Introduction
Adrenocortical carcinoma (ACC) is a rare endocrine
malignancy that arises from the adrenal cortex, with an occurrence
rate of 0.7–2.0 cases per 1,000,000 each year (1–3). The
overall 5-year survival rate for patients in the Netherlands is
~32%, and patients with stage III and IV have a particularly lower
survival rate, due to its highly aggressive biological behavior
(2,4). ACC-associated mortality accounts for
0.02–0.20% of all cancer-associated mortalities (5,6). At
present, complete surgical resection with or without mitotane
adjuvant treatment is the only treatment option (7,8).
However, 40–70% of patients with ACC present with metastasis at the
time of diagnosis (9). Therefore, it
is important to explore the molecular mechanisms underlying ACC and
identify candidate biomarkers for its diagnosis and treatment.
At present, the diagnosis and classification of
adrenocortical cancers relies on histological examination of tumor
sections and immunohistochemical markers, such as Ki-67, IGF2 and
SF-1, are used to support the diagnosis of ACC (10). The Weiss or modified Weiss score
systems are most often used in diagnosis as the primary
determinants of malignancy in adrenocortical tumors (10,11);
however, the diagnosis of these tumors remains challenging,
particularly for rare subtypes of ACC such as oncocytic, myxoid and
sarcomatoid subtypes (12).
Following developments in microarray technology,
several studies have demonstrated that abnormal expressed and
mutated genes are involved in the tumorigenesis and progression of
ACC (9,13–15). For
example, using DNA microarray analysis, Giordano et al
(13) demonstrated that several cell
cycle and proliferation genes, such as Cyclin B2 (CCNB2),
Abnormal Spindle Microtubule Assembly, Ribonucleotide Reductase
Regulatory Subunit M2 (RRM2), DNA Topoisomerase II a and
Cyclin Dependent Kinase Inhibitor 3, as well as genes associated
with tumor invasion, such as Secreted Phosphoprotein 1 may serve as
potential diagnostic biomarkers that could be developed into useful
immunohistochemical tools. Kulshrestha and Suman (14) identified a total of 53 genes as
common hubs of the disease system, which may exert important
biological functions in pediatric adrenocortical adenoma and
carcinoma. Cyclin Dependent Kinase 1 (CDK1), Cyclin B1
(CCNB1), Cell Division Cycle 20 and BUB1 Mitotic Checkpoint
Serine/Threonine Kinase B may serve as potential biomarkers of
pediatric ACC and as potential targets for its treatment (16). Yuan et al (15) analyzed 12 hub genes associated with
the progression and prognosis of ACC by weighted gene co-expression
network analysis. In addition, Duregon et al (17) assessed the expression of miRNAs
associated with the regulation of the IGF2 gene and hypoxia induced
microRNA in histological variants including 35 classical, 6 myxoid
and 10 oncocytic cases of ACC and reported that miR-483-3p,
miR-483-5p and miR-210, which were identified as candidates for
tumor aggressiveness and poor prognosis in ACC, are differentially
expressed in ACC variants. However, the latent molecular and
pathway interactions of ACC have yet to be completely elucidated.
There is a need for additional studies with comprehensive and
integrated genomic characterization, combined with clinical data to
explore the molecular mechanisms and identify candidate biomarkers
for the diagnosis of ACC, as well as advance the presently
available understanding of the tumorigenesis mechanism.
The aim of the present study was to analyze the
differentially expressed genes (DEGs) in ACC by combining two mRNA
microarray datasets from the Gene Expression Omnibus (GEO)
database. Subsequently, Gene Ontology (GO), Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway enrichment and protein-protein
interaction (PPI) network analyses were performed to provide
detailed insights into the biological mechanisms in ACC. GEO and
Oncomine databases were subsequently combined to validate the
importance of the hub genes. In conclusion, using bioinformatics
methods, the present study identified 14 hub genes which provided
significant diagnostic and prognostic value and may serve as
candidate biomarkers for ACC.
Materials and methods
Data resources
The gene expression data was retrieved from the GEO
database (ncbi.nlm.nih.gov/geo/). Two gene expression datasets,
GSE12368 (18) and GSE19750
(19) were downloaded from GEO using
GPL570 Affymetrix Human Genome U133 Plus 2.0 Array. The GSE12368
dataset contained 12 ACC and 6 normal samples. The GSE19750 dataset
contained 44 ACC and 4 normal adrenal glands samples.
Identification of DEGs
The DEGs between ACC and normal adrenal gland
samples were screened using GEO2R (ncbi.nlm.nih.gov/geo/geo2r). GEO2R is an interactive
web tool using limma R packages (version R 3.2.3; limma 3.26.8)
(20), which allows users to compare
two or more datasets in a GEO series, to identify DEGs across
experimental conditions (21,22). The
adjusted P-values (adj. P) and Benjamini and Hochberg false
discovery rate provided a balance between the discovery of
statistically significant genes and the limitations of
false-positive results. Probe sets without corresponding gene
symbols or genes with >1 probe sets were removed or averaged,
respectively. The cut-off criteria of |logFC(fold-change)|>1 and
adj. P<0.05 were considered statistically significant.
KEGG and GO enrichment analysis of
DEGs
The Database for Annotation, Visualization and
Integrated Discovery (DAVID; http://david.ncifcrf.gov; version 6.8) is an online
biological information database that integrates biological data and
analysis tools, and provides a comprehensive set of functional
annotation information of genes and proteins for users to extract
biological information (23). KEGG
(http://www.genome.ad.jp/kegg) is a
knowledge base for systematic analysis of gene functions (24). GO analysis predicts the function of
the target genes in three aspects, including biological processes
(BPs), cellular components (CCs) and molecular functions (MFs)
(25). To analyze the possible
functions of DEGs, functional annotation was performed using the
DAVID database; P<0.05 was considered statistically
significant.
PPI network construction and module
analysis
The PPI network was predicted using the Search Tool
for the Retrieval of Interacting Genes (STRING; http://string-db.org; version 10.5) database (26). Analyzing the functional interactions
between proteins can predict the interaction relationship involved
in the development and progression of ACC (27,28). In
the present study, a PPI network of DEGs was constructed using the
STRING database, and an interaction with a combined score of
>0.4 was considered statistically significant. Cytoscape
(version 3.6.1; http://cytoscape.org) is an open
source bioinformatics platform for visualizing molecular
interaction networks (29). The
Molecular Complex Detection (MCODE; version 1.4.2) Cytoscape plugin
allows for clustering a given network based on topology to identify
densely connected regions (30). The
PPI networks were constructed using Cytoscape and the most
significant module in the PPI networks was identified using MCODE.
The selection criteria were as follows: Degree cut-off=2, node
score cut-off=0.2, max depth=100 and k-score=2. Subsequently, KEGG
and GO analyses for genes in this module were performed using
DAVID. Data analysis of biological processes in the hub genes was
performed using R (http://www.r-project.org, version 3.2.4).
Hub gene selection and analysis
Hub genes with a degree of ≥10 were selected, and
the network of the genes and their co-expression genes was analyzed
using cBioPortal (http://www.cbioportal.org) (31,32).
Hierarchical clustering of hub genes was constructed using UCSC
Cancer Genomics Browser (http://genome-cancer.ucsc.edu) (33). To evaluate the prognostic value of
the selected hub genes in ACC, overall and disease-free survival
based on expression of the hub genes were performed using
Kaplan-Meier curves in cBioPortal. In addition, the association
between hub gene expression and tumor Weiss grade in patients with
ACC was analyzed using the Oncomine database (www.oncomine.com). mRNA expression analyses of
thymidylate synthetase (TYMS), GINS complex subunit 1
(GINS1), ribonucleotide reductase regulatory subunit M2
(RRM2), ZW10 interacting kinetochore protein (ZWINT)
and structural maintenance of chromosomes 4 (SMC4) genes in
ACC vs. normal tissues was performed in the Giordano Adrenal and
Giordano Adrenal 2 datasets from the Oncomine database (13,34).
Results
Identification of DEGs in ACC
Following standardization of the microarray results
by GEO2R, 970 and 998 DEGs were identified in the GSE12368 and
GSE19750 gene expression profile datasets, respectively. The
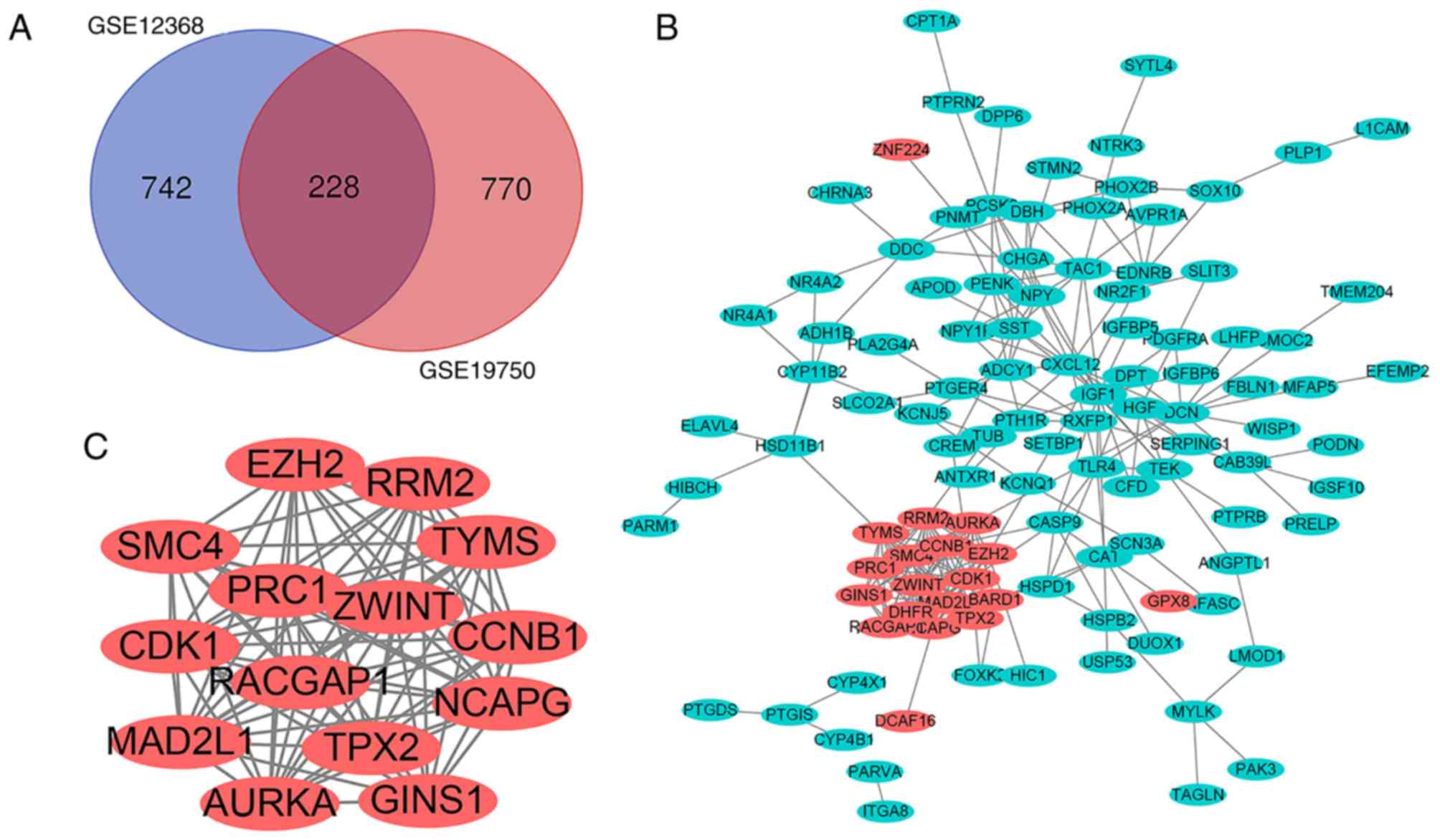
overlap between the 2 datasets contained 228 genes, as illustrated
in the Venn diagram (Fig. 1A),
consisting of 29 up and 199 downregulated genes in ACC tissue
compared with normal tissues.
KEGG and GO enrichment analyses of
DEGs
To investigate the functional annotation of the
DEGs, GO terms and pathway enrichment analysis were performed using
DAVID. The results indicated that the BPs of DEGs were
significantly enriched in ‘Cell division’, ‘Regulation of
transcription involved in G1/S transition of mitotic cell cycle’,
‘G1/S transition of mitotic cell cycle’, ‘aging’ and ‘signal
transduction’ (Table I). Changes in
MFs were primarily enriched in protein kinase binding, protein
binding and calcium ion binding (Table
I). Changes in the DEG CCs were primarily enriched in the
nucleus and extracellular space (Table
I). In addition, KEGG pathway analysis showed that the
upregulated DEGs were mainly involved in the ‘p53 signaling
pathway’, ‘Oocyte meiosis’ and ‘Progesterone-mediated oocyte
maturation’, whereas the downregulated DEGs were mainly involved in
‘Tyrosine metabolism’, ‘Focal adhesion’ and the ‘Ras signaling
pathway’ (Table I).
 | Table I.GO and KEGG pathway enrichment
analysis of DEGs in ACC samples. |
Table I.
GO and KEGG pathway enrichment
analysis of DEGs in ACC samples.
| A, Upregulated |
|---|
| ID | Description | Count | P-value |
|---|
| GO:0051301 | Cell division | 8 |
5.60×10−7 |
| GO:0000083 | Regulation of
transcription involved in G1/S transition of mitotic cell
cycle | 4 |
5.06×10−6 |
| GO:0000082 | G1/S transition of
mitotic cell cycle | 5 |
1.47×10−5 |
| GO:0005634 | Nucleus | 23 |
1.26×10−7 |
| GO:0005654 | Nucleoplasm | 17 |
4.07×10−7 |
| GO:0072686 | Mitotic
spindle | 4 |
3.33×10−5 |
| GO:0019901 | Protein kinase
binding | 5 |
2.83×10−3 |
| GO:0035173 | Histone kinase
activity | 2 |
6.38×10−3 |
| GO:0005515 | Protein
binding | 21 |
1.64×10−2 |
| Hsa04115 | p53 signaling
pathway | 3 |
3.19×10−3 |
| Hsa04914 |
Progesterone-mediated oocyte
maturation | 3 |
3.19×10−3 |
| Hsa04114 | Oocyte meiosis | 3 |
5.33×10−3 |
|
| B,
Downregulated |
|
| ID |
Description | Count | P-value |
|
| GO:0014068 | Positive regulation
of phosphatidylinositol 3-kinase signaling | 6 |
4.30×10−4 |
| GO:0006006 | Glucose metabolic
process | 6 |
4.95×10−4 |
| GO:0007165 | Signal
transduction | 22 |
4.58×10−3 |
| GO:0005615 | Extracellular
space | 33 |
1.37×10−6 |
| GO:0005578 | Proteinaceous
extracellular matrix | 13 |
2.60×10−4 |
| GO:0005576 | Extracellular
region | 30 |
6.40×10−4 |
| GO:0005509 | Calcium ion
binding | 16 |
3.02×10−3 |
| Hsa00350 | Tyrosine
metabolism | 4 |
4.57×10−3 |
| Hsa04510 | Focal adhesion | 7 |
1.44×10−2 |
| Hsa04014 | Ras signaling
pathway | 7 |
2.17×10−2 |
PPI network construction and module
analysis
A total of 113 DEGs, consisting of 19 up and 94
downregulated genes, were filtered into the PPI network using the
STRING database. The network contained 113 nodes and 272 edges, and
were visualized using Cytoscape software (Fig. 1B). The most significant module was
obtained using Cytoscape and it contained 14 nodes and 88 edges
(Fig. 1C). Functional analysis of
the genes in the most significant module was performed using DAVID
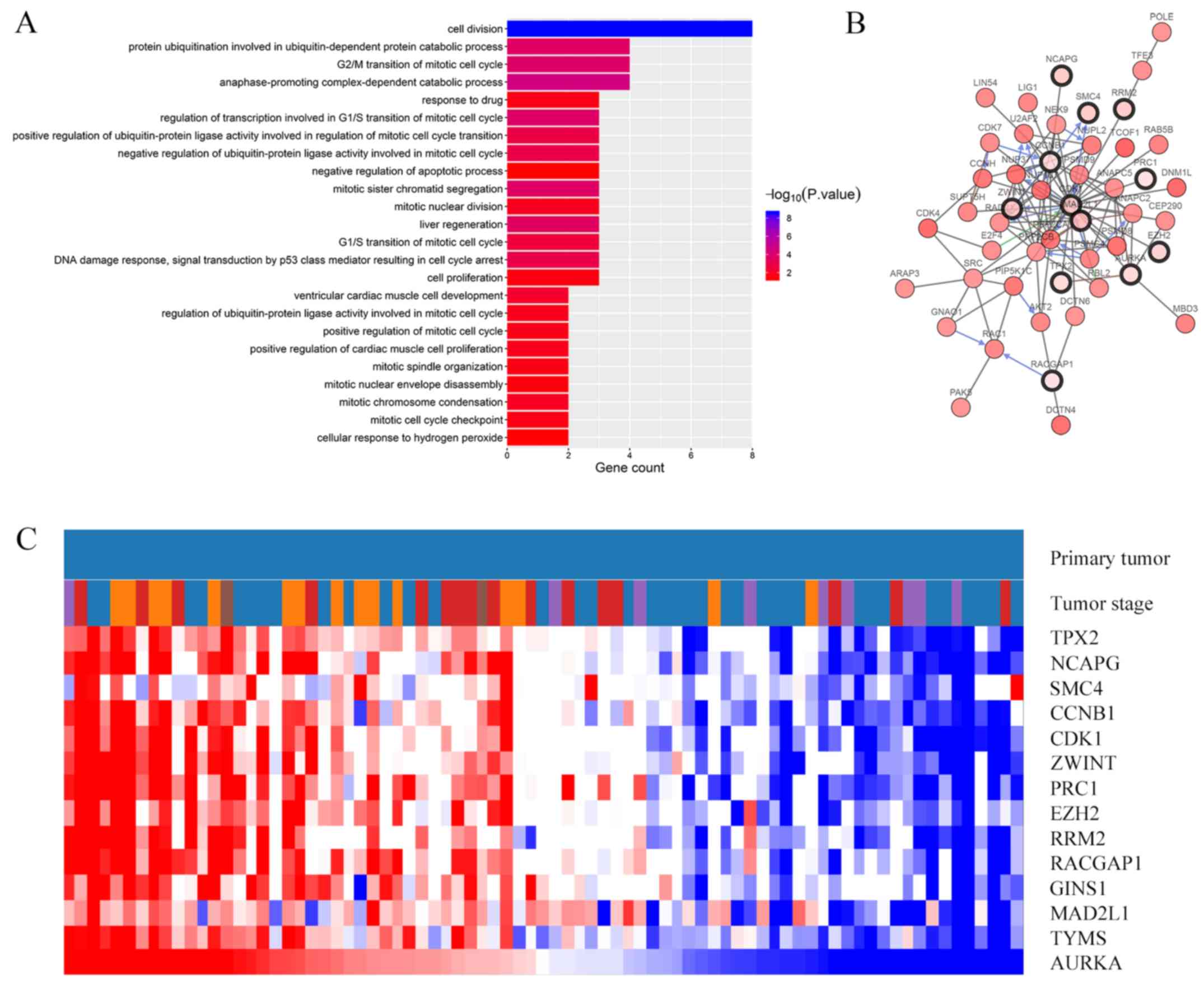
(Fig. 2A; Table II). BP analysis revealed that genes
in this module were mainly enriched in ‘Cell division’,
‘Anaphase-promoting complex-dependent catabolic process’,
‘Regulation of transcription involved in G1/S transition of mitotic
cell cycle’, ‘G2/M transition of mitotic cell cycle’ and ‘Protein
ubiquitination involved in ubiquitin-dependent protein catabolic
process’ (Fig. 2A). The CC, MF and
KEGG pathway analyses of hub genes are presented in Table II. KEGG pathway analysis revealed
that those genes were predominantly enriched in the p53 signaling
pathway, progesterone-mediated oocyte maturation, oocyte meiosis
and cell cycle (Table II).
| Table II.GO and KEGG pathway enrichment
analysis of DEGs in the most significant module. |
Table II.
GO and KEGG pathway enrichment
analysis of DEGs in the most significant module.
| ID | Description | Count in gene
set | P-value |
|---|
| GO:0005634 | Nucleus | 14 |
1.39×10−7 |
| GO:0005829 | Cytosol | 12 |
3.86×10−7 |
| GO:0005654 | Nucleoplasm | 11 |
1.25×10−6 |
| GO:0019901 | Protein kinase
binding | 5 |
1.48×10−4 |
| GO:0035173 | Histone kinase
activity | 2 |
3.08×10−3 |
| GO:0005515 | Protein
binding | 12 |
1.62×10−2 |
| Hsa04115 | P53 signaling
pathway | 3 |
1.36×10−3 |
| Hsa04914 |
Progesterone-mediated oocyte
maturation | 3 |
2.28×10−3 |
| Hsa04114 | Oocyte meiosis | 3 |
3.55×10−3 |
| Hsa04110 | Cell cycle | 3 |
4.57×10−3 |
Hub gene selection and analysis
A total of 14 genes were identified as hub genes
(degrees ≥10). A network of the hub and their co-expression genes
was analyzed using the cBioPortal online platform and is
illustrated in Fig. 2B. Hierarchical
clustering showed that the hub genes were associated with a high
clinical stage of ACC (Fig. 2C). In
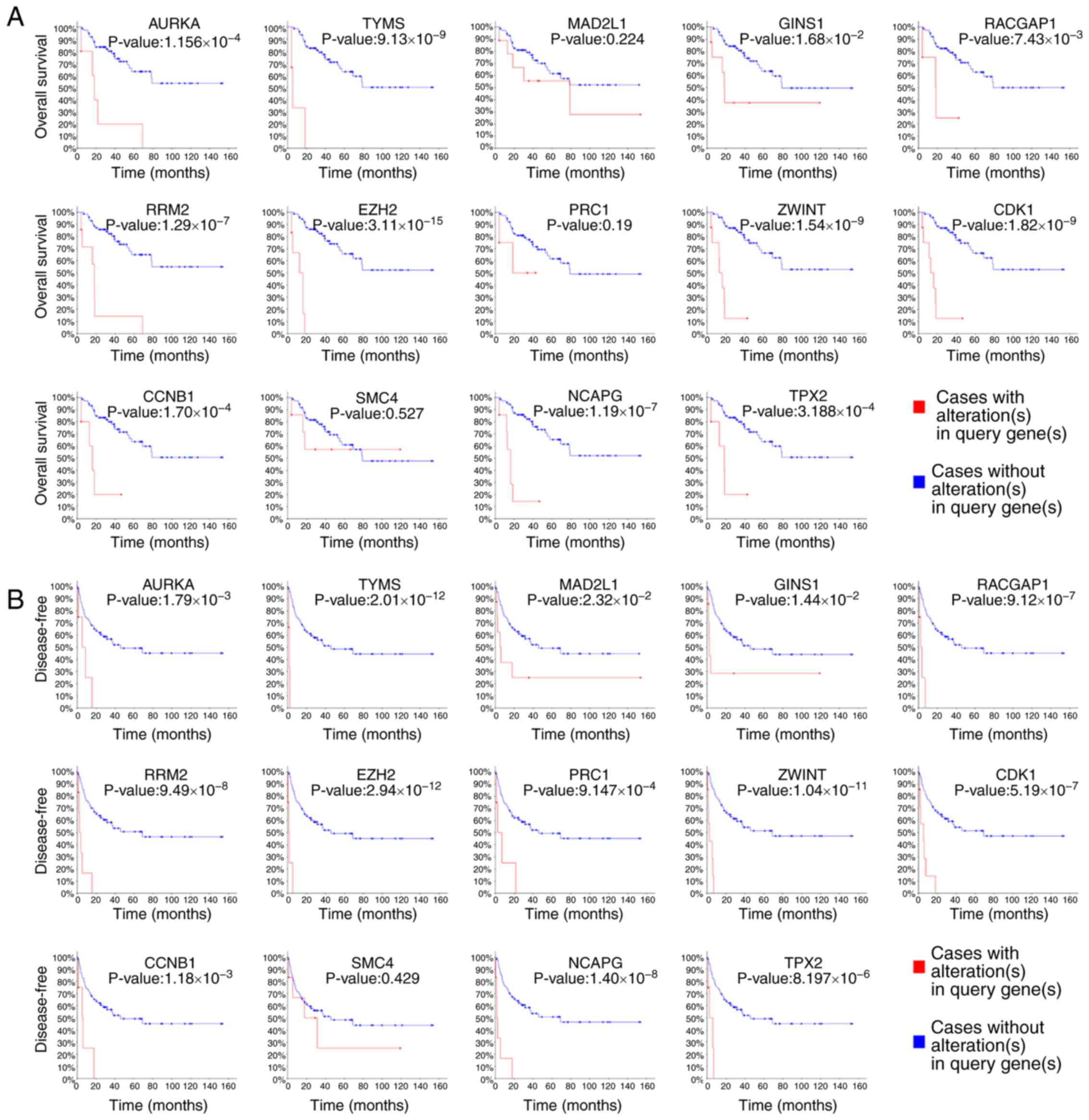
addition, the overall survival analysis of the hub genes performed
using Kaplan-Meier analysis in cBioPortal demonstrated that
patients with ACC with upregulation of AURKA, TYMS, GINS1,
RACGAP1, RRM2, EZH2, ZWINT, CDK1, CCNB1, NCAPG and TPX2
presented with a decreased overall and disease-free survival
(Fig. 3). Patients with ACC with
upregulation of Mitotic arrest deficient 2 like 1 (MAD2L1)
and PRC1 genes presented with a worse disease-free survival
(Fig. 3B). Nonetheless, no change
was observed in the patients with ACC with SMC4 alterations,
according to the cBioPortal data (P=0.527 for overall survival and
P=0.429 for disease-free survival; Fig.
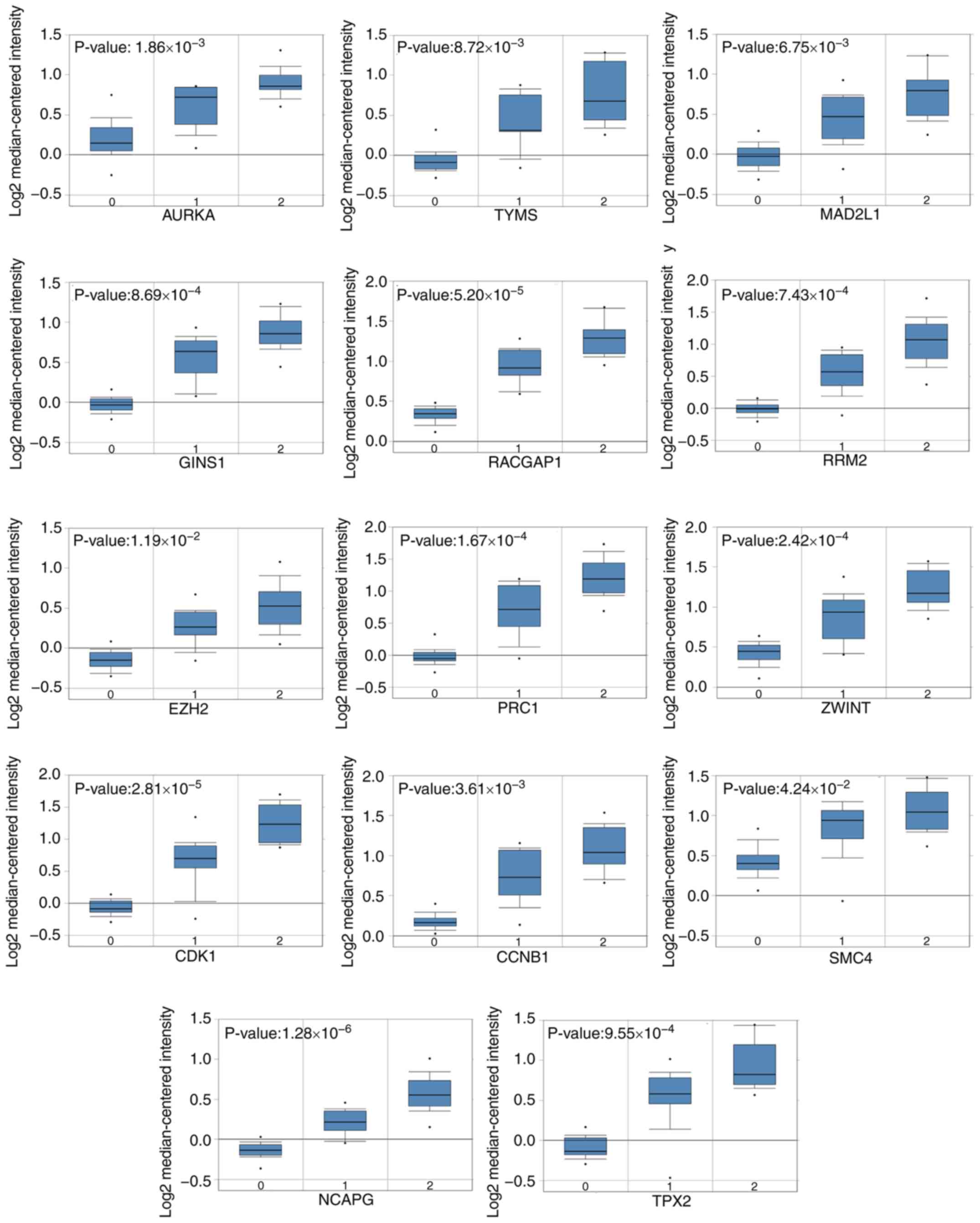
3). In addition, Oncomine analysis of cancer vs. normal tissue
showed that higher mRNA expression levels of those hub genes
increased tumor Weiss grade in the Giordano Adrenal 2 dataset
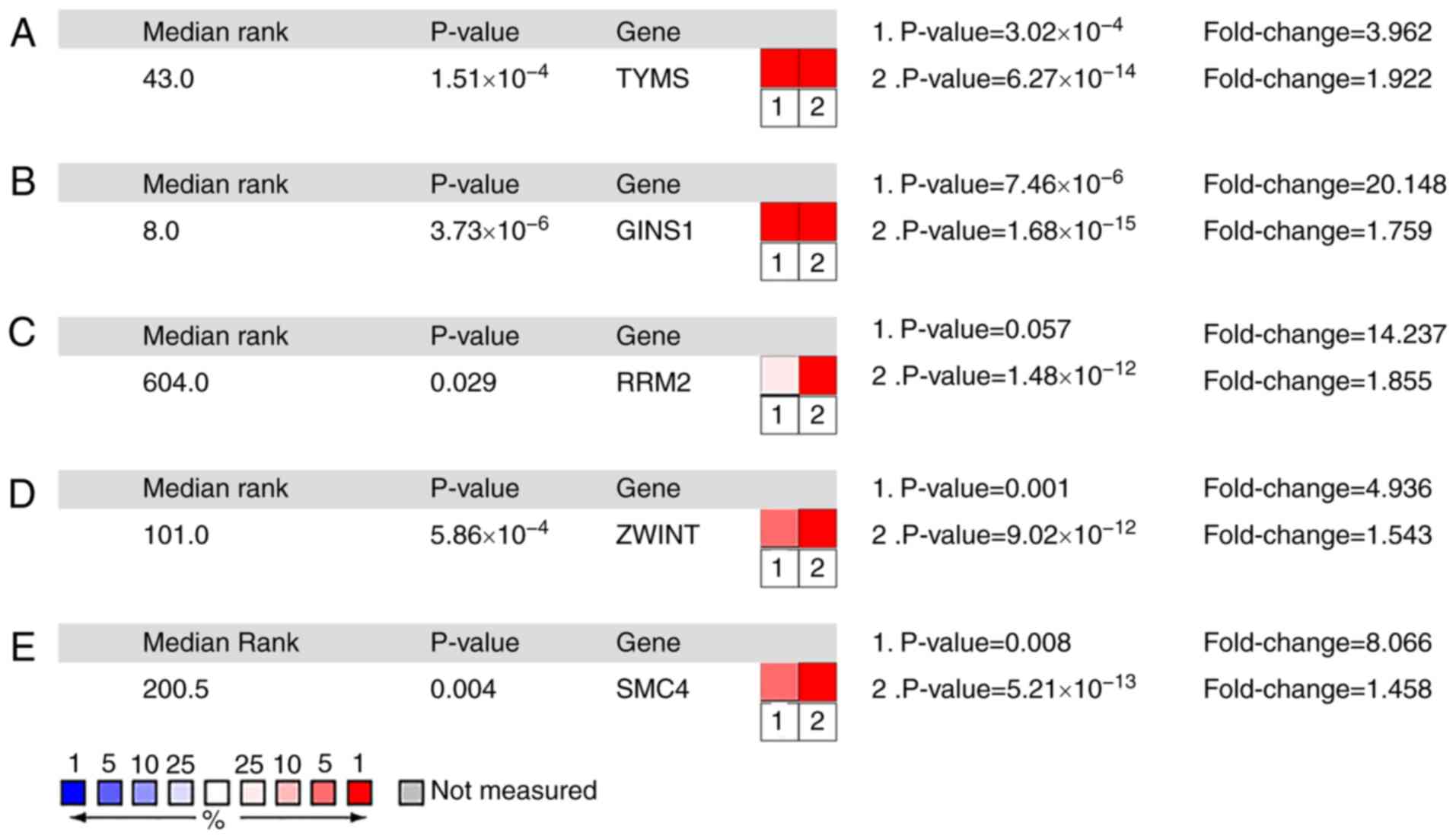
(Fig. 4). Of note, the mRNA
expression levels of TYMS, GINS1, RRM2, ZWINT and
SMC4 analyzed in the Oncomine database were observed to be
significantly increased in ACC compared with normal tissues
(Fig. 5).
Discussion
ACC is associated with a poor prognosis, limited
treatment options and high tumor recurrence rates (1–3). The
pathogenetic mechanisms of ACC includes alterations of the
Insulin-like Growth Factor system (35), Wnt/β-catenin pathway activation
(36), TP53 mutations and
prognostic molecular markers involved in cancer cell invasion
properties and angiogenesis, appear to be very promising in
elucidating of tumorigenesis and progression of ACC (37,38).
However, the molecular mechanisms of ACC remain poorly understood.
The identification of biomarkers associated with ACC tumorigenesis,
progression and prognosis are urgently required.
Microarray technology combined with bioinformatics
analysis has enabled researchers to explore genetic alterations,
and has been proven to be a useful approach in identifying novel
biomarkers in several diseases, such as hepatocellular carcinoma
and adrenocortical tumors (22,35). In
the present study, a total of 228 DEGs were identified, 14 of which
were selected as hub genes (degrees ≥10). BP analysis suggested
that these hub genes were significantly enriched in cell division
and the mitotic cell cycle, which indicated that the deregulation
of the cell cycle may serve a key role in the tumorigenesis and
development of ACC. The present study additionally combined various
databases to identify and validate the diagnostic and prognostic
value of hub genes in ACC. Kaplan-Meier analysis revealed that the
expressions of AURKA, TYMS, GINS1, RACGAP1, RRM2, EZH2, ZWINT,
CDK1, CCNB1, NCAPG and TPX2 were negatively associated
with overall and disease-free survival, suggesting these genes may
exert pivotal functions in the progression of ACC.
Some of these hub genes have previously been
identified as biomarkers for ACC (18,39–41). For
example, AURKA, which regulates cell cycle and meiotic
division, was overexpressed in pediatric adrenocortical tumors,
suggesting it may be associated with more aggressive disease and
poor prognosis, and could help develop an interesting therapeutic
approach against ACC (39,40). MAD2L1 and CCNB1 have
also been reported as potential markers for differentiating ACCs
from adenomas (18). In particular,
overexpressed CCNB1 dysregulated the cell cycle in the G2-M
phase transition, with poor survival in the majority of solid
tumors (41). Similarly, in the
present study, upregulation of MAD2L1 and CCNB1 in
tumor tissues predicted a worse overall and disease-free survival
in patients with ACC using the cBioPortal platform, which indicated
a poor prognosis. CDK1 serves an important role in
regulating cell cycle progression and mediating the phosphorylation
of Bcl-2 by binding with cyclin B to form a complex called cyclin
B-CDK1 (42). In adrenocortical
tumors, CDK1 overexpression is associated with tumor
suppressor miR-7 downregulation, which may serve as a target for
inhibiting the progression of ACC (43,44).
EZH2 was significantly associated with poorer outcomes in
ACC (45). A recent study by Drelon
et al (46) reported that
EZH2, as a deregulated histone modifier, deregulated
activity of the P53/RB/E2F pathway and WNT signaling modulation to
promote cell proliferation, which may be a new therapeutic target
for ACC. In addition, Yuan et al (15) underlined the potential of TPX2,
PRC1 and RACGAP1 as markers for the diagnosis and
prognosis of ACC, which was consistent with the hypothesis of the
present study.
Although six other hub genes (TYMS, RRM2, ZWINT,
GINS1, SMC4 and NCAPG) have not been extensively
reported to participate in ACC progression, they were observed to
be involved in various tumors. Using Oncomine evaluation, the
present study identified that the mRNA expression levels of
TYMS, GINS1, RRM2, ZWINT and SMC4 were higher in ACC
compared with normal tissues. In addition, Oncomine analysis of ACC
vs. normal tissues revealed that the upregulation of hub genes was
significantly associated with a higher Weiss grade.
Among these six hub genes, the elevated expression
of TYMS has also been reported in lung (47), gastric (48), colorectal (49,50),
renal cell (51) and prostate cancer
(52), suggesting it may serve as a
valuable biomarker for the diagnosis, treatment and prognosis of
tumors. In the present study, the PPI network demonstrated that
TYMS directly interacted with other hub genes, such as
CDK1, AURKA and PRC1, and that it may affect cell
proliferation through modulation of the cell cycle and multiple
signaling pathways. In addition, overexpression of TYMS was
significantly associated with a shorter survival time and higher
tumor Weiss grade, indicating a key role in the tumorigenesis or
progression of ACC. RRM2 serves a key regulatory role in DNA
synthesis and cell proliferation (53). RRM2 has been hypothesized to
promote angiogenesis by producing reactive oxygen species to
activate the ERK1/2 signaling pathway, and inducing HIF-1α and VEGF
expression in human cervical cancer, which is associated with a
poor outcome in certain types of cancer (54). ZWINT is essential for mitotic
checkpoint signaling (55,56). It was recently demonstrated that
ZWINT was significantly correlated with the expression of
cell-cycle proteins such as PCNA, cyclin B1, Cdc25C and CDK1, and
may be considered as potential therapeutic targets for
hepatocellular carcinoma (55).
GINS1, also known as PSF1 (57), was significantly associated with a
worse overall survival, but not disease-free survival in the
present study. However, PSF1 was highly expressed in several
types of cancer (58–61). In addition, previous studies have
demonstrated that the transcriptional activity of the PSF1
gene was associated with cancer cell malignancy by affecting the
cell cycle and proliferation, highlighting it as a potentially
useful biomarker for the identifying patients who may have
unfavorable prognoses (59,61). SMC4 has been reported to be
involved in tumor cell growth, migration and invasion (62,63).
However, its role in ACC has yet to be completely elucidated. In
the present study, although SMC4 alteration was not
significantly associated with a worse overall and disease-free
survival, hierarchical clustering for hub genes and data from
Oncomine indicates that it serves a crucial role in ACC
tumorigenesis. NCAPG organizes the coiling topology of
individual chromatids during cell mitosis and meiosis, which is
involved in the progression of liver carcinoma (64). Therefore, it was speculated that they
may serve a critical role in the carcinogenesis and progression of
ACC.
In conclusion, the present study identified and
analyzed key biomarkers in ACC using bioinformatics analysis. Two
databases were combined to screen 228 DEGs, and 14 hub genes, which
may be regarded as powerful and promising biomarkers for predicting
tumorigenesis and progression of ACC, were identified. These hub
genes were associated with tumor cell proliferation and cell cycle
regulation. Of note, candidate hub gene upregulation was associated
with a worse survival rate and higher Weiss grade; and this may
provide a basis for further clinical molecular target therapy
experiments and diagnostic approaches for ACC, if these potential
genes are developed as novel useful diagnostic as well as
prognostic markers and the underlying pathological causative
pathways or involved signaling targets are elucidated. However,
further studies are required to confirm the biological functions
and mechanisms of action of these genes in ACC.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Science Foundation of China (grant nos. 81660138 and
81860146).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
ZX and ZL conceived and designed the study, analyzed
the data and drafted the manuscript. HY and ZH collected the data
and performed the statistical analysis. XL was responsible for
drawing the figures, and help designed the bioinformatics study.
All authors read and commented on the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kebebew E, Reiff E, Duh QY, Clark OH and
McMillan A: Extent of disease at presentation and outcome for
adrenocortical carcinoma: Have we made progress? World J Surg.
30:872–878. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kerkhofs TM, Verhoeven RH, Van der Zwan
JM, Dieleman J, Kerstens MN, Links TP, Van de Poll-Franse LV and
Haak HR: Adrenocortical carcinoma: A population-based study on
incidence and survival in the Netherlands since 1993. Eur J Cancer.
49:2579–2586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sharma E and Dahal S, Sharma P, Bhandari
A, Gupta V, Amgai B and Dahal S: The characteristics and trends in
adrenocortical carcinoma: A united states population based study. J
Clin Med Res. 10:636–640. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bourdeau I, MacKenzie-Feder J and Lacroix
A: Recent advances in adrenocortical carcinoma in adults. Curr Opin
Endocrinol Diabetes Obes. 20:192–197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wandoloski M, Bussey KJ and Demeure MJ:
Adrenocortical cancer. Surg Clin North Am. 89:1255–1267. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Else T, Kim AC, Sabolch A, Raymond VM,
Kandathil A, Caoili EM, Jolly S, Miller BS, Giordano TJ and Hammer
GD: Adrenocortical carcinoma. Endocr Rev. 35:282–326. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grubbs EG, Callender GG, Xing Y, Perrier
ND, Evans DB, Phan AT and Lee JE: Recurrence of adrenal cortical
carcinoma following resection: Surgery alone can achieve results
equal to surgery plus mitotane. Ann Surg Oncol. 17:263–270. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Postlewait LM, Ethun CG, Tran TB, Prescott
JD, Pawlik TM, Wang TS, Glenn J, Hatzaras I, Shenoy R, Phay JE, et
al: Outcomes of adjuvant mitotane after resection of adrenocortical
carcinoma: A 13-institution study by the us adrenocortical
carcinoma group. J Am Coll Surg. 222:480–490. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stephan EA, Chung TH, Grant CS, Kim S, Von
Hoff DD, Trent JM and Demeure MJ: Adrenocortical carcinoma survival
rates correlated to genomic copy number variants. Mol Cancer Ther.
7:425–431. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lam AK: Update on adrenal tumours in 2017
world health organization (WHO) of endocrine tumours. Endocr
Pathol. 28:213–227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Erickson LA: Challenges in surgical
pathology of adrenocortical tumours. Histopathology. 72:82–96.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Papotti M, Libè R, Duregon E, Volante M,
Bertherat J and Tissier F: The weiss score and
beyond-histopathology for adrenocortical carcinoma. Horm Cancer.
2:333–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giordano TJ, Kuick R, Else T, Gauger PG,
Vinco M, Bauersfeld J, Sanders D, Thomas DG, Doherty G and Hammer
G: Molecular classification and prognostication of adrenocortical
tumors by transcriptome profiling. Clin Cancer Res. 15:668–676.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kulshrestha A and Suman S: Common module
analysis reveals prospective targets and mechanisms of pediatric
adrenocortical adenoma and carcinoma. Oncol Lett. 15:3267–3272.
2018.PubMed/NCBI
|
|
15
|
Yuan L, Qian G, Chen L, Wu CL, Dan HC,
Xiao Y and Wang X: Co-expression network analysis of biomarkers for
adrenocortical carcinoma. Front Genet. 9:3282018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kulshrestha A, Suman S and Ranjan R:
Network analysis reveals potential markers for pediatric
adrenocortical carcinoma. OncoTargets Ther. 9:4569–4581. 2016.
View Article : Google Scholar
|
|
17
|
Duregon E, Rapa I, Votta A, Giorcelli J,
Daffara F, Terzolo M, Scagliotti GV, Volante M and Papotti M:
MicroRNA expression patterns in adrenocortical carcinoma variants
and clinical pathologic correlations. Hum Pathol. 45:1555–1562.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Soon PS, Gill AJ, Benn DE, Clarkson A,
Robinson BG, McDonald KL and Sidhu SB: Microarray gene expression
and immunohistochemistry analyses of adrenocortical tumors identify
IGF2 and Ki-67 as useful in differentiating carcinomas from
adenomas. Endocr Relat Cancer. 16:573–583. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Demeure MJ, Coan KE, Grant CS, Komorowski
RA, Stephan E, Sinari S, Mount D and Bussey KJ: PTTG1
overexpression in adrenocortical cancer is associated with poor
survival and represents a potential therapeutic target. Surgery.
154:1405–1416. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clough E and Barrett T: The gene
expression omnibus database. Methods Mol Biol. 1418:93–110. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res 41 (Database Issue).
D991–D995. 2013.
|
|
22
|
Li L, Lei Q, Zhang S, Kong L and Qin B:
Screening and identification of key biomarkers in hepatocellular
carcinoma: Evidence from bioinformatic analysis. Oncol Rep.
38:2607–2618. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kanehisa M: The KEGG database. Novartis
Found Symp. 247:91–103, 119-128, 244–252. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alshabi AM, Vastrad B, Shaikh IA and
Vastrad C: Identification of important invasion and proliferation
related genes in adrenocortical carcinoma. Med Oncol. 36:732019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xia WX, Yu Q, Li GH, Liu YW, Xiao FH, Yang
LQ, Rahman ZU, Wang HT and Kong QP: Identification of four hub
genes associated with adrenocortical carcinoma progression by
WGCNA. PEERJ. 7:e65552019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bandettini WP, Kellman P, Mancini C,
Booker OJ, Vasu S, Leung SW, Wilson JR, Shanbhag SM, Chen MY and
Arai AE: Multicontrast delayed enhancement (MCODE) improves
detection of subendocardial myocardial infarction by late
gadolinium enhancement cardiovascular magnetic resonance: A
clinical validation study. J Cardiovasc Magn Reson. 14:832012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang AM, Song H, Shen YH and Liu Y:
Construction of a gene-gene interaction network with a combined
score across multiple approaches. Genet Mol Res. 14:7018–7030.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Goldman M, Craft B, Swatloski T, Cline M,
Morozova O, Diekhans M, Haussler D and Zhu J: The UCSC cancer
genomics browser: Update 2015. Nucleic Acids Res. 43:D812–D817.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Giordano TJ, Thomas DG, Kuick R, Lizyness
M, Misek DE, Smith AL, Sanders D, Aljundi RT, Gauger PG, Thompson
NW, et al: Distinct transcriptional profiles of adrenocortical
tumors uncovered by DNA microarray analysis. Am J Pathol.
162:521–531. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Altieri B, Colao A and Faggiano A: The
role of insulin-like growth factor system in the adrenocortical
tumors. Minerva Endocrinol. 44:43–57. 2019.PubMed/NCBI
|
|
36
|
Gaujoux S, Grabar S, Fassnacht M, Ragazzon
B, Launay P, Libé R, Chokri I, Audebourg A, Royer B, Sbiera S, et
al: β-catenin activation is associated with specific clinical and
pathologic characteristics and a poor outcome in adrenocortical
carcinoma. Clin Cancer Res. 17:328–336. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dworakowska D, Drabarek A, Wenzel I,
Babińska A, Świątkowska-Stodulska R and Sworczak K: Adrenocortical
cancer (ACC)-literature overview and own experience. Endokrynol
Pol. 65:492–502. 2014.PubMed/NCBI
|
|
38
|
Varghese J and Habra MA: Update on
adrenocortical carcinoma management and future directions. Curr
Opin Endocrinol Diabetes Obes. 24:208–214. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Borges KS, Moreno DA, Martinelli CE Jr,
Antonini SR, de Castro M, Tucci S Jr, Neder L, Ramalho LN,
Seidinger AL, Cardinalli I, et al: Spindle assembly checkpoint gene
expression in childhood adrenocortical tumors (ACT): Overexpression
of aurora kinases A and B is associated with a poor prognosis.
Pediatr Blood Cancer. 60:1809–1816. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Damodaran AP, Vaufrey L, Gavard O and
Prigent C: Aurora a kinase is a priority pharmaceutical target for
the treatment of cancers. Trends Pharmacol Sci. 38:687–700. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ye C, Wang J, Wu P, Li X and Chai Y:
Prognostic role of cyclin B1 in solid tumors: A meta-analysis.
Oncotarget. 8:2224–2232. 2017.PubMed/NCBI
|
|
42
|
Terrano DT, Upreti M and Chambers TC:
Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation
acts as a functional link coupling mitotic arrest and apoptosis.
Mol Cell Biol. 30:640–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Glover AR, Zhao JT, Gill AJ, Weiss J,
Mugridge N, Kim E, Feeney AL, Ip JC, Reid G, Clarke S, et al:
MicroRNA-7 as a tumor suppressor and novel therapeutic for
adrenocortical carcinoma. Oncotarget. 6:36675–36688. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nilubol N, Boufraqech M, Zhang L, Gaskins
K, Shen M, Zhang YQ, Gara SK, Austin CP and Kebebew E: Synergistic
combination of flavopiridol and carfilzomib targets commonly
dysregulated pathways in adrenocortical carcinoma and has
biomarkers of response. Oncotarget. 9:33030–33042. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ip JC, Pang TC, Glover AR, Soon P, Zhao
JT, Clarke S, Robinson BG, Gill AJ and Sidhu SB:
Immunohistochemical validation of overexpressed genes identified by
global expression microarrays in adrenocortical carcinoma reveals
potential predictive and prognostic biomarkers. Oncologist.
20:247–256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Drelon C, Berthon A, Mathieu M, Ragazzon
B, Kuick R, Tabbal H, Septier A, Rodriguez S, Batisse-Lignier M,
Sahut-Barnola I, et al: EZH2 is overexpressed in adrenocortical
carcinoma and is associated with disease progression. Hum Mol
Genet. 25:2789–2800. 2016.PubMed/NCBI
|
|
47
|
Kotoula V, Krikelis D, Karavasilis V,
Koletsa T, Eleftheraki AG, Televantou D, Christodoulou C, Dimoudis
S, Korantzis I, Pectasides D, et al: Expression of DNA repair and
replication genes in non-small cell lung cancer (NSCLC): A role for
thymidylate synthetase (TYMS). BMC Cancer. 12:3422012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Formentini A, Henne-Bruns D and Kornmann
M: Thymidylate synthase expression and prognosis of patients with
gastrointestinal cancers receiving adjuvant chemotherapy: A review.
Langenbecks Arch Surg. 389:405–413. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Popat S, Matakidou A and Houlston RS:
Thymidylate synthase expression and prognosis in colorectal cancer:
A systematic review and meta-analysis. J Clin Oncol. 22:529–536.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang YC, Wu GC, Jin L, Wang KL, Bai ZG,
Wang J and Zhang ZT: Association of thymidylate synthase
polymorphisms with the tumor response to preoperative
chemoradiotherapy in rectal cancer: A systematic review and
meta-analysis. Pharmacogenomics J. 17:265–273. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mizutani Y, Wada H, Yoshida O, Fukushima
M, Nonomura M, Nakao M and Miki T: Significance of thymidylate
synthase activity in renal cell carcinoma. Clin Cancer Res.
9:1453–1460. 2003.PubMed/NCBI
|
|
52
|
Burdelski C, Strauss C, Tsourlakis MC,
Kluth M, Hube-Magg C, Melling N, Lebok P, Minner S, Koop C, Graefen
M, et al: Overexpression of thymidylate synthase (TYMS) is
associated with aggressive tumor features and early PSA recurrence
in prostate cancer. Oncotarget. 6:8377–8387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Grolmusz VK, Karászi K, Micsik T, Tóth EA,
Mészáros K, Karvaly G, Barna G, Szabó PM, Baghy K, Matkó J, et al:
Cell cycle dependent RRM2 may serve as proliferation marker and
pharmaceutical target in adrenocortical cancer. Am J Cancer Res.
6:2041–2053. 2016.PubMed/NCBI
|
|
54
|
Wang N, Zhan T, Ke T, Huang X, Ke D, Wang
Q and Li H: Increased expression of RRM2 by human papillomavirus E7
oncoprotein promotes angiogenesis in cervical cancer. Br J Cancer.
110:1034–1044. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ying H, Xu Z, Chen M, Zhou S, Liang X and
Cai X: Overexpression of Zwint predicts poor prognosis and promotes
the proliferation of hepatocellular carcinoma by regulating
cell-cycle-related proteins. Onco Targets Ther. 11:689–702. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Woo Seo D, Yeop You S, Chung WJ, Cho DH,
Kim JS and Su Oh J: Zwint-1 is required for spindle assembly
checkpoint function and kinetochore-microtubule attachment during
oocyte meiosis. Sci Rep. 5:154312015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cottineau J, Kottemann MC, Lach FP, Kang
YH, Vély F, Deenick EK, Lazarov T, Gineau L, Wang Y, Farina A, et
al: Inherited GINS1 deficiency underlies growth retardation along
with neutropenia and NK cell deficiency. J Clin Invest.
127:1991–2006. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nakahara I, Miyamoto M, Shibata T,
Akashi-Tanaka S, Kinoshita T, Mogushi K, Oda K, Ueno M, Takakura N,
Mizushima H, et al: Up-regulation of PSF1 promotes the growth of
breast cancer cells. Genes Cells. 15:1015–1024. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tahara H, Naito H, Kise K, Wakabayashi T,
Kamoi K, Okihara K, Yanagisawa A, Nakai Y, Nonomura N, Morii E, et
al: Evaluation of PSF1 as a prognostic biomarker for prostate
cancer. Prostate Cancer Prostatic Dis. 18:56–62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang J, Wu Q, Wang Z, Zhang Y, Zhang G,
Fu J and Liu C: Knockdown of PSF1 expression inhibits cell
proliferation in lung cancer cells in vitro. Tumour Biol.
36:2163–2168. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhou L, Sun XJ, Liu C, Wu QF, Tai MH, Wei
JC, Lei L, Meng FD, Qu K and Xu J: Overexpression of PSF1 is
correlated with poor prognosis in hepatocellular carcinoma
patients. Int J Biol Markers. 30:e56–e64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhou B, Yuan T, Liu M, Liu H, Xie J, Shen
Y and Chen P: Overexpression of the structural maintenance of
chromosome 4 protein is associated with tumor de-differentiation,
advanced stage and vascular invasion of primary liver cancer. Oncol
Rep. 28:1263–1268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Feng XD, Song Q, Li CW, Chen J, Tang HM,
Peng ZH and Wang XC: Structural maintenance of chromosomes 4 is a
predictor of survival and a novel therapeutic target in colorectal
cancer. Asian Pac J Cancer Prev. 15:9459–9465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu W, Liang B, Liu H, Huang Y, Yin X,
Zhou F, Yu X, Feng Q, Li E, Zou Z and Wu L: Overexpression of
non-SMC condensin I complex subunit G serves as a promising
prognostic marker and therapeutic target for hepatocellular
carcinoma. Int J Mol Med. 40:731–738. 2017. View Article : Google Scholar : PubMed/NCBI
|