. The nude mice (n=20; age, 5–6 weeks;
female; mean weight, 25 g) were obtained from Beijing Vital River
Laboratory Animal Technology Co., Ltd. (Beijing, China). The mice
were housed in a pathogen-free animal facility at 25°C with a 12-h
light/dark cycle, and randomly assigned to the control or
experimental group (four mice per group) (20). HEC1B cells (8×106) were
suspended in 0.1 ml DMEM, which was injected subcutaneously into
the right flank of each mouse (20).
The mice were divided into five groups and injected with control
(transfected with vector), Taxol, miR-23a con, miR-23a or
miR-23a+Taxol once every 3 or 4 days from day 14 (17). The final concentration of control,
miR-23a con and miR-23a for each intradermal injection of HEC1B
cells (0.01 mol) was 100 nM. The tumor volume was measured using
Vernier calipers every 3 or 4 days. The maximum tumor size measured
was 14 mm, and the maximum number of tumors observed in a single
mouse was 20. The following formula was used for calculation of
tumor volume: Tumor volume (mm3)=tumor length (mm) ×
tumor width (mm2/2). Only stable miR-23a mimics, not
miR-23a ASO, and cell lines were used for the in vivo
experiments.
Immunohistochemistry (IHC)
Following ethics committee approval, tumor samples
were obtained from 16 patients (aged 31–49 years) undergoing
surgical resection at Tianjin Medical University General Hospital
between July 2010 and May 2012, and IHC diagnosis was performed in
the hospital at the Department of Gynecology and Obstetrics. The
specimens were fixed with 10% formaldehyde at room temperature for
24 h. Specimens were embedded in paraffin, sectioned (5-µm thick)
and deparaffinized via the addition of alcohol at decreasing
concentrations (100, 95, 85 and 75%) for 5 min per step. For
antigen retrieval, a 96°C water-bath was used for antigen retrieval
in 0.01 mol/l sodium citrate buffer (10 mM, pH 6.0) for 20 min.
Subsequently, the sections were treated with 5 mM citrate buffer
and 3% H2O2 for 15 min, 5% goat serum (cat.
no. ZLI-9022; OriGene Technologies, Inc.) was used to block
sections at 37°C for 30 min, which were then incubated with a SIX1
antibody (1:200 dilution; cat. no. ab211359; Abcam) for 12 h at
4°C. Subsequently, the sections were incubated at 37°C for 30 min
with a secondary antibody (goat anti-rabbit immunoglobulin G; cat.
no. ZB-2301; ZSGB-BIO; OriGene Technologies, Inc.). All sections
were counterstained with hematoxylin at room temperature for 30
sec. The sections were observed under a light microscope (Olympus
Corporation, Tokyo, Japan). The frequencies of positive cells were
scored in degrees: 1 (low), <33; 2 (medium) 34–67%; and 3 (high)
>67%. The cases were further classified into positive groups
(2–5, low; >5, high) by the intensity and proportion of the
cancer cells immunostained for SIX1 (21,22). PBS
instead of the primary antibody was used for creating the negative
control for IHC staining (data not shown).
Statistical analysis
All experiments were repeated at least three times,
independently. Analyses were performed using SPSS v.22.0 software
(IBM Corp, Armonk, NY, USA). A Student's t-test was used to compare
between two groups. A two-way ANOVA followed by a post hoc Tukey's
test was used to compare between three or more groups. All data are
expressed as the means ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-23a expression is decreased in
endometrial cancer tissues compared with in para-carcinoma tissues,
and the overexpression of miR-23a may inhibit cell proliferation
and invasion in Ishikawa and HEC1B cells
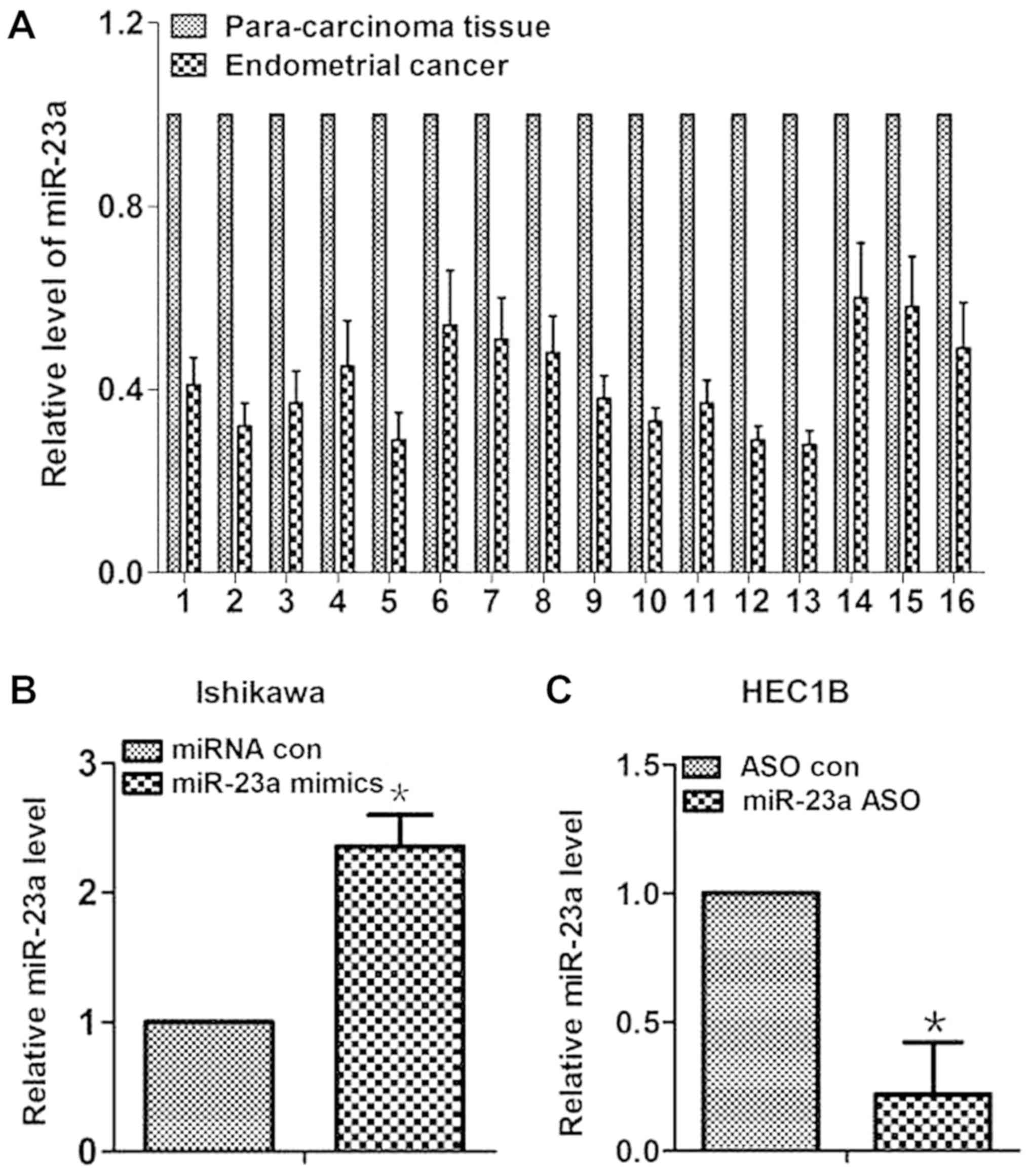
First, expression levels of miR-23a were measured in
16 pairs of human tissues. As shown in Fig. 1A, miR-23a expression levels were
reduced by 42% on average in endometrial cancer tissues compared
with in paired para-carcinoma tissues. To test the effect of
miR-23a in endometrial cancer cells, miR-23a mimics were
transfected into the Ishikawa and HEC1B cells to increase miR-23a
expression (Fig. 1B and C). The
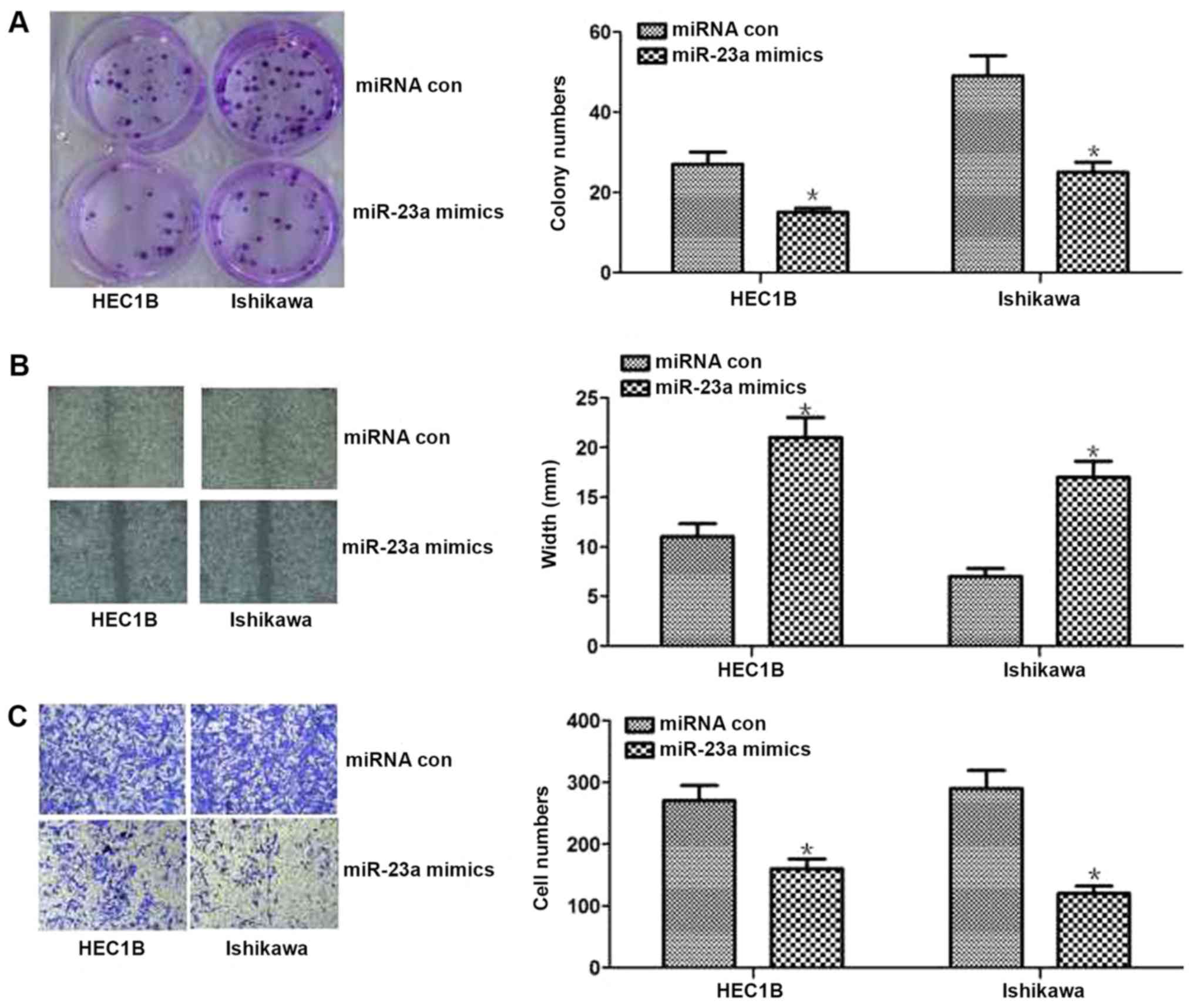
results of cell function experiments revealed that increased
miR-23a expression could inhibit proliferation and invasion in
vitro (P<0.05; Fig. 2). The
results of the colony formation assay revealed that proliferation
was significantly decreased in miR-23a mimic-transfected cells
compared with control cells (P<0.05; Fig. 2A). The results of the wound healing
assay demonstrated significantly less wound healing in miR-23a
mimic-transfected cells compared with the control cells (P<0.05;
Fig. 2B). Additionally, the results
of the Transwell assay revealed significantly lower numbers of
invasive cells in the miR-23a mimic-transfected group compared with
the control group (P<0.05; Fig.
2C). Overall, these results indicated that miR-23a expression
was decreased in endometrial cancer tissues, and overexpression of
miR-23 inhibited cell proliferation and invasion in endometrial
cancer cells.
SIX1 is a direct target of miR-23a,
and miR-23a downregulates SIX1 expression in endometrial cancer
cells
SIX1 was on the list of miR-23a targets suggested by
microrna software. To test the possibility of a direct link between
miR-23a and SIX1, the SIX1 3′UTR with either a wild-type or a
mutant miR-23a target sequence downstream of the firefly luciferase
gene was inserted (Fig. 3A). The
pGL3-SIX1 3′UTR wild-type, pGL3-SIX1 3′UTR Mut and pGL3 constructs
were individually transfected into HEC1B cells. The results
indicated that miR-23a may directly bind to SIX1 3′UTR (P<0.05;
Fig. 3B). In conclusion, the SIX1
gene may be a downstream post-transcriptional target of
miR-23a.
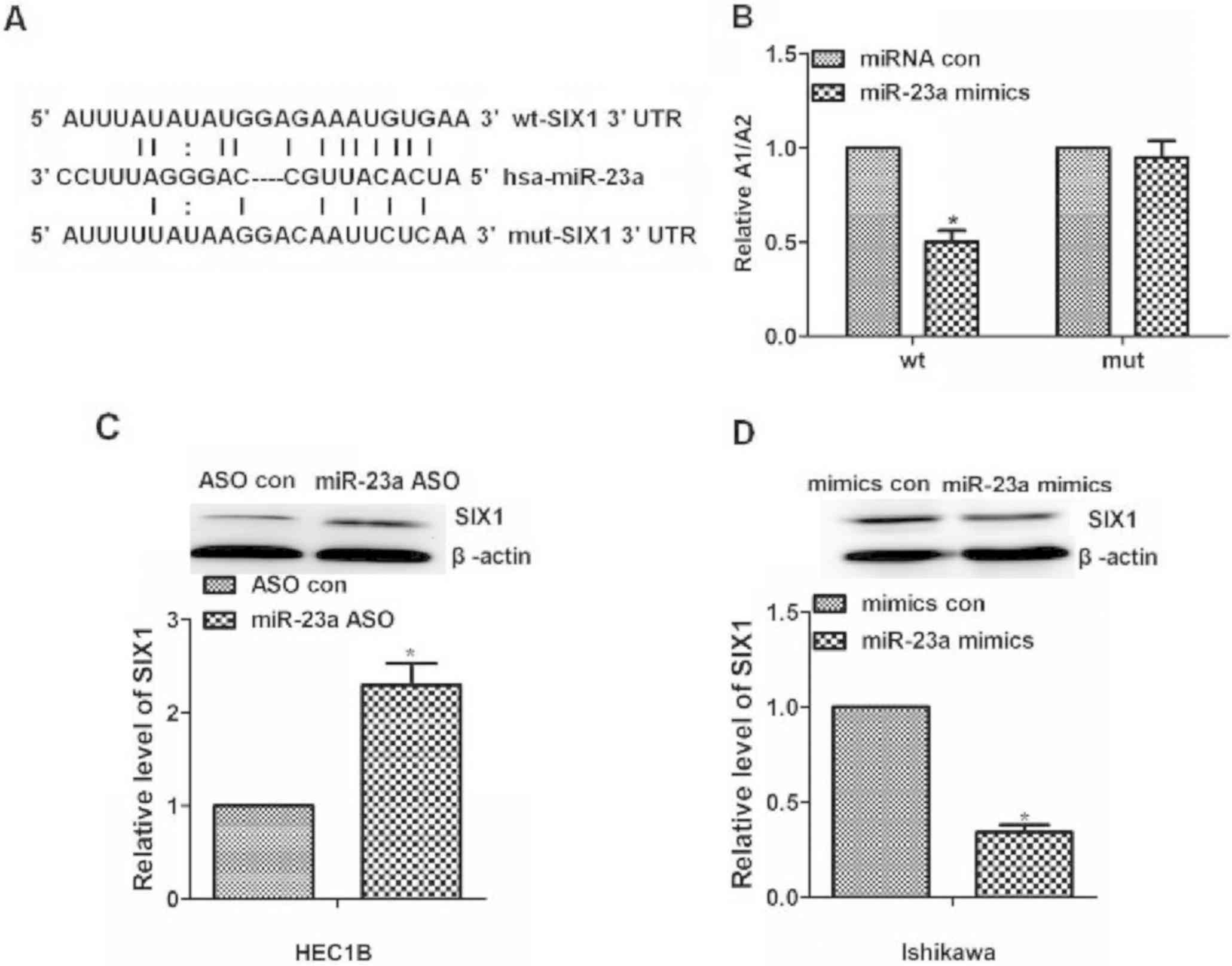 | Figure 3.miR-23a downregulates SIX1 expression
in vitro. (A) Bioinformatics results. The 3′UTR of SIX1
contains a potential miRNA-binding site for miR-23a. SIX1 UTR with
either a wt or mut miR-23a target sequence downstream of the
firefly luciferase gene was inserted into the pGL3-control vector
to create the pGL3-SIX1 UTR wt or the pGL3-SIX1 UTR mut construct,
respectively. (B) Luciferase reporter assay results. pGL3-SIX1 UTR
WT, pGL3-SIX1 UTR Mut and pGL3 constructs were individually
transfected into HEC1B cells. miR-23a significantly decreased the
relative luciferase activity of the wild-type SIX1 3′UTR compared
with the control, but miR-23a could not decrease the relative
luciferase activity of the mutant SIX1 3′UTR. (C) SIX1 protein
expression following different miR-23a treatments in endometrial
cancer HEC1B cells, semi-quantified by western blotting. The SIX1
protein expression level was higher in the group treated with
miR-23a ASO than that in the group treated with ASO con. (D) SIX1
protein expression following different miR-23a treatments in
endometrial cancer Ishikawa cells, semi-quantified by western
blotting. The SIX1 protein expression level was lower in the group
treated with miR-23a mimics than that in the group treated with
mimics con in the Ishikawa cells. *P<0.05, compared with wt
miRNA con group in (B). 3′UTR, 3′untranslated region; ASO,
antisense oligonucleotide; con, control; hsa, homo sapiens;
miR-23a, microRNA-23a; miRNA, microRNA; mut, mutant; SIX1, sine
oculis homeobox homolog 1; wt, wild-type. |
To further investigate the mechanism of miR-23a in
the chemoresistance of endometrial cancer via its ability to
repress its downstream gene SIX1, SIX1 protein expression following
different miR-23a treatment in two types of endometrial cancer
cells was detected by western blotting. According to a previous
study, SIX1 protein expression levels are high in Ishikawa cells
and low in HEC1B cells (22).
Consistent with the previous study, through the different miR-23a
treatment in the two types of endometrial cancer cells, the results
of the present study demonstrated that SIX1 protein expression was
increased in HEC1B cells treated with miR-23a ASO compared with
cells treated with ASO control (P<0.05; Fig. 3C). In addition, the present study
revealed that the SIX1 protein expression level was lower in
Ishikawa cells treated with miR-23a mimics than in cells treated
with miRNA con (P<0.05; Fig. 3D).
Overall, miR-23a may downregulate SIX1 expression in endometrial
cancer cells.
SIX1 may inhibit cell proliferation,
migration and invasion in endometrial cancer cells
To investigate the influence of SIX1 on the
biological behavior of endometrial cancer cells, SIX1 plasmid and
siRNA were used to increase or decrease the expression levels of
SIX1 in HEC1B or Ishikawa cells. Due to the differing levels of
SIX1 expression in the two cell lines, the knockdown and
overexpression experiments were performed in two different cell
lines. As shown in Fig. 4A, the
expression levels of SIX1 in HEC1B or Ishikawa cells were
significantly increased or decreased, when treated with plasmid and
siRNA, respectively (P<0.05). The results of the colony
formation assay revealed that capacity for proliferation caused by
the increased or reduced expression levels of SIX1 were
significantly enhanced or weakened compared with the control in
HEC1B or Ishikawa cells, respectively (P<0.05; Fig. 4B). The results of the wound healing
assay indicated the significantly smaller or bigger width caused by
the raised or reduced SIX1 than that in the control of HEC1B or
Ishikawa cells, respectively (P<0.05; Fig. 4C). The results of the Transwell assay
demonstrated significantly higher or lower numbers of invasive
cells in association with increased or decreased expression levels
of SIX1 compared with the control in HEC1B or Ishikawa cells
(P<0.05; Fig. 4D). Overall, the
results indicated that increased or decreased SIX1 expression
promoted or inhibited cell proliferation, migration and invasion
in vitro.
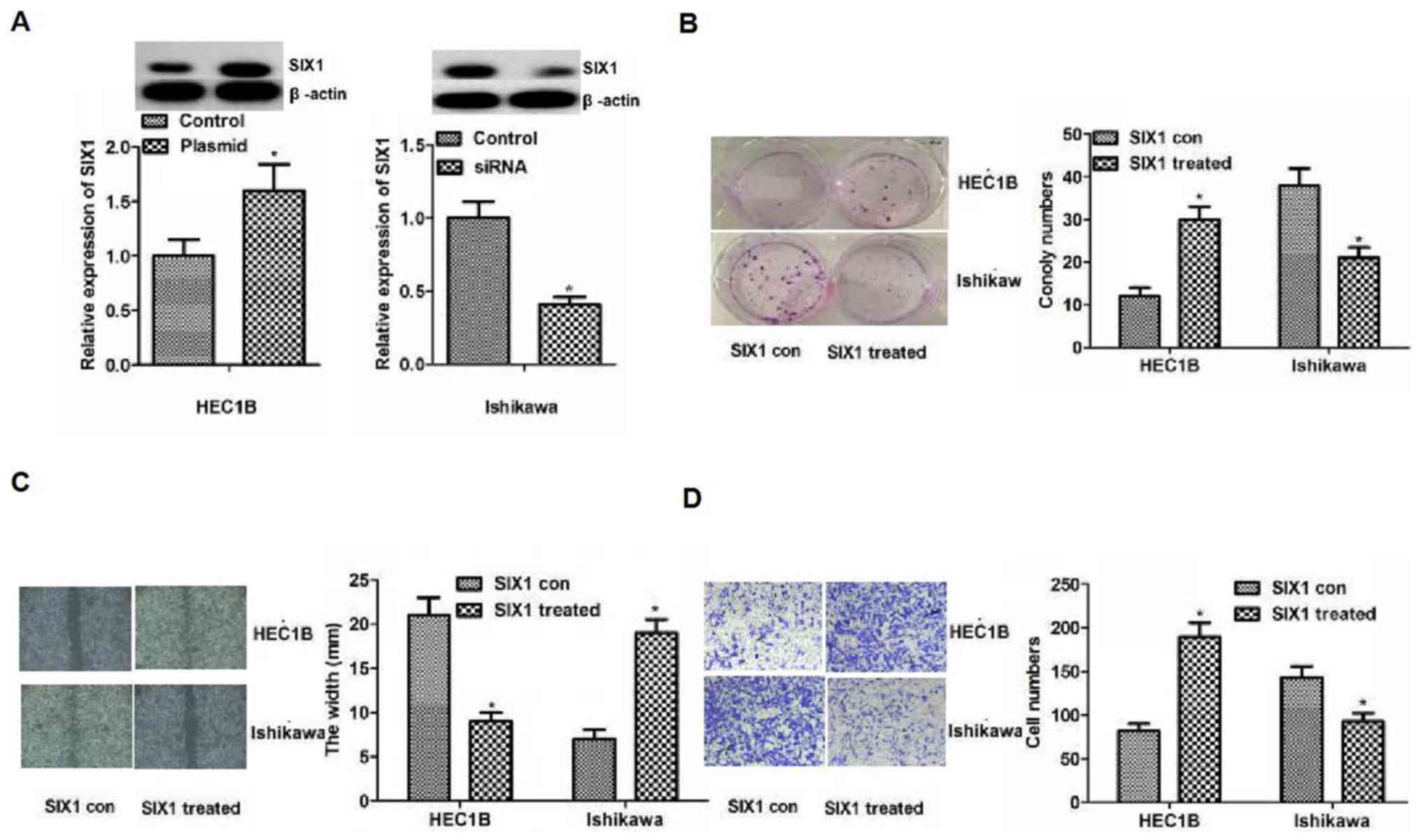 | Figure 4.Influence of SIX1 on the biological
behavior of endometrial cancer cell lines. (A) SIX1 plasmid or
siRNA were used to increase or reduce the protein expression levels
of SIX1 in HEC1B or Ishikawa cells. Differences were detected using
western blotting. Protein expression levels of SIX1 in HEC1B or
Ishikawa cells were increased or reduced in the SIX1 plasmid or
siRNA group compared with the control group, respectively. (B)
Colony formation assay. The results of colony formation assay
demonstrated that proliferation was significantly enhanced or
weakened compared with the control in HEC1B or Ishikawa cells,
respectively. Magnification, ×200. (C) Migration detected by wound
healing assays. The results revealed significantly smaller or
bigger width in the treated group compared with the control group
in HEC1B or Ishikawa cells, respectively. (D) Invasion detected by
Transwell assays. Significantly more or fewer invasive cells were
observed in the treated group compared with in the control group in
HEC1B or Ishikawa cells, respectively. Magnification, ×200.
*P<0.05 vs. con. Con, control; siRNA, small interfering RNA;
SIX1, sine oculis homeobox homolog 1; SIX1 treated,
overexpression in the HEC1B cell line or knockdown in the Ishikawa
cell line. |
miR-23a inhibits the development of
endometrial cancer by targeting SIX1
The aforementioned results revealed the possibility
that miR-23a inhibited endometrial cancer cell proliferation,
migration and invasion by downregulating SIX1. To further
demonstrate this, cell proliferation, migration and invasion
abilities were reversed by adding SIX1 siRNA or plasmid at the same
time as performing knockdown or overexpression of miR-23a (Fig. 5). As shown in Fig. 5A, miR-23a expression was reduced in
the miR-23a ASO group in HEC1B cells (P<0.05), while no
significant difference in miR-23a expression was identified between
the SIX1 siRNA + miR-23a ASO group and the miR-23a ASO group
(P>0.05). Similarly, miR-23a expression was increased in
Ishikawa cells treated with miR-23a mimics (P<0.05; Fig. 5A), while no significant difference
was observed in the increase in miR-23a expression between the SIX1
plasmid + miR-23a mimics group and the miR-23a mimics group
(P>0.05; Fig. 5A). The results
demonstrated that the knockdown or overexpression of miR-23a was
not altered by adding SIX1 siRNA or plasmid. Additionally, SIX1
expression was increased or decreased following the knockdown or
overexpression of miR-23a in the miR-23a ASO or mimics group
(P<0.05, respectively; Fig. 5B),
and SIX1 expression levels were reduced or increased by adding SIX1
siRNA or plasmid in HEC1B or Ishikawa cells (P<0.05,
respectively; Fig. 5B). The results
demonstrated that miR-23a directly targeted SIX1 in endometrial
cancer cells. Besides, the results of clone formation, wound
healing and Transwell assays revealed that proliferation, migration
and invasion abilities were strengthened or weakened, following the
knockdown or overexpression of miR-23a in the miR-23a ASO or
miR-23a mimics group in HEC1B or Ishikawa cells (P<0.05;
Fig. 5C-E), but the altered
proliferation, migration and invasion abilities following the
knockdown or overexpression of miR-23a could be reversed by the
downregulation or upregulation of SIX1 in HEC1B and Ishikawa cells
(P<0.05; 5C-E). Overall, the aforementioned results indicated
that miR-23a may promote the development of endometrial cancer by
targeting SIX1.
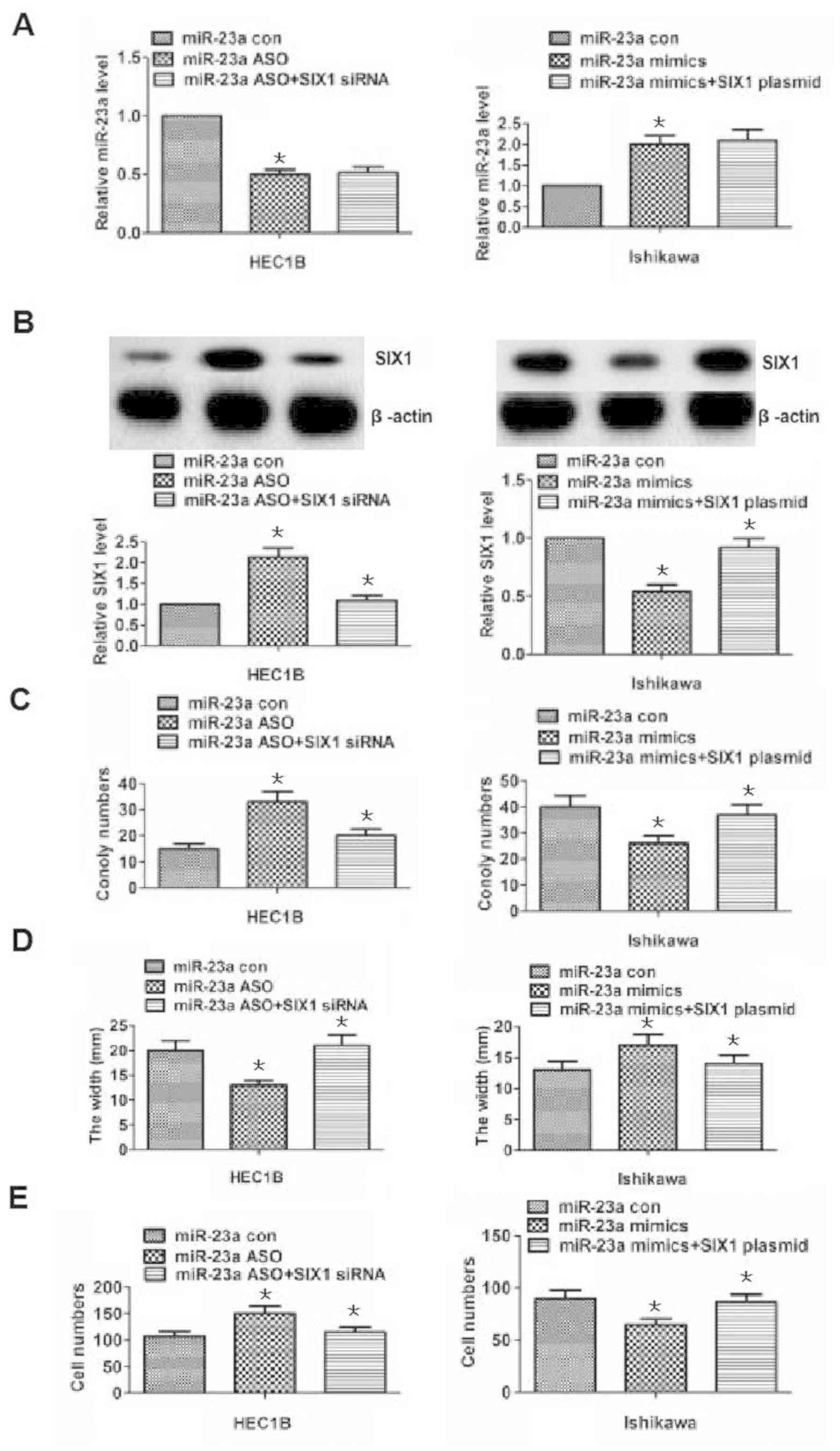 | Figure 5.Alterations in proliferation,
migration and invasion following miR-23a knockdown or
overexpression are reversed by SIX1 downregulation or upregulation.
(A) miR-23a expression was assessed using reverse
transcription-quantitative polymerase chain reaction in HEC1B and
Ishikawa cells for the differently treated groups. miR-23a
expression was decreased or increased in the miR-23a ASO or mimic
groups following the knockdown or the overexpression of miR-23a,
compared with in the associated control groups. However, the
decreased or increased miR-23a expression was not statistically
different between the SIX1 siRNA + miR-23a ASO or SIX1 plasmid +
miR-23a mimics groups compared with the miR-23a ASO or miR-23a
mimics groups following the knockdown or overexpression of miR-23a
in HEC1B or Ishikawa cells. (B) SIX1 protein expression was
detected by western blotting in HEC1B or Ishikawa cells in the
differently treated groups. SIX1 expression was increased or
decreased in the miR-23a ASO or mimics following the knockdown or
the overexpression of miR-23a compared with in the associated
control groups. Additionally, SIX1 expression was significantly
decreased or increased in the SIX1 siRNA + miR-23a ASO or SIX1
plasmid + miR-23a mimics groups compared with the miR-23a ASO or
miR-23a mimics groups following the knockdown or overexpression of
miR-23a in HEC1B or Ishikawa cells. (C) Results of the colony
formation assays in HEC1B and Ishikawa cells. The number of
colonies was increased or decreased in the miR-23a ASO or mimics
group following the knockdown or the overexpression of miR-23a
compared with the associated control group. Additionally, the
number of colonies was significantly decreased or increased between
the SIX1 siRNA + miR-23a ASO group or SIX1 plasmid + miR-23a mimics
group compared with the miR-23a ASO group or miR-23a mimics group,
following the knockdown or overexpression of miR-23a in HEC1B or
Ishikawa cells, respectively. (D and E) Results of wound healing
and Transwell assays for the different groups in HEC1B and Ishikawa
cells. The results revealed that the migration and invasion
abilities were altered in the miR-23a ASO and mimics groups
following the knockdown or overexpression of miR-23a compared with
in the associated control group. Additionally, migration and
invasion were significantly altered in the SIX1 siRNA + miR-23a ASO
group or SIX1 plasmid + miR-23a mimics group compared with in the
miR-23a ASO group or miR-23a mimics group following the knockdown
or overexpression of miR-23a in HEC1B and Ishikawa cells.
*P<0.05, compared with the associated control group. ASO,
antisense oligonucleotide; con, control; miR-23a, microRNA-23a;
siRNA, small interfering RNA; SIX1, sine oculis homeobox
homolog 1. |
Inhibitory effect of miR-23a mimics
and/or Taxol by reducing SIX1 expression in endometrial cancer in
vivo
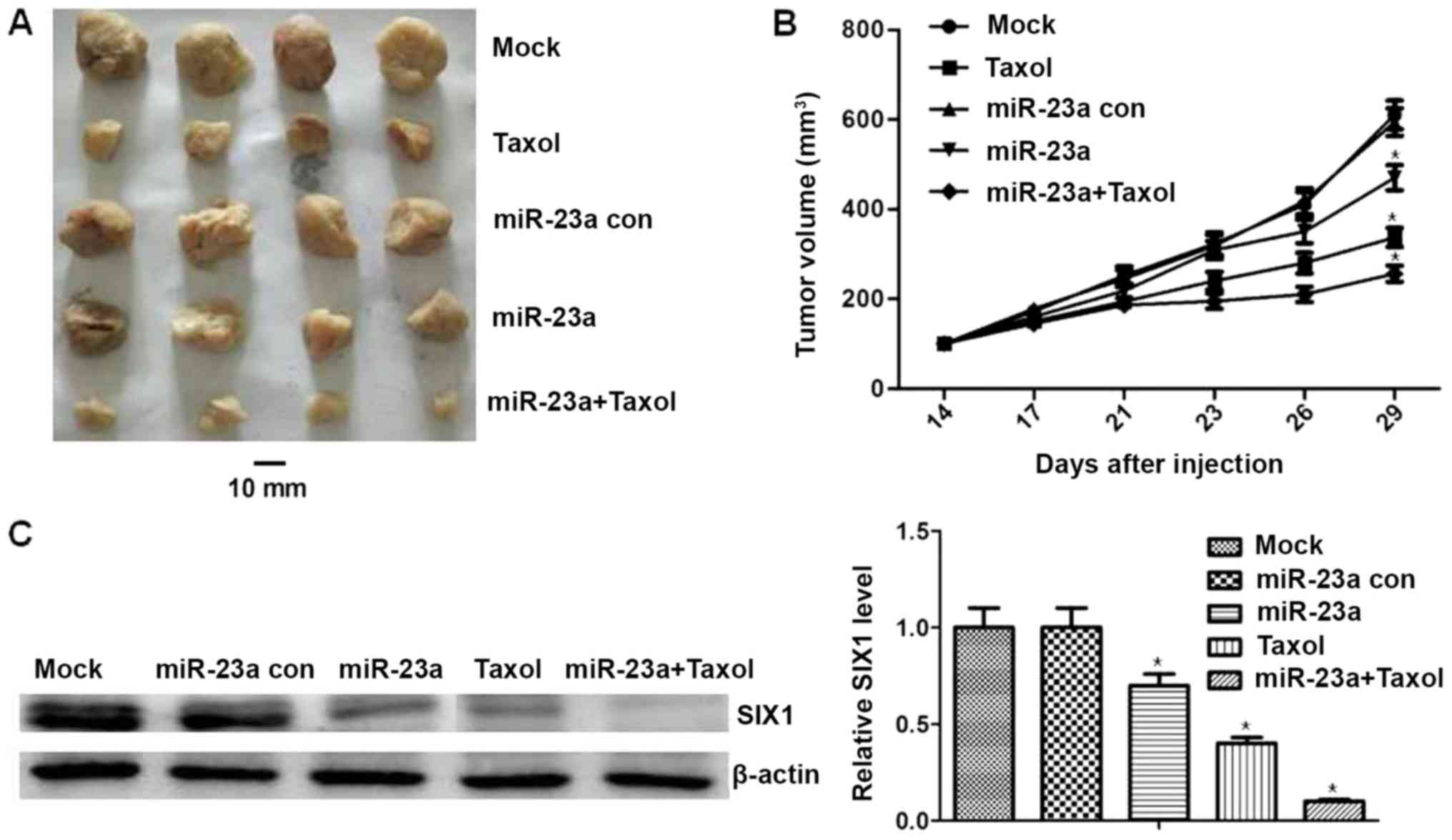
To verify the inhibitory effect of miR-23a mimics
and/or Taxol and to determine whether miR-23a regulates SIX1
expression in vivo, Ishikawa cells were injected
subcutaneously into the right flank of nude mice. The results
revealed that the mean tumor volume in the Taxol group was
significantly lower compared with in the control group (P<0.05;
Fig. 6A and B). The mean tumor
volume was significantly lower for the miR-23a mimics and miR-23a
mimics + Taxol group compared with the mimics control group
(P<0.05; Fig. 6A and B). Besides,
the mean tumor volume was significantly lower in the miR-23a +
Taxol group compared with in the Taxol group (P<0.05; Fig. 6A and B). In addition, SIX1 expression
was assessed in mice tumors by western blotting. This revealed that
the expression of SIX1 was decreased in the miR-23a mimics and/or
Taxol groups (P<0.05; Fig. 6C).
Additionally, compared with in the Taxol group, the expression
levels of SIX1 in the miR-23a mimics + Taxol group were
significantly decreased (P<0.05; Fig.
6C). There was no significant difference identified in the body
weight of mice treated with miR-23a mimics and/or Taxol, none of
the mice tested exhibited signs of other adverse effects, and no
toxic effects were observed through blood counts or the observation
of liver and renal function (data not shown). These results
demonstrated the antitumor effects and safety of miR-23a mimics
and/or Taxol. Overall, the aforementioned results indicated that
miR-23a could inhibit chemoresistance of endometrial cancer in
vivo by targeting SIX1.
Association between miR-23a and SIX1
expression in endometrial cancer tissues
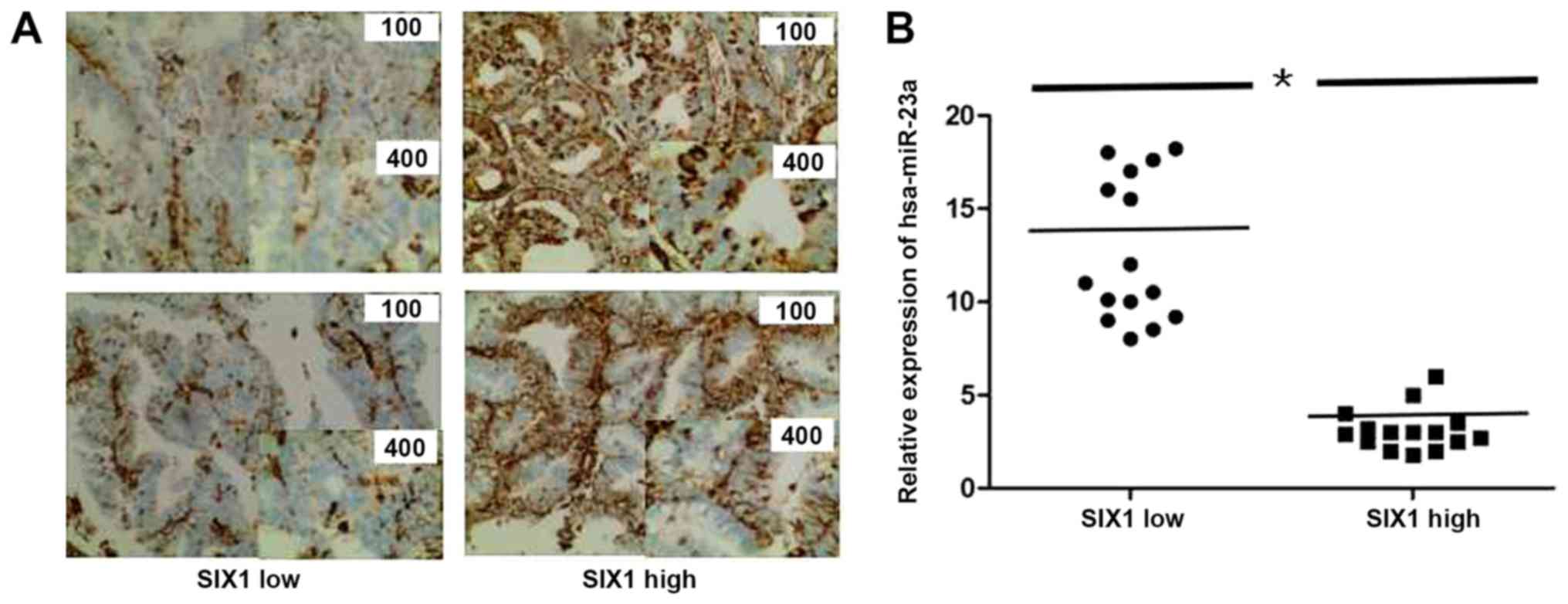
Finally, to explore the association between miR-23a
and SIX1 in endometrial cancer tissues, their expression levels in
30 endometrial cancer cases were assessed. SIX1 expression was
assessed by IHC, the 30 cases were divided into two groups: SIX1
low and high groups (n=15 for each group; Fig. 7A). Additionally, miR-23a expression
was tested by RT-qPCR in the two groups, and a significant
difference between the SIX1 low and high tissue samples was
identified (P<0.05). Therefore, the results suggested a negative
association between miR-23a and SIX1 expression in endometrial
cancer tissues.
Discussion
miRNAs serve different roles (promote or suppress
cancer development) in cancer cellular processes, including in
endometrial cancer (2,5–13).
Previously, miR-23a has been linked to chemoresistance in cancer
(10,12,15–17,19), and
there have been three studies investigating the association between
overexpression of miR-23a in cancer and chemoresistance (15,16,19).
However, thus far, only one study investigated the association
between miR-23a and endometrial cancer, and indicated that miR-23a
is downregulated in cancer tissue and that miR-23a may inhibit EMT
in endometrial endometrioid adenocarcinoma by targeting SMAD3
(14). In the present study, to
investigate the function and mechanism of miR-23a, miR-23a was
first demonstrated to be decreased in endometrial cancer tissues
compared with in para-carcinoma tissues, and overexpression of
miR-23a inhibited cell proliferation or invasion in Ishikawa cells
and HEC1B cells. In addition, in the present study, overexpression
of miR-23a inhibited proliferation, migration and invasion, which
was different from the results reported in previous studies
(15,16,19), and
a possible explanation for this phenomenon was that this was due to
the different types of cancer being investigated. Subsequently, it
was identified that SIX1 was a target gene of miR-23a by different
types of software, including TargetScan. Additionally, it was
demonstrated that SIX1 was a functional target of miR-23a in
endometrial cancer cells.
SIX1, a transcription factor, belongs to the SIX
family of hemoproteins, and it has been reported to be less
expressed in human normal tissue but expressed in mouse dental
follicle, human periodontal ligament-derived, mouse skeletal muscle
and cephalic neural crest cells; particularly, SIX1 overexpression
has been reported to occur in several human types of cancer,
including breast cancer, cervical cancer, ovarian cancer, oral
squamous cell carcinoma, gastric cancer, hepatocellular carcinoma
and pancreatic cancer, which leads to cancer cell proliferation,
invasion and metastasis (21–38).
Consistent with these results, the present study demonstrated that
SIX1 promoted cell proliferation, migration and invasion in
endometrial cancer. In addition, it has been reported that SIX1
upregulation induces cancer cell EMT (39–41).
SIX1 upregulation leads to EMT via the activation of zinc-finger
E-box binding homeobox1 (41).
miR-23a could inhibit EMT in endometrial adenocarcinoma by
targeting SMAD3 (14). This may
provide clues for the association between miR-23a and SIX1.
Additionally, SIX1 may mediate resistance to paclitaxel in breast
cancer cells (27), which indicates
that a similar mechanism may exist in endometrial cancer. Previous
studies have identified an association between overexpression of
miR-23a in cancer and chemoresistance (15,16,19).
Particularly, a recent study demonstrated that SIX1 is
overexpressed in endometrial carcinoma and promotes the malignant
behavior of cancer cells via ERK and AKT signaling, the expression
levels of SIX1 were high or low in Ishikawa or HEC1B cell lines
(22), which is consistent with the
results of the present study. Overall, the aforementioned studies
and results revealed indirect evidence for the hypothesis that
miR-23a may promote endometrial cancer development by targeting
SIX1. In addition, the direct targeting association was further
supported by the results of the luciferase reporter assay.
Additionally, expression levels of miR-23a and SIX1 were assessed
in 30 endometrial cancer cases, and this revealed a negative
association between miR-23a and SIX1 expression in endometrial
cancer tissues.
The present study demonstrated that miR-23a may be a
suppressor gene in endometrial cancer cells, and alterations in the
miR-23a-SIX1 interaction may be associated with the development of
endometrial cancer. Furthermore, in contrast to the results of
other studies, possibly due to different types of cancer and
different perspectives, the results of the present study indicated
that SIX1 was targeted by miR-23a, similar methods could be used to
further explore the mechanism (24):
For example, miR-188 is downregulated in oral squamous cell
carcinoma and inhibits proliferation and invasion by targeting
SIX1. The bioinformatics analysis, dual-luciferase reporter assay
and the regulation results, with the same tendency of the
associations between the upstream and downstream genes, indicated
that SIX1 was a downstream target gene of miR-23a in vitro,
and upregulation or downregulation of cell proliferation, migration
and invasion, following the knockdown or overexpression of miR-23a,
were reversed by adding SIX1 siRNA or plasmid in vitro.
As demonstrated in the present study, the inhibitory
effect of miR-23a mimics, which reduces SIX1 expression in
endometrial cancer, may be observed in vitro and in
vivo. However, future studies should determine the
aforementioned mechanism in various types of endometrial cancer
cells in vitro and in vivo. Perhaps the association
between genes known to be associated with endometrial cancer and
the miR-23a-SIX1 interaction should be explored. A recent study
indicated that it may be useful to investigate the association
between miR-23a and SIX1 in the EMT and resistance mechanism
(14). In conclusion, the results of
the present study suggested that miR-23a may inhibit endometrial
cancer cell development by targeting SIX1, and that miR-23a may
serve as a therapeutic target for endometrial cancer.
Acknowledgements
Not applicable.
Funding
This work was supported by Tianjin Medical
University Second Hospital Youth Fund (grant no. 2017ydey03).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HLL and JJS carried out the molecular biology
experiments and drafted the manuscript. HM and SJL performed the
animal experiments. NL, SJG and YS participated in the sequence
alignment. HLL and JJS participated in the design of the study and
performed the statistical analysis. YYX, ZYQ, YQW, FW, RMG and DL
participated in the design and coordination of the study, and
helped to draft the manuscript. FXX conceived the study,
participated in its design and coordination, and helped to draft
the manuscript. All authors read and approved the final
manuscript
Ethics approval and consent to
participate
All applicable international, national, and/or
institutional guidelines for the care and use of human specimens
and animals were followed. The animal study was carried out in
accordance with the guidelines approved by the Animal
Experimentation Ethics Committee of the Secondary Hospital of
Tianjin Medical University. The protocol was approved by the
committee, all surgery was performed under sodium pentobarbital
anesthesia, and all efforts were made to minimize suffering. The
use of human samples was approved by the Ethics Committee of
Secondary Hospital of Tianjin Medical University. The patients or
their families provided written informed consent for the use of
these tissues.
Patient consent for publication
The patients or parents provided written informed
consent for the publication of any associated data and accompanying
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vogel RI, Pulver T, Heilmann W, Mooneyham
A, Mullany S, Zhao X, Shahi M, Richter J, Klein M, Chen L, et al:
USP14 is a predictor of recurrence in endometrial cancer and a
molecular target for endometrial cancer treatment. Oncotarget.
7:30962–30976. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tran AQ and Gehrig P: Recent advances in
endometrial cancer. F1000Res. 6:812017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kang S, Kang WD, Chung HH, Jeong DH, Seo
SS, Lee JM, Lee JK, Kim JW, Kim SM, Park SY and Kim KT:
Preoperative identification of a low-risk group for lymph node
metastasis in endometrial cancer: A Korean gynecologic oncology
group study. J Clin Oncol. 30:1329–1334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wright JD, Burke WM, Wilde ET, Lewin SN,
Charles AS, Kim JH, Goldman N, Neugut AI, Herzog TJ and Hershman
DL: Comparative effectiveness of robotic versus laparoscopic
hysterectomy for endometrial cancer. J Clin Oncol. 30:783–791.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brasseur K, Gévry N and Asselin E:
Chemoresistance and targeted therapies in ovarian and endometrial
cancers. Oncotarget. 8:4008–4042. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nakamura K, Sawada K, Yoshimura A, Kinose
Y, Nakatsuka E and Kimura T: Clinical relevance of circulating
cell-free microRNAs in ovarian cancer. Mol Cancer. 15:482016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang D, Sun Y, Hu L, Zheng H, Ji P, Pecot
CV, Zhao Y, Reynolds S, Cheng H, Rupaimoole R, et al: Integrated
analyses identify a master microRNA regulatory network for the
mesenchymal subtype in serous ovarian cancer. Cancer Cell.
23:186–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou Y, Wang M, Wu J, Jie Z, Chang S and
Shuang T: The clinicopathological significance of miR-1307 in
chemotherapy resistant epithelial ovarian cancer. J Ovarian Res.
8:232015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Záveský L, Jandáková E, Turyna R,
Langmeierová L, Weinberger V, Záveská Drábková L, Hůlková M,
Hořínek A, Dušková D, Feyereisl J, et al: Evaluation of cell-free
urine microRNAs expression for the use in diagnosis of ovarian and
endometrial cancers. A pilot study. Pathol Oncol Res. 21:1027–1035.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ran X, Yang J, Liu C, Zhou P, Xiao L and
Zhang K: miR-218 inhibits HMGB1-mediated autophagy in endometrial
carcinoma cells during chemotherapy. Int J Clin Exp Pathol.
8:6617–6626. 2015.PubMed/NCBI
|
|
13
|
Dong P, Kaneuchi M, Watari H, Hamada J,
Sudo S, Ju J and Sakuragi N: MicroRNA-194 inhibits epithelial to
mesenchymal transition of endometrial cancer cells by targeting
oncogene BMI-1. Mol Cancer. 10:992011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu P, Wang C, Ma C, Wu Q, Zhang W and Lao
G: MicroRNA-23a regulates epithelial-to-mesenchymal transition in
endometrial endometrioid adenocarcinoma by targeting SMAD3. Cancer
Cell Int. 16:672016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li X, Li X, Liao D, Wang X, Wu Z, Nie J,
Bai M, Fu X, Mei Q and Han W: Elevated microRNA-23a expression
enhances the chemo-resistance of colorectal cancer cells with
microsatellite instability to 5-fluorouracil by directly targeting
ABCF1. Curr Protein Pept Sci. 16:301–309. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guan L, Hu X, Liu L, Xing Y, Zhou Z, Liang
X, Yang Q, Jin S, Bao J, Gao H, et al: Bta-miR-23a involves in
adipogenesis of progenitor cells derived from fetal bovine skeletal
muscle. Sci Rep. 7:437162017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu X, Wang Y, Liang H, Fan Q, Zhu R, Cui
J, Zhang W, Zen K, Zhang CY, Hou D, et al: miR-23a/b promote tumor
growth and suppress apoptosis by targeting PDCD4 in gastric cancer.
Cell Death Dis. 8:e30592017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shang J, Yang F, Wang Y, Wang Y, Xue G,
Mei Q, Wang F and Sun S: MicroRNA-23a antisense enhances
5-fluorouracil chemosensitivity through APAF-1/caspase-9 apoptotic
pathway in colorectal cancer cells. J Cell Biochem. 115:772–784.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun KX, Jiao JW, Chen S, Liu BL and Zhao
Y: MicroRNA-186 induces sensitivity of ovarian cancer cells to
paclitaxel and cisplatin by targeting ABCB1. J Ovarian Res.
8:802015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He Z, Li G, Tang L and Li Y: SIX1
overexpression predicts poor prognosis and induces radioresistance
through AKT signaling in esophageal squamous cell carcinoma. Onco
Targets Ther. 10:1071–1079. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xin X, Li Y and Yang X: SIX1 is
overexpressed in endometrial carcinoma and promotes the malignant
behavior of cancer cells through ERK and AKT signaling. Oncol Lett.
12:3435–3440. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kawasaki T, Takahashi M, Yajima H, Mori Y
and Kawakami K: Six1 is required for mouse dental follicle cell and
human periodontal ligament-derived cell proliferation. Dev Growth
Differ. 58:530–545. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L and Liu H: microRNA-188 is
downregulated in oral squamous cell carcinoma and inhibits
proliferation and invasion by targeting SIX1. Tumour Biol.
37:4105–4113. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu D, Li L, Zhang XX, Wan DY, Xi BX, Hu
Z, Ding WC, Zhu D, Wang XL, Wang W, et al: SIX1 promotes tumor
lymphangiogenesis by coordinating TGFβ signals that increase
expression of VEGF-C. Cancer Res. 74:5597–5607. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feng GW, Dong LD, Shang WJ, Pang XL, Li
JF, Liu L and Wang Y: HDAC5 promotes cell proliferation in human
hepatocellular carcinoma by up-regulating Six1 expression. Eur Rev
Med Pharmacol Sci. 18:811–816. 2014.PubMed/NCBI
|
|
27
|
Li Z, Tian T, Hu X, Zhang X, Nan F, Chang
Y, Lv F and Zhang M: Six1 mediates resistance to paclitaxel in
breast cancer cells. Biochem Biophys Res Commun. 441:538–543. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hetzler KL, Collins BC, Shanely RA, Sue H
and Kostek MC: The homoeobox gene SIX1 alters myosin heavy chain
isoform expression in mouse skeletal muscle. Acta Physiol (Oxf).
210:415–428. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garcez RC, Le Douarin NM and Creuzet SE:
Combinatorial activity of Six1-2-4 genes in cephalic neural crest
cells controls craniofacial and brain development. Cell Mol Life
Sci. 71:2149–2164. 2014.PubMed/NCBI
|
|
30
|
Li Z, Tian T, Lv F, Chang Y, Wang X, Zhang
L, Li X, Li L, Ma W, Wu J and Zhang M: Six1 promotes proliferation
of pancreatic cancer cells via upregulation of cyclin D1
expression. PLoS One. 8:e592032013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sato S, Ikeda K, Shioi G, Ochi H, Ogino H,
Yajima H and Kawakami K: Conserved expression of mouse Six1 in the
pre-placodal region (PPR) and identification of an enhancer for the
rostral PPR. Dev Biol. 344:158–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ng KT, Lee TK, Cheng Q, Wo JY, Sun CK, Guo
DY, Lim ZX, Lo CM, Poon RT, Fan ST and Man K: Suppression of
tumorigenesis and metastasis of hepatocellular carcinoma by shRNA
interference targeting on homeoprotein Six1. Int J Cancer.
127:859–872. 2010.PubMed/NCBI
|
|
33
|
Plant KE, Anderson E, Simecek N, Brown R,
Forster S, Spinks J, Toms N, Gibson GG, Lyon J and Plant N: The
neuroprotective action of the mood stabilizing drugs lithium
chloride and sodium valproate is mediated through the up-regulation
of the homeodomain protein Six1. Toxicol Appl Pharmacol.
235:124–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu Y, Davicioni E, Triche TJ and Merlino
G: The homeoprotein six1 transcriptionally activates multiple
protumorigenic genes but requires ezrin to promote metastasis.
Cancer Res. 66:1982–1989. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Coletta RD, Christensen K, Reichenberger
KJ, Lamb J, Micomonaco D, Huang L, Wolf DM, Müller-Tidow C, Golub
TR, Kawakami K and Ford HL: The Six1 homeoprotein stimulates
tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci USA.
101:6478–6483. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reichenberger KJ, Coletta RD, Schulte AP,
Varella-Garcia M and Ford HL: Gene amplification is a mechanism of
Six1 overexpression in breast cancer. Cancer Res. 65:2668–2675.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng XH, Liang PH, Guo JX, Zheng YR, Han
J, Yu LL, Zhou YG and Li L: Expression and clinical implications of
homeobox gene Six1 in cervical cancer cell lines and cervical
epithelial tissues. Int J Gynecol Cancer. 20:1587–1592.
2010.PubMed/NCBI
|
|
38
|
Behbakht K, Qamar L, Aldridge CS, Coletta
RD, Davidson SA, Thorburn A and Ford HL: Six1 overexpression in
ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and
is associated with poor survival. Cancer Res. 67:3036–3042. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Smith AL, Iwanaga R, Drasin DJ, Micalizzi
DS, Vartuli RL, Tan AC and Ford HL: The miR-106b-25 cluster targets
Smad7, activates TGF-b signaling, and induces EMT and tumor
initiating cell characteristics downstream of Six1 in human breast
cancer. Oncogene. 31:5162–5171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Radisky DC: Defining a role for the
homeoprotein Six1 in EMT and mammary tumorigenesis. J Clin Invest.
119:2528–2531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ono H, Imoto I, Kozaki K, Tsuda H, Matsui
T, Kurasawa Y, Muramatsu T, Sugihara K and Inazawa J: SIX1 promotes
epithelial-mesenchymal transition in colorectal cancer through ZEB1
activation. Oncogene. 31:4923–4934. 2012. View Article : Google Scholar : PubMed/NCBI
|