Introduction
Myelodysplastic syndrome (MDS) is a group of
acquired clonal disorders that originate in the hematopoietic
stem/progenitor cells, and are characterized by ineffective
erythropoiesis of the bone marrow, long-term progressive refractory
cytopenia, and high risk of conversion to acute leukemia (1–3).
With the continuous progress in the field of life
sciences, researchers have begun to examine the pathogenesis of MDS
at the gene level and have reported that more and more gene
abnormalities are associated with MDS pathogenesis (4–8). Studies
on MDS genetic alterations have revealed that 70–80% of the
patients with MDS have at least one mutation (4–6,9). With the development of sequencing
technology and its broad applications, gene mutations and other
molecular biological markers have been included in the guidelines
for the diagnosis and treatment of MDS (9,10). In
November 2016, the National Comprehensive Cancer Network released
the Clinical Practice Guidelines in Oncology: Myelodysplastic
Syndromes (version 2.2017), which proposed that frequent mutations
in MDS-associated genes may be suggestive of the presence of clonal
hematopoiesis (11). A previous
study reported that there are ~60 MDS-affected genes, which are
subdivided into RNA splicing, DNA methylation, chromatin
remodeling, transcription, receptors/kinases, cohesion, RAS pathway
and DNA repair (9). The completion
of whole-genome sequencing and targeted gene sequencing in patients
with MDS has preliminarily revealed the molecular mechanism
underlying the pathogenesis of MDS (12,13). In
the present study, the mutant genes of patients with a novel MDS
diagnosis were determined by the next-generation sequencing
technology to analyze the association between the mutant genes and
clinicopathological features of the patients.
Materials and methods
Diagnosis and classification
criteria
The diagnostic criteria, classification criteria,
and international prognostic scoring system for MDS were based on
the 2007 Vienna standards for the diagnosis of MDS (14), the 2008 World Health Organization
(WHO) classification criteria for MDS (15), and the Revised International
Prognostic Scoring System (IPSS-R) (16), respectively.
Sample collection
The subjects were 47 patients with a novel MDS
diagnosis in the Department of Hematology of Xiyuan Hospital of
China Academy of Chinese Medical Sciences (Beijing, China) between
July 15th, 2017 and December 31st, 2017. The sample included 26
females (55.3%) and 21 males (44.7%) with median age of 56 years
(range, 19–82 years). The median peripheral white blood cell count,
hemoglobin level, platelet count, neutrophil count and bone marrow
blast percentage were 2.52 (1.9–8.2×109/l), 77 (35–168
g/l), 44 (3–540×109/l), 1.12 (0–8.48×109/l)
and 2% (0–17.2%), respectively. According to the 2008 WHO
classification criteria, 1 case of refractory anemia (RA), 27 cases
of refractory cytopenia with multilineage dysplasia (RCMD), 12
cases of type 1 RA with excess blasts (RAEB-1), 5 cases of type 2
RAEB (RAEB-2), and 2 cases of MDS-unclassified (MDS-U) were
included. According to the cytogenetic risk classification, 36
cases of good-prognosis karyotype, 10 cases of
intermediate-prognosis karyotype, and 1 case of poor-prognosis
karyotype were detected. According to the IPSS-R, 8 low-risk, 24
intermediate-risk, 10 high-risk, and 5 very high-risk cases were
noted (Table I). The study protocol
was approved by the Clinical Research Ethics Committee of Xiyuan
Hospital, China Academy of Chinese Medical Sciences (Beijing,
China). All patients provided written informed consent to
participate in the study.
 | Table I.Characteristics of patients. |
Table I.
Characteristics of patients.
| Parameters | n (%) |
|---|
| Age, years | 56 (19–82) |
| Male | 21 (44.7) |
| Female | 26 (55.3) |
| WBC,
×109/l | 2.52 (1.9–8.2) |
| PLT,
×109/l | 44 (3–540) |
| Hb, g/l | 77 (35–168) |
| ANC,
×109/l | 1.12 (0–8.48) |
| Bone marrow blasts,
% | 2 (0–17.2) |
| WHO classification
(10) |
|
| RA | 1 (2.13) |
|
RCMD | 27 (55.45) |
|
RAEB-1 | 12 (25.53) |
|
RAEB-2 | 5 (10.64) |
|
MDS-U | 2 (4.25) |
| Cytogenetic
risk |
|
|
Good | 36 (76.59) |
|
Intermediate | 10 (21.28) |
| Very
Poor | 1 (2.13) |
| IPSS-R risk group
(11) |
|
|
Low | 8 (17.02) |
|
Intermediate | 24 (51.06) |
|
High | 10 (21.28) |
| Very
high | 5 (10.64) |
Next-generation sequencing
The genomic DNA (gDNA) was extracted following bone
marrow or peripheral blood sample collection from patients. The
concentration of gDNA was >10 ng/l, with optical density
(OD)260/OD280=1.7–1.9, and the total amount
was >1,000 ng. If quality inspection yielded good results, the
gDNA was subsequently used for the construction of an Illumina
standard library (Illumina, Inc.), and the Roche NimbleGen liquid
phase hybrid capture chip was employed to perform 127-target gene
sequencing (Table SI). The captured
exon library was sequenced on the Illumina NextSeq 550AR platform
(Illumina, Inc.), and each sample was required to have an average
effective depth ≥1,000× in the target area. Using the
Burrows-Wheeler Alignment algorithm version 0.7.12 (17) to compare the sequence data with the
human genome (version: GRCh37), Picard version 1.115 (https://github.com/broadinstitute/picard) was used to
mark the polymerase chain reaction duplicates, and the quality
value of the sequence alignment results was corrected by means of
BaseRecalibrator in Genome Analysis Toolkit version 3.5 (18). The MuTect2 version 3.5 software
(18) was employed for mutation
detection, and all test results were annotated in the Annovar
version 0722 software (19). The
types of analysis included single-nucleotide variants and
insertions and deletions (indels). Single-nucleotide polymorphisms
described in dbSNP version 135 database (https://www.ncbi.nlm.nih.gov/snp/) were excluded.
Variant allele frequency (VAF) was calculated as the number of the
variant reads divided by the total number of reads for the mutation
position. Circos plot was performed in Circos version 0.69–6
software (http://www.circos.ca/), corresponding to
the relative frequency and pairwise co-occurrence of gene
mutations, and the threshold was a patient with paired
mutations.
Karyotype analysis
Bone marrow cells were cultured for short-term (24
h), G-banding of chromosomes was performed, and karyotypes were
determined according to the International Nomenclature System for
Human Cytogenetics (11). For the
G-banding of chromosomes 0.2–0.4 ml bone marrow was absorbed and
injected into 10 ml preheated (37°C) culture medium, which
consisted of RPMI-1640 medium (Thermo Fisher Scientific, Inc.),
fetal bovine serum (Hangzhou Sijiqing Biological Engineering
Materials Co., Ltd.) and penicillin-streptomycin double antibiotics
(Beijing Solarbio Science & Technology Co., Ltd.) with the
ratio 100:20:1. This was followed by treatment with 0.1 ml from 5
ug/ml colchicine (Hubei DiBo Chemical Co., Ltd.) and incubation at
37°C for 4–6 h. The cells were harvested after 24–48 h. The cell
suspension was centrifuged at 402 × g at 25°C for 10 min. 10 ml of
acetic acid and methanol mixture (ratio 3:1) was added to the
suspension for 30 min, followed by centrifugation of the cell
suspension at 402 × g at 25°C for 10 min. The process was repeated
three times. The obtained cell suspension was used to prepare
chromosome slices. Saline (45 ml) and 2% trypsin solution were
added to the container and heated in a water bath at 37°C. The
chromosome slices were immersed in the aforementioned trypsin
working solution at 37°C and shaken for 30 sec. Staining was
performed with Giemsa solution (Giemsa stock solution and
phosphoric acid buffer solution with a ratio of 1:10) at room
temperature for 8–10 min and rinsed with tap water, followed by the
removal of the specimen.
Statistical analysis
Data analysis was performed in the Python 3.5.2
statistical software (https://www.python.org/) using the χ2 test
or Fisher's exact test. The raw values (Fig. 1), the median (minimum to maximum), if
appropriate (Table I), or the
maximum, upper quartile, median, lower quartile, and minimum values
(Fig. 4) are presented. The odds
ratio was calculated as the ratio of mutation frequency between two
different groups. P<0.05 was considered to indicate a
statistically significant difference.
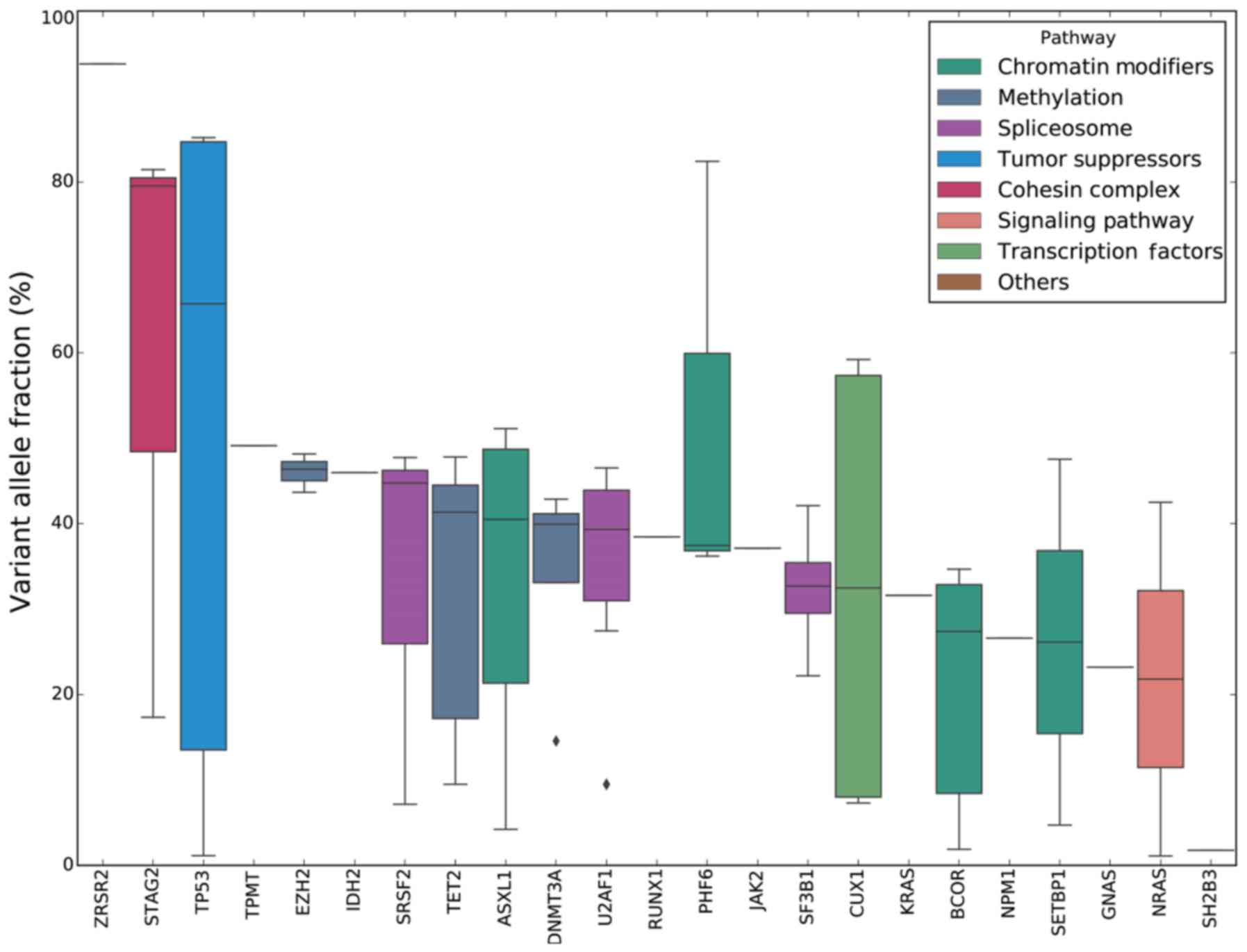 | Figure 4.VAF analysis of different mutated
genes. The boxplot indicates the median, 25th, and 75th percentiles
of VAF observed across the entire cohort of 47 patients. For
ZRSR2, TPMT, IDH2, RUNX1, JAK2, KRAS, NPM1, GNAS and
SH2B3, each of these genes has been detected in only one
patient. All bars are colored according to the different functional
groups assigned to each mutated gene. VAF, variant allele
fraction. |
Results
Analysis of mutant genes in patients
with MDS
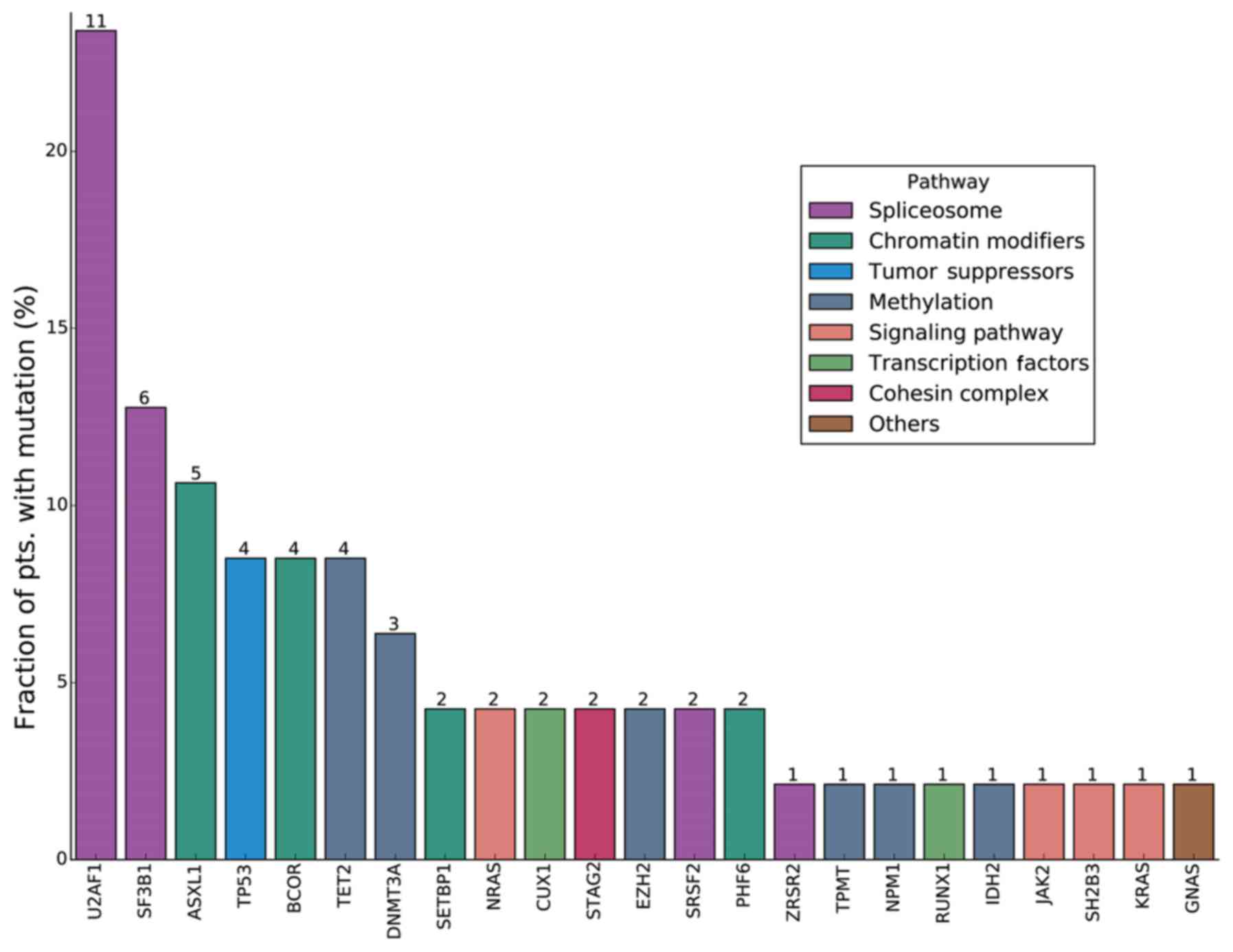
Among the 47 patients with a novel MDS diagnosis, 31
patients had a gene mutation(s), and the overall rate of mutation
prevalence was 66.0% (31/47). A total of 23 mutant genes of
clinical significance were detected. According to the descending
order of the detection frequency among the 47 patients with MDS,
there were 11 cases with a U2 small nuclear RNA auxiliary factor 1
(U2AF1) mutation (23.4%); 6 cases with a splicing factor 3b
subunit 1 (SF3B1) mutation (12.8%); 5 cases with an ASXL
transcriptional regulator 1 (ASXL1) mutation (10.6%); 4
cases each with a mutation in tet methylcytosine dioxygenase 2
(TET2), BCL6 corepressor (BCOR) or TP53
(8.5%); 3 cases with a DNA methyltransferase 3α (DNMT3A)
mutation (6.8%); 2 cases each with a mutation in serine and
arginine rich splicing factor 2 (SRSF2), enhancer of zeste 2
polycomb repressive complex 2 subunit (EZH2), PHD finger
protein 6 (PHF6), SET binding protein 1 (SETBP1),
stromal antigen 2 (STAG2), cut like homeobox 1 or NRAS
proto-oncogene, GTPase (NRAS) (4.3%); and 1 case each with a
mutation in zinc finger CCCH-type, RNA binding motif and
serine/arginine rich 2 (ZRSR2), thiopurine
S-methyltransferase, isocitrate dehydrogenase [NADP(+)] 2,
mitochondrial (IDH2), nucleophosmin 1 (NPM1), RUNX
family transcription factor 1 (RUNX1), KRAS, SH2B
adaptor protein 3 (SH2B3), Janus kinase 2 (JAK2) or
GNAS complex locus (2.1%). Each of seven genes (U2AF1, SF3B1,
ASXL1, TET2, BCOR, TP53 and DNMT3A) had mutation
prevalence of >5% in the present study cohort (Fig. 1).
Analysis of mutant genes in patients
with MDS according to genetic functional groups
According to the classification based on the genetic
functional groups and the descending order of population mutation
frequency, 19, 11, 11, 5, 4, 3, and 2 cases were respectively
associated with the following categories among the 47 patients with
a novel MDS diagnosis: RNA splicing (40.4%), chromatin remodeling
(23.4%), DNA methylation (23.4%), a signaling pathway (10.6%), a
tumor suppressor (8.5%), a transcription factor (6.4%), and the
cohesin complex (4.26%) (Fig.
2).
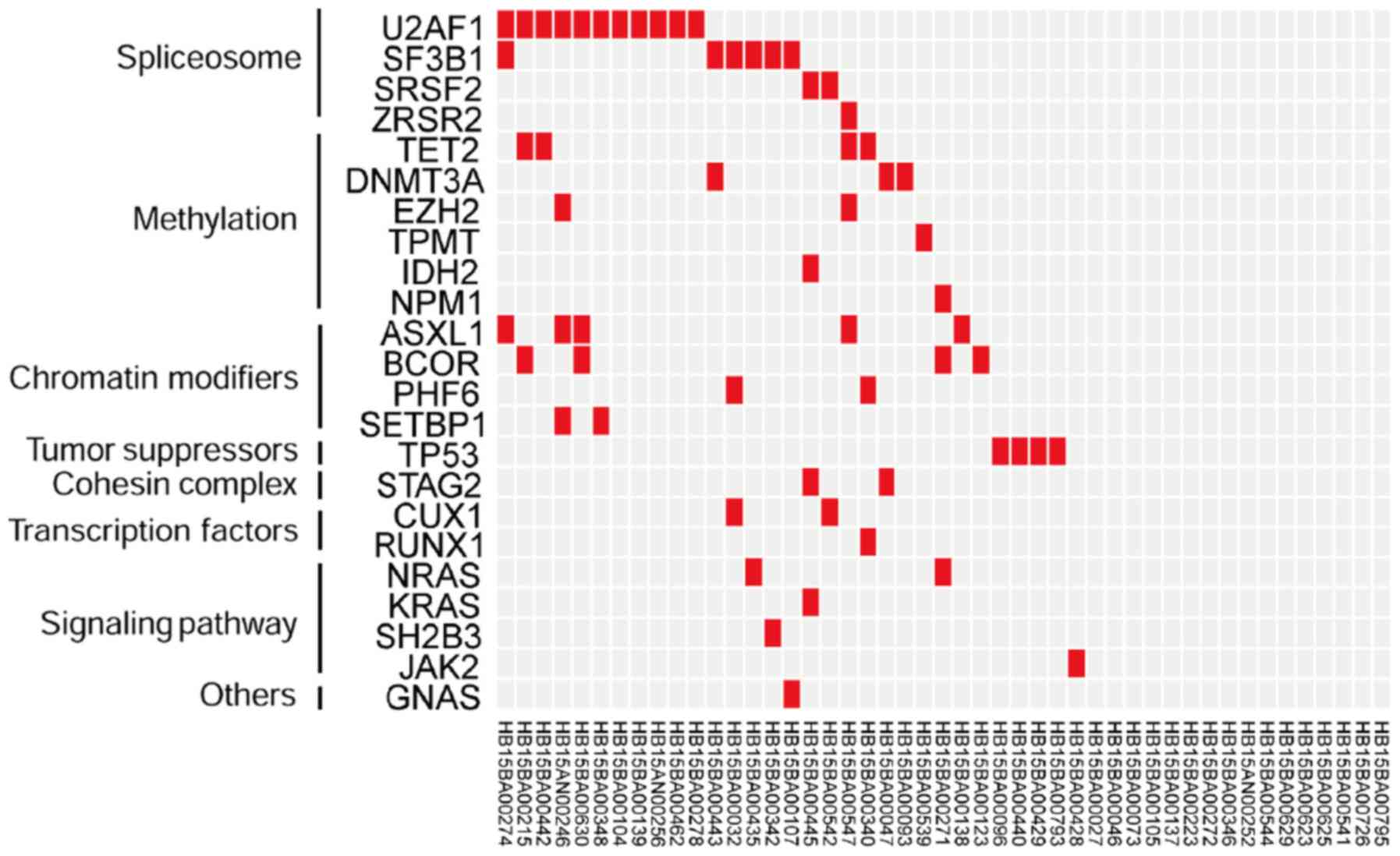
Of the 31 patients with mutations, 16 patients had
≥2 mutations (51.6%). Among them, 7 patients had two genetic
alterations, 6 patients had three genetic alterations, and 3
patients had four genetic alterations (Fig. 2). Of the 16 patients with ≥2
mutations, 4 (25%) had a synergistic mutation within the same
functional group and the remaining 12 (75%) had mutations in
different genetic functional groups. The prevalence of synergistic
mutations in different functional groups was significantly higher
compared with that in the single functional group (P=0.036;
Fig. 2).
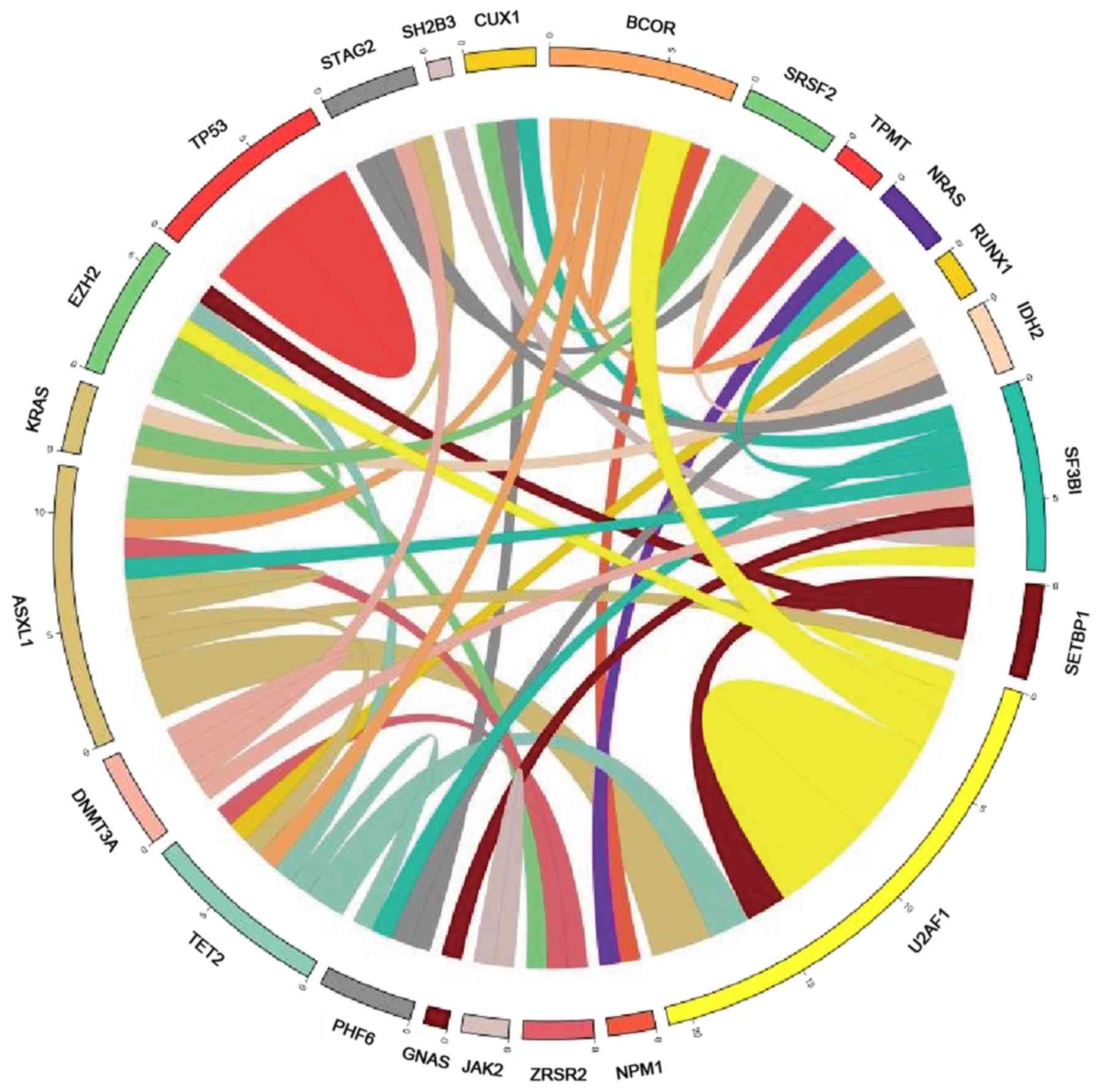
Association analysis of the mutant
genes with MDS
The results indicated that the mutations in genes
EZH2 and ASXL1 (P=0.009), IDH2 and KRAS
(P=0.021), IDH2 and STAG2 (P=0.043), IDH2 and
SRSF2 (P=0.043), KRAS and STAG2 (P=0.043),
RUNX1 and PHF6 (P=0.043), NPM1 and NRAS
(P=0.043), and EZH2 and ZRSR2 (P=0.043) co-occurred
and these associations were statistically significant (Fig. 3).
VAF analysis
In the present study, 23 mutant genes were detected,
and the median VAFs were compared and sorted in descending order.
The results revealed that the four genes associated with ‘signaling
pathway’, JAK2, KRAS, NRAS and SH2B3, had a low VAF,
which suggested that the corresponding mutations were acquired
relatively late during the evolution of the leukemic clones
(Fig. 4).
Analysis of the association between
genetic alterations and clinicopathological features of the
patients
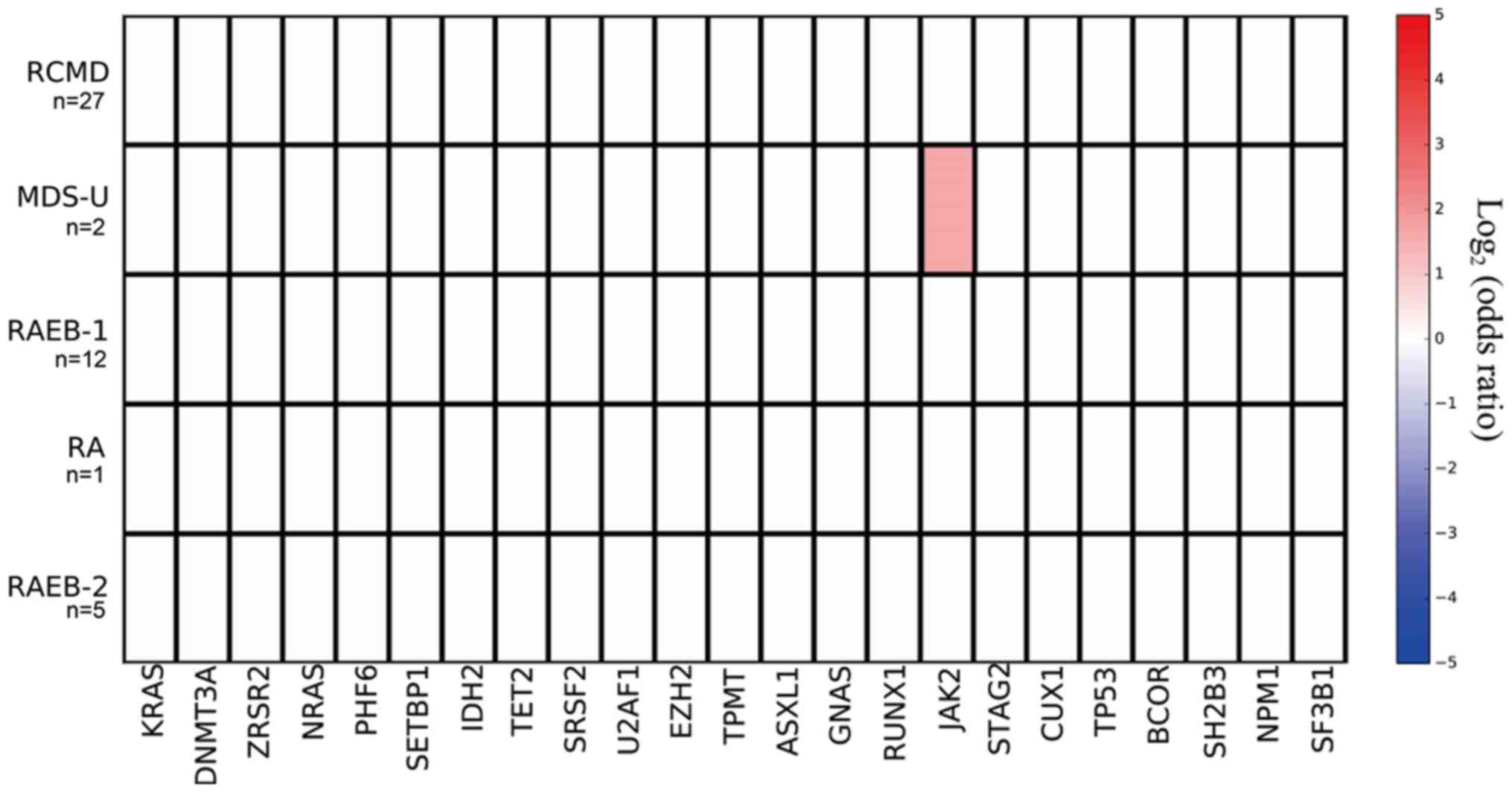
Mutant genes and MDS subtypes
The mutation prevalence rates of the JAK2 gene in
subtypes RA (0/1), RCMD (0/27), RAEB-1 (0/12), and RAEB-2 (0/5)
were all 0%, and in the MDS-U subtype, the mutation prevalence was
50% (1/2); the mutation prevalence rate of the JAK2 gene was
significantly higher in the MDS-U subtype compared with non MDS-U
subtypes (P=0.043; Fig. 5).
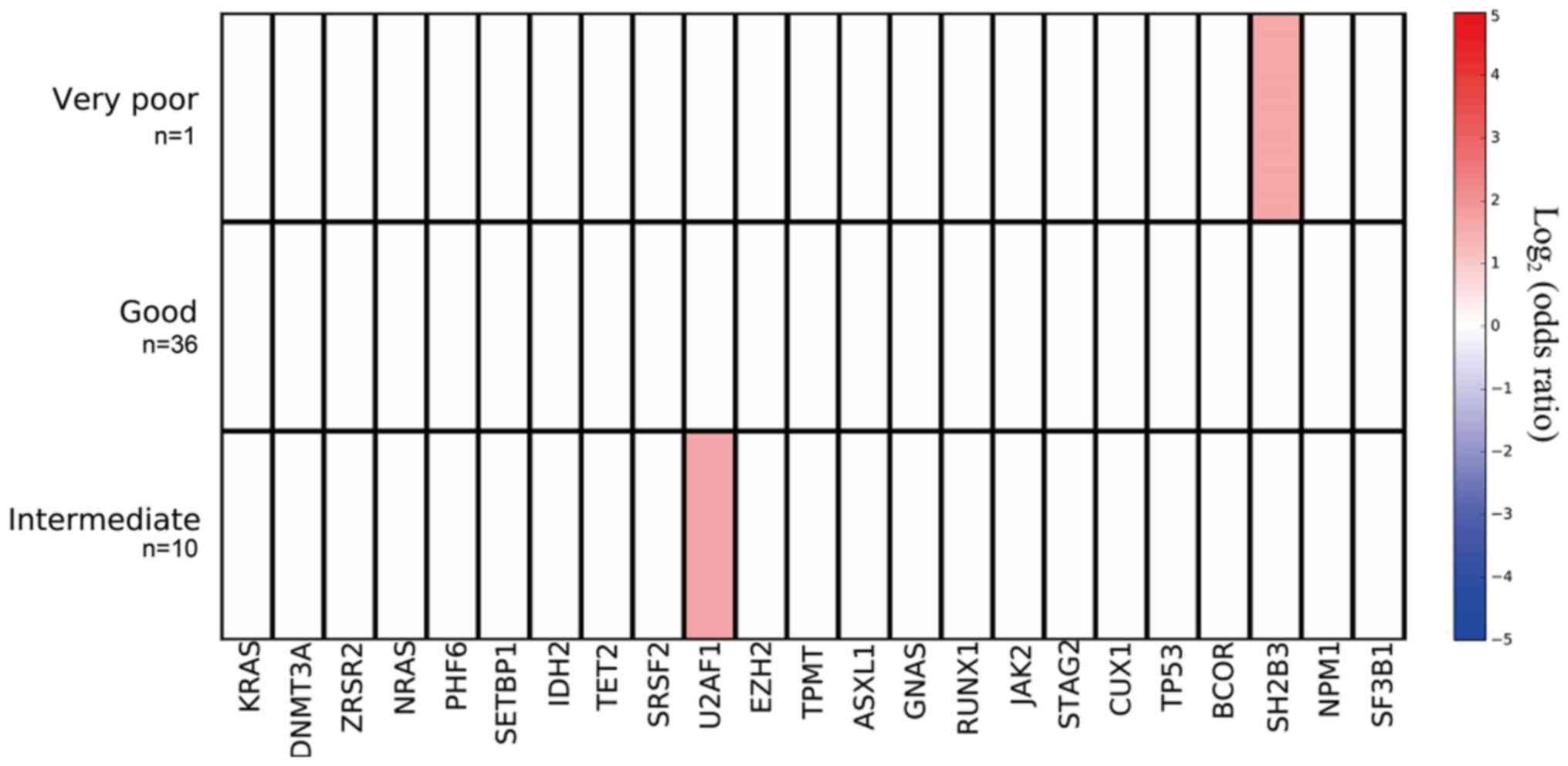
Mutant genes and karyotype
The mutation prevalence rates of the SH2B3
gene in the patients with good, intermediate and very poor
prognosis karyotypes were 0 (0/36), 0 (0/10), and 100% (1/1),
respectively; mutation prevalence was significantly higher in the
patients with the very poor prognosis karyotype (P=0.021). The
mutation prevalence rates of the U2AF1 gene in the patients
with good, intermediate and very poor prognosis karyotypes were
16.7 (6/36), 50.0 (5/10) and 0 (0/1), respectively; mutation
prevalence tended to be highest in the patients with the
intermediate prognosis karyotype (P=0.07; Fig. 6).
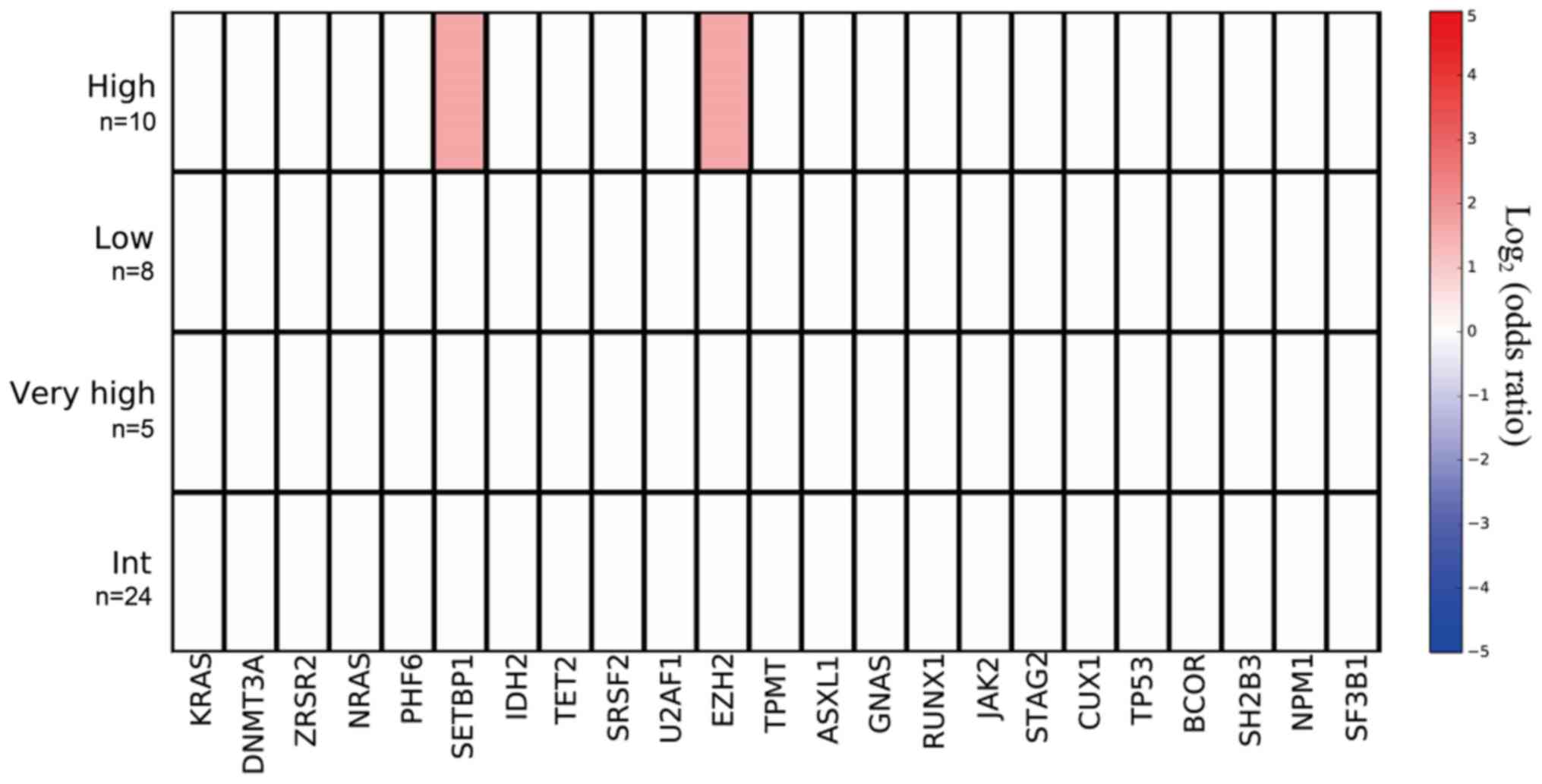
Mutant genes and IPSS-R
The mutation prevalence rates of the SETBP1
gene among low-risk, intermediate-risk, high-risk, and very
high-risk patients were 0 (0/8), 0 (0/24), 20 (2/10), and 0 (0/5),
respectively; and mutation prevalence was significantly higher in
the high-risk group as defined by IPSS-R (P=0.042). The mutation
rates of EZH2 among low-risk, intermediate- risk, high-risk,
and very high-risk patients were 0 (0/8), 0 (0/24), 20 (2/10), and
0 (0/5), respectively; and mutation prevalence was significantly
the highest in the high-risk group on the basis of the IPSS-R score
(P=0.042; Fig. 7).
Discussion
The positive gene mutation detection rates in the
study by Haferlach et al (9)
in a 104-target gene panel, Xu et al (20) in a 28-target gene panel, and the
present study in a 127-target gene panel were 89.5, 84.0, and
66.0%, respectively, suggesting that mutations in patients with
newly diagnosed MDS are relatively common. Twelve genes,
TET2 (33.3%), SF3B1 (32.9%), ASXL1 (23.4%),
SRSF2 (17.5%), DNMT3A (13.1%), RUNX1 (10.6%),
U2AF1 (7.7%), ZRSR2 (7.6%), STAG2 (7.5%),
TP53 (6.4%), EZH2 (5.5%) and Cbl proto-oncogene
(5.1%%), with a mutation frequency prevalence >5% in the MDS
population have been previously reported (9). In the present study, seven genes with a
mutation prevalence >5% were detected, including U2AF1
(23.4%), SF3B1 (12.8%), ASXL1 (10.6%), TET2
(8.5%), BCOR (8.5%), TP53 (8.5%) and DNMT3A
(6.8%).
The pathogenesis of MDS is associated with genetic
alterations. Previous studies from China reported that the
U2AF1 mutation has one of highest prevalence rates among
other mutations in the Chinese MDS population (20–22). The
mutation prevalence of U2AF1 according to different Chinese
research groups was 16.8% among 511 patients (21), 9.4% among 320 patients (22) and 8.0% among 125 patients (20). Furthermore, U2AF1 mutations
are more common among patients with trisomy 8 (21–23). The
results of the present study are in accordance with the
aforementioned results, since U2AF1 mutations had the
highest prevalence (23.4%) among the 47 Chinese patients with MDS
and tended to occur in the patients with the intermediate-prognosis
karyotype. In addition, the genetic alterations with clear clinical
significance and poor prognosis were prone to be accompanied by
poor prognostic clinical (objective) indicators.
Furthermore, in the present study population, it was
also indicated that the prevalence of SETBP1 mutations
(4.3%) was relatively low, similar to the result (4.7%) obtained by
Xu et al (22); however, a
mutation in this gene has not been reported in patients with MDS in
western countries (9). The
prevalence of SRSF2 mutations (17.5%) is reported to be
higher in patients with MDS in western countries (9). The results of the present study
revealed that the prevalence of SRSF2 mutations (4.3%) was
lower in Chinese patients with MDS, which is in accordance with the
findings (3.4%) of Xu et al (22) in China. In the present study, the
prevalence rates of mutations in genes, including IDH2, TP53,
BCOR and EZH2, resemble those reported by the other two
groups in China (22) and in western
country (9).
The most common mutant genes in these 47 patients
were the splicing genes, followed by the methylation genes;
consistent with previous literature data (9). Among the 16 patients with ≥2 mutations,
the mutations that co-occurred were detected in gene pairs
IDH2-SRSF2, IDH2-STAG2 and EZH2-ASXL1, which were
distributed among different functional groups. This result
suggested that genes in different functional groups may undergo
synergistic mutations. Among the patients with newly diagnosed MDS,
the median VAFs of the genes associated with ‘signaling pathway’
were relatively low, suggesting that mutations in
signaling-pathway-associated genes appeared later in the clonal
evolution of MDS.
In this study, a 31-year-old patient had a
synergistic interaction of mutations in SETBP1, ASXL1, EZH2
and U2AF1; according to the RAEB-1 subtype with a blast
percentage of 6%, this patient belonged to the high-risk group on
the basis of IPSS-R and had agranulocytosis status. The percentages
of WT1 and PRAME quantitative gene detection were
12.8 and 387.8, respectively, which were all associated with acute
leukemia. This result is consistent with the findings of Inoue
et al (24) who concluded
that the SETBP1 gene mutation can trigger the ASXL1
mutation in patients with MDS and the conversion of MDS to
leukemia. Since the patient with synergistic genetic alterations
had a worse prognosis, synergistic genetic alterations were
targeted by therapeutic interventions in the present study. In the
2016 WHO classification system, JAK2 gene mutation was the
main indicator of chronic myeloproliferative neoplasms (25). The mutation prevalence of JAK2
was significantly higher in the MDS-U subtype compared with non
MDS-U subtypes in this study, suggesting that JAK2 could
help with the differential diagnosis of this disease.
In myeloid neoplasms, EZH2 gene mutations
often occur in MDS and myeloproliferative neoplasms, and have been
associated with a poor prognosis (15). A knockout mouse model revealed that
after EZH2 undergoes inactivating mutations, the number of
modifications of H3k27me3 sharply diminishes, and the transcription
of oncogenes, including target genes Hmga2, Pbx3, Lmo1 and
Myc, is inhibited, leading to MDS or myeloproliferative
neoplasm-like phenotypes (26). In
the present study, the EZH2 gene mutation mostly occurred in
the high-risk group on the basis of the IPSS-R score, suggesting
that EZH2 mutations are associated with a poor prognosis
among patients with MDS, a finding that is in accordance with
previous literature (15).
In summary, 66.0% of 47 Chinese patients with a
novel MDS diagnosis were indicated to have a genetic mutation, as
detected by the highly promising next-generation sequencing
technology. The results for gene mutations in this study will
supplement the database of patients with MDS in China. Due to the
small sample size, the results concerning the association between
genetic alterations and clinicopathological features of patients
with MDS in this study require further confirmation with a larger
cohort.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81673821), the Beijing
Municipal Science & Technology Commission (grant no.
Z141100006014003) and the Special Research Foundation of Central
Level Public Scientific Research Institutes (grant no.
ZZ10-016).
Availability of data and materials
All data and materials analyzed during the current
study are included in this published article.
Authors' contributions
XH contributed to the study design; PZ, JQ and XH
wrote the manuscript; XH, PZ, WL, RQ, HX, CL, LL, YL, QZ, HW and XG
conducted the clinical research; JQ and JW performed the
next-generation sequencing; PZ and JQ performed the data processing
and statistical analysis. All authors have read and agreed to the
final version of the manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Clinical
Research Ethics Committee of Xiyuan Hospital, China Academy of
Chinese Medical Sciences (approval no. 2017XLA019-2). All patients
provided written informed consent to participate in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Della Porta MG, Galli A, Bacigalupo A,
Zibellini S, Bernardi M, Rizzo E, Allione B, van Lint MT, Pioltelli
P, Marenco P, et al: Clinical effects of driver somatic mutations
on the outcomes of patients with myelodysplastic syndromes treated
with allogeneic hematopoietic stem-cell transplantation. J Clin
Oncol. 34:3627–3637. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cazzola M and Malcovati L: Myelodysplastic
syndromes-coping with ineffective hematopoiesis. N Engl J Med.
352:536–538. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ades L, Itzykson R and Fenaux P:
Myelodysplastic syndromes. Lancet. 383:2239–2252. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang L, Padron E and Lancet J: The
molecular basis and clinical significance of genetic mutations
identified in myelodysplastic syndromes. Leuk Res. 39:6–17. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Papaemmanuil E, Gerstung M, Malcovati L,
Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC,
Pellagatti A, et al: Clinical and biological implications of driver
mutations in myelodysplastic syndromes. Blood. 122:3616–3627. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cazzola M, Della Porta MG and Malcovati L:
The genetic basis of myelodysplasia and its clinical relevance.
Blood. 122:4021–4034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ogawa S: Genetics of MDS. Blood.
7:1049–1059. 2019. View Article : Google Scholar
|
|
8
|
Tefferi A and Vardiman JW: Myelodysplastic
syndromes. N Engl J Med. 361:1872–1885. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haferlach T, Nagata Y, Grossmann V, Okuno
Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T,
et al: Landscape of genetic lesions in 944 patients with
myelodysplastic syndromes. Leukemia. 28:241–247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McGranahan N and Swanton C: Clonal
heterogeneity and tumor evolution: Past, present, and the future.
Cell. 168:613–628. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Greenberg PL, Stone RM, Al-Kali A, Barta
SK, Bejar R, Bennett JM, Carraway H, De Castro CM, Deeg HJ, DeZern
AE, et al: Myelodysplastic syndromes, version 2.2017, NCCN clinical
practice guidelines in oncology. J Natl Compr Canc Netw. 15:60–87.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Walter MJ, Shen D, Shao J, Ding L, White
BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, et al:
Clonal diversity of recurrently mutated genes in myelodysplastic
syndromes. Leukemia. 27:1275–1282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ruffalo M, Husseinzadeh H, Makishima H,
Przychodzen B, Ashkar M, Koyuturk M, Maciejewski JP and LaFramboise
T: Whole-exome sequencing enhances prognostic classification of
myeloid malignancies. J Biomed Inform. 58:104–113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Valent P, Horny HP, Bennett JM, Fonatsch
C, Germing U, Greenberg P, Haferlach T, Haase D, Kolb HJ, Krieger
O, et al: Definitions and standards in the diagnosis and treatment
of the myelodysplastic syndromes: Consensus statements and report
from a working conference. Leuk Res. 31:727–736. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ernst T, Chase AJ, Score J, Hidalgo-Curtis
CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, et al:
Inactivating mutations of the histone methyltransferase gene EZH2
in myeloid disorders. Nat Genet. 42:722–726. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gonzalez Garcia JR and Meza-Espinoza JP:
Use of the international system for human cytogenetic nomenclature
(ISCN). Blood. 108:3952–3953. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A mapreduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu Y, Li Y, Xu Q, Chen Y, Lv N, Jing Y,
Dou L, Bo J, Hou G, Guo J, et al: Implications of mutational
spectrum in myelodysplastic syndromes based on targeted
next-generation sequencing. Oncotarget. 8:82475–82490.
2017.PubMed/NCBI
|
|
21
|
Li B, Liu J, Jia Y, Wang J, Xu Z, Qin T,
Shi Z, Song Z, Peng S, Huang H, et al: Clinical features and
biological implications of different U2AF1 mutation types in
myelodysplastic syndromes. Genes Chromosomes Cancer. 57:80–88.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu F, Wu LY, He Q, Wu D, Zhang Z, Song LX,
Zhao YS, Su JY, Zhou LY, Guo J, et al: Exploration of the role of
gene mutations in myelodysplastic syndromes through a sequencing
design involving a small number of target genes. Sci Rep.
7:431132017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu L, Song L, Xu L, Chang C, Xu F, Wu D,
He Q, Su J, Zhou L, Xiao C, et al: Genetic landscape of recurrent
ASXL1, U2AF1, SF3B1, SRSF2, and EZH2 mutations in 304 chinese
patients with myelodysplastic syndromes. Tumour Biol. 37:4633–4640.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Inoue D, Kitaura J, Matsui H, Hou HA, Chou
WC, Nagamachi A, Kawabata KC, Togami K, Nagase R, Horikawa S, et
al: SETBP1 mutations drive leukemic transformation in ASXL1-mutated
MDS. Leukemia. 29:847–857. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the world health organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Muto T, Sashida G, Oshima M, Wendt GR,
Mochizuki-Kashio M, Nagata Y, Sanada M, Miyagi S, Saraya A, Kamio
A, et al: Concurrent loss of Ezh2 and Tet2 cooperates in the
pathogenesis of myelodysplastic disorders. J Exp Med.
210:2627–2639. 2013. View Article : Google Scholar : PubMed/NCBI
|