Introduction
Cancer is considered to be one of the most dangerous
factors to human life. The global cancer statistics for 2018
demonstrated that breast cancer exhibits the highest morbidity and
mortality rates in females worldwide compared with other types of
cancer (1). Several therapeutic
strategies have been developed for breast cancer treatment,
including surgery, chemotherapy, radiotherapy, hormone therapy and
newly improved immunotherapy (2).
However, due to the high heterogeneity among different types of
breast cancer, the prognosis for a number of patients is still
poor, especially for patients with distant metastases, who are
usually diagnosed at a late stage (3). As a result, it is very important to
identify the basic molecular mechanisms leading to the occurrence
and development of breast cancer. Improved knowledge on breast
cancer may result in more efficient treatment strategies.
With the development of new research techniques,
bioinformatics analysis is considered as one of the most important
methods used to study cancer, especially the underlying molecular
mechanisms (4). A number of studies
have used bioinformatics to analyze the gene expression profiles of
various types of cancer by identifying, comparing or clustering
differentially expressed genes in cancer cells compared with
healthy cells. Candidate genes involved in the occurrence and
development of cancer can be identified and further studied to be
developed as promising therapeutic targets (5–8).
Bioinformatics analysis is considered to be an important technique
in breast cancer study and has already helped achieve promising
improvements, such as identifying new prognostic factors or
pathways and genes associated with breast cancer (9–12).
The present study analyzed the expression data from
GSE20711 and revealed that the RAD54B gene was associated with the
Tumor-Node-Metastasis (TNM) stage, which may be used as a signature
for predicting the overall survival time of patients with breast
cancer. This signature may contribute to the precise treatment and
prognostic monitoring of patients with breast cancer. Furthermore,
the present study identified a compound (Japonicone A) from the
traditional herb Inula japonica Thunb that could decrease
the proliferation of breast cancer cells by inhibiting the
expression of RAD54B. The present study identified a novel
candidate gene and a candidate compound as promising therapeutic
targets for the treatment of breast cancer.
Materials and methods
Gene expression datasets
The gene expression datasets GSE20711 and GSE85871
were downloaded from the Gene Expression Omnibus database
(https://www.ncbi.nlm.nih.gov/geo).
GSE20711 was comprised of 88 breast cancer samples and 2 normal
breast tissue samples, and used the platform GPL570 (Affymetrix
Human Genome U133 Plus 2.0 Array) (13). GSE85871 was comprised of the gene
expression profiles of MCF-7 cells, which were treated with 102
different molecules used in traditional Chinese medicine, and used
the GPL571 platform (Affymetrix Human Genome U133A 2.0 Array)
(14). The Cancer Genome Atlas
(TCGA) Breast Invasive Carcinoma dataset (including high-throughput
sequencing (HTSeq) and clinical data of 1,104 breast cancer tissue
samples and 113 normal breast samples) was downloaded using the R
package ‘TCGAbiolinks (version 2.10.0)’ (15).
Screening for differentially expressed
genes (DEGs)
The ‘limma (version 3.36.2)’ package was used to
load normalized data into R (version 3.3.3; http://www.r-project.org) software and screen the DEGs
between breast cancer and non-tumor tissues (16). The genes with fold-change ≥2 and an
adjusted P-value (false discovery rate) <0.05 were identified as
DEGs (17).
Co-expression network construction and
module identification
Weighted correlation network analysis (WGCNA) is a
commonly used systemic biological data mining method for describing
the correlation patterns among genes and identifying the modules of
highly correlated genes; it uses average linkage hierarchical
clustering coupled with topological overlap dissimilarity based on
high-throughput chip or RNA-Seq data (18). The ‘WGCNA (version 1.63)’ package in
R was used to construct the co-expression network for the DEGs in
the 88 breast cancer samples in GSE20711 (18). β is a soft-thresholding parameter
that emphasizes strong correlations between genes and depreciates
weak correlations (19). In the
present study, β=18 (scale-free R2=0.8) was used to
ensure a scale-free network. A cut height of 0.85 and effect size
of ≥10 were used to identify the modules. Pearson's correlation
matrices were calculated for the modules (20).
Enrichment analysis
The Gene Ontology (GO) and Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway enrichment analysis of the genes
in the modules were performed using the ‘clusterProfiler (version
3.9.1)’ R package based on hypergeometric distribution algorithm
(P<0.05), and ‘GOplot (version 1.0.2)’ was used for further
analysis (21,22).
Survival analysis
To validate the genes in the turquoise modules, the
largest modules, the clinical information and RNA sequencing data
(HTSeq-FPKM) of breast cancer were obtained from TCGA Project
database (https://cancergenome.nih.gov). Kaplan-Meier survival
analysis with the log-rank test was conducted to evaluate the
association between the genes in the turquoise module and patient
survival. P<0.05 was considered to indicate a statistically
significant association. Univariate Cox analysis was used to test
whether the genes may be used as independent prognostic factors.
The data were randomly divided into two groups: A discovery cohort
(n=458) and an internal testing cohort (n=457). The genes and
clinicopathological characteristics that were significant in the
univariate Cox analysis and the Kaplan-Meier survival analysis were
used for the multivariate Cox regression analysis. The Akaike
information criterion (AIC) value, which was calculated based on
different influencing factors by the multivariate Cox regression
analysis, was used to remove the confounding factors to obtain the
best variable for data fitting, where the minimum AIC value has the
best fit (23). Subsequently, a
prognostic mRNA and clinical trait signature with min AUC value was
constructed, which may be used to calculate a risk score for each
individual patient with breast cancer. According to the median of
the risk score (median value, 1.915), the patients were stratified
into low-risk and high-risk groups. The risk groups from the two
cohorts were evaluated using Kaplan-Meier analysis and the log-rank
test (24). A nomogram combining the
risk score with two other clinical factors (age and sex) was
constructed to provide a graphic representation of the prediction
model using the R package rms (version 5.1–3.1) (25).
Protein-protein interaction (PPI)
network construction
The search tool for the retrieval of interacting
genes/proteins (STRING) database (https://string-db.org/) was used to construct the PPI
networks, and the results exported from STRING were imported into
Cytoscape (version 3.4.0) for visualization (24).
Immunohistochemistry
The Human Protein Atlas (http://www.proteinatlas.org/) was used to validate the
expression of the three genes in breast cancer tissue (26). The direct links to these images are
as follows: RAD54B, https://www.proteinatlas.org/ENSG00000197275-RAD54B/pathology/tissue/breast+cancer#imid_2186145;
KIF21A, https://www.proteinatlas.org/ENSG00000139116-KIF21A/pathology/tissue/breast+cancer#imid_17124866;
and C8orf76, https://www.proteinatlas.org/ENSG00000189376-C8orf76/pathology/tissue/breast+cancer#imid_6235831.
Gene Set Enrichment Analysis
(GSEA)
GSEA was performed using KEGG pathway annotation
data from the KEGG database (27).
According to the median value of RAD54B expression, patients from
the GSE20711 and TCGA datasets were divided into two groups. The
clusterProfiler (version 3.9.1) package was used to analyze the
data and construct the ridge plot (28).
Cell culture and stimulation
The MCF-7 cell line was obtained from the American
Type Culture Collection. The cells were cultured in complete
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.) in 5% CO2 stored at
37°C. The cell cultures were checked for Mycoplasma
contamination using a Mycoplasma PCR Detection kit
(Sigma-Aldrich; Merck KGaA) every 3 months. The cell line was not
listed in the database of commonly misidentified cell lines
(https://iclac.org/databases/cross-contaminations).
Japonicone A (>97% purity) was kindly gifted by
Professor Weidong Zhang (School of Pharmacy, Second Military
Medical University, Shanghai, China) (29–32).
Japonicone A was dissolved in dimethyl sulfoxide (DMSO) and the
solution was diluted in the cell culture media to ensure that the
concentration of DMSO was <0.1%, as described in a previous
study (31). The working
concentration (10 µM) of Japonicone A was the same as the one
applied in the GSE85871 dataset, which was initially used to
identify that Japonicone A inhibited the expression of RAD5B in
MCF-7 cells (15). In addition, Hu
et al (29) used 10 µM
Japonicone A in a treatment assay. Preliminary experiments were
performed with 10 µM Japonicone A on the proliferation of MCF-7
cells in the present study, and the results suggested that the
concentration was effective (data not shown). Therefore, MCF-7
cells were treated with 10 µM Japonicone A for 24 h prior to cell
collection and analysis. DMSO without Japonicone A was used as the
solvent control. The volume of the solvent control was the same
with DMSO-dissolved Japonicone A used in the experimental
group.
Reverse transcription-quantitative
PCR
Total RNA was extracted from MCF-7 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RNA was reverse-transcribed into cDNA with High-Capacity
cDNA Reverse Transcription kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.). qPCR was performed using SYBR® Premix
Ex Taq II (Takara Bio, Inc.). Relative mRNA expression levels were
calculated by normalizing the relative quantitation cycle value to
the control group following standardization to the internal control
β-actin (33). The primers used were
as follows: Human RAD54B forward, 5′-AAGAACCTGACTGCCTCACG-3′ and
reverse, 5′-TCCACCACAGGTAAACCAGC-3′; and human β-actin forward,
5′-CAGGGCGTGATGGTGGGCA-3′ and reverse,
5′-CAAACATCATCTGGGTCATCTTCTC-3′. The thermocycling conditions for
the RT-qPCR was as follows: 2 min at 95°C, 40 cycles at 95°C for 10
sec, 59.5°C for 10 sec, 68°C for 15 sec, and 72°C for 10 sec.
Receiver operating characteristic
(ROC) curve analysis
The R package ‘survivalROC (version 1.0.3)’ was used
for the ROC curve analysis (34).
The ROC curve was used to test the sensitivity and specificity of
the variables in predicting overall survival, and to assess the
predictive ability of the calculated prognostic signature for
5-year patient survival (25).
Decision curve analysis (DCA)
R package ‘rmda (version 1.6)’ was used for DCA,
which estimates the net benefit of a signature by subtracting the
false-positives from the true-positives (35,36).
Flow cytometry
Anti-mouse marker of proliferation Ki-67
(ki-67)-FITC flow cytometry antibody was purchased from Miltenyi
Biotec (diluted 1:100 with 1X permeabilization buffer; catalog no.
130-117-691). The intracellular staining of ki-67 was performed
using the Foxp3/Transcription Factor Staining Buffer Set
(eBioscience; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Briefly, MCF-7 cells were collected
and centrifuged at 150 × g for 5 min at room temperature. The cell
pellets were mixed with 1 ml fixation-permeabilization buffer (a
1:3 mixture of fixation-permeabilization concentrate and diluent,
which were included in the kit) at 4°C for 1 h. The cells were
washed twice with 2 ml 1X permeabilization buffer (10X
permeabilization buffer diluted to 1X with ddH2O). The
fixed cells were stained with the anti-ki-67-FITC antibody for 30
min at room temperature in the dark. Finally, the washed cells were
resuspended in PBS prior to detection using BD FACSVerse (BD
Biosciences). The flow cytometric data were analyzed with FlowJo
software (version 10.3; FlowJo LLC).
MTT assay
A total of 5×103 MCF-7 cells were seeded
in 96-well plates and treated with the aforementioned amount of
control (DMSO) or 10 µM Japonicone A for 24 h. Subsequently, 20 µl
MTT (5 mg/ml) was added to the wells and incubated for 4 h. The
wells were supplemented with 150 µl DMSO to dissolve the formazan
crystals prior to optical density measurement by a microplate
reader at 490 nm. There were three duplicate wells for each group,
and the cell viability was normalized to the control group in each
experiment.
Statistical analysis
The statistical analyses were performed using R
(version 3.3.3). Student's t-test was used for comparisons between
two independent groups. P<0.05 was considered to indicate a
statistically significant difference. The experiments presented in
Fig. 7 were replicated four times
and each dot in the histogram represents one independent
experiment; lines and error bars represent the mean ± SD.
Results
Co-expression network construction and
key module identification
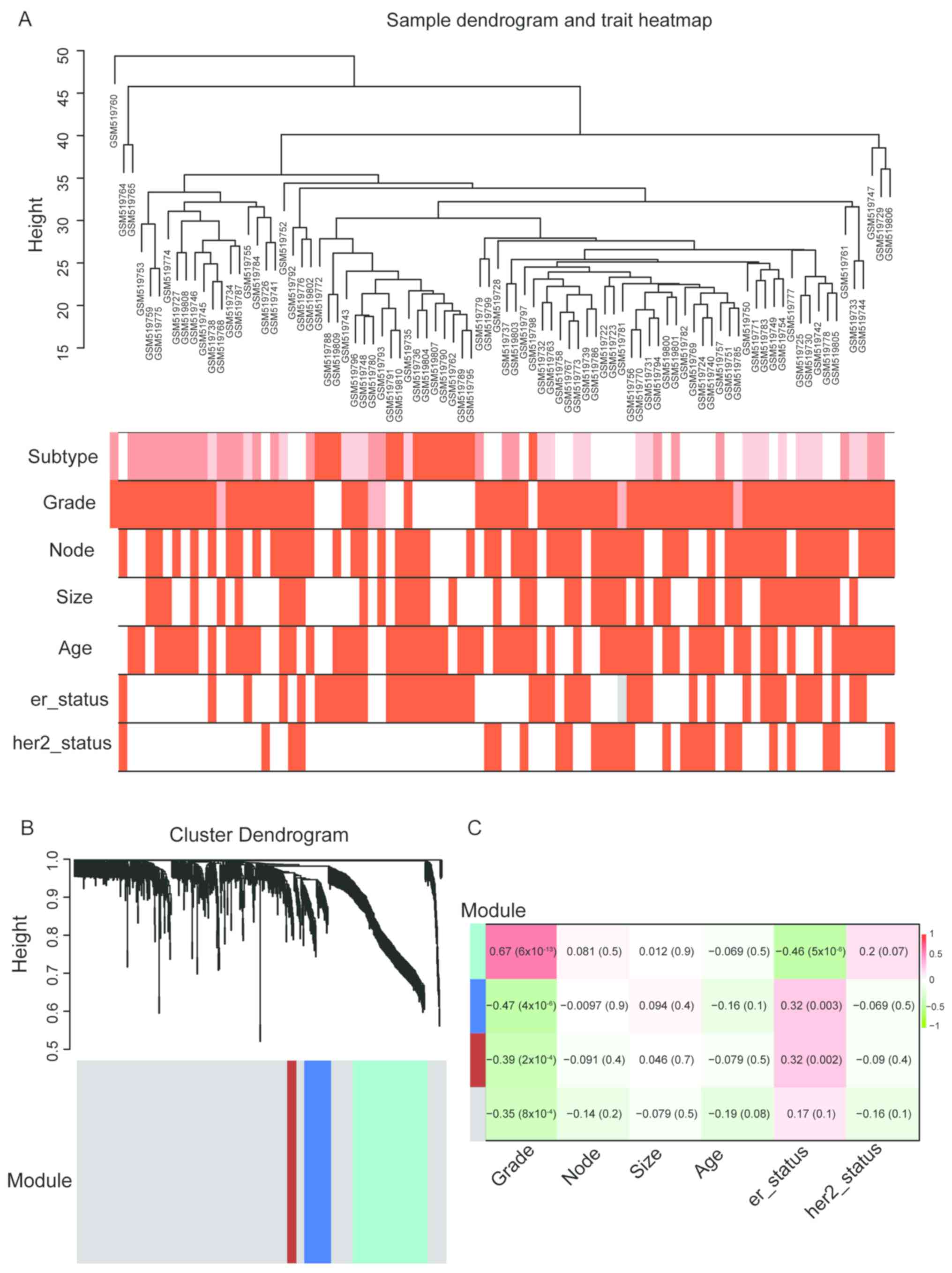
The GSE20711 samples with complete clinical data
were included in the WGCNA analysis (Fig. 1A). DEGs between breast cancer tissues
and non-tumor tissues were identified (Table SI). Based on the DEGs, a
co-expression network was constructed, and the modules were
identified by WGCNA. The results revealed that the most significant
DEGs could be grouped into three major modules (turquoise, blue and
brown modules) (Fig. 1B). Further
analysis demonstrated that the turquoise module exhibited the
highest positive correlation with TNM stage and negative
correlation with ER status compared with the other modules
(Fig. 1C). Thus, this module was
identified as the clinically significant module for subsequent
analysis.
Enrichment analysis and PPI network of
the turquoise module
GO enrichment analysis was performed for the genes
in the turquoise module, and the results were categorized into
three functional groups: Biological process (BP), molecular
function (MF) and cellular component (CC). The genes in the BP
group were mainly enriched in ‘mitotic cell cycle process’, ‘cell
cycle’, ‘cell cycle process’, ‘mitotic cell cycle’, ‘cell division’
and ‘nuclear division’ (Fig. 2A);
the genes in the CC group were enriched in ‘condensed chromosome’,
‘chromosome’, ‘spindle’ and ‘chromosomal region’ (Fig. 2B); the genes in the MF group were
mainly enriched in ‘protein binding’, ‘ATP binding’, ‘catalytic
activity’ and ‘microtubule binding’ (Fig. 2C). In addition, the KEGG pathway
analysis revealed that the DEGs were mainly involved in ‘cell
cycle’, ‘oocyte meiosis’, ‘pyrimidine metabolism’, ‘p53 signaling
pathway’ and ‘DNA replication’ (Fig.
2D). The PPI network based on the genes from the turquoise
module demonstrated that the majority of the genes closely
interacted with each other (Fig.
2E). Taken together, the results demonstrated that the DEGs in
the clinically significant module mainly participated in the
regulation of cancer cell proliferation, and that the majority of
them interacted with each other.
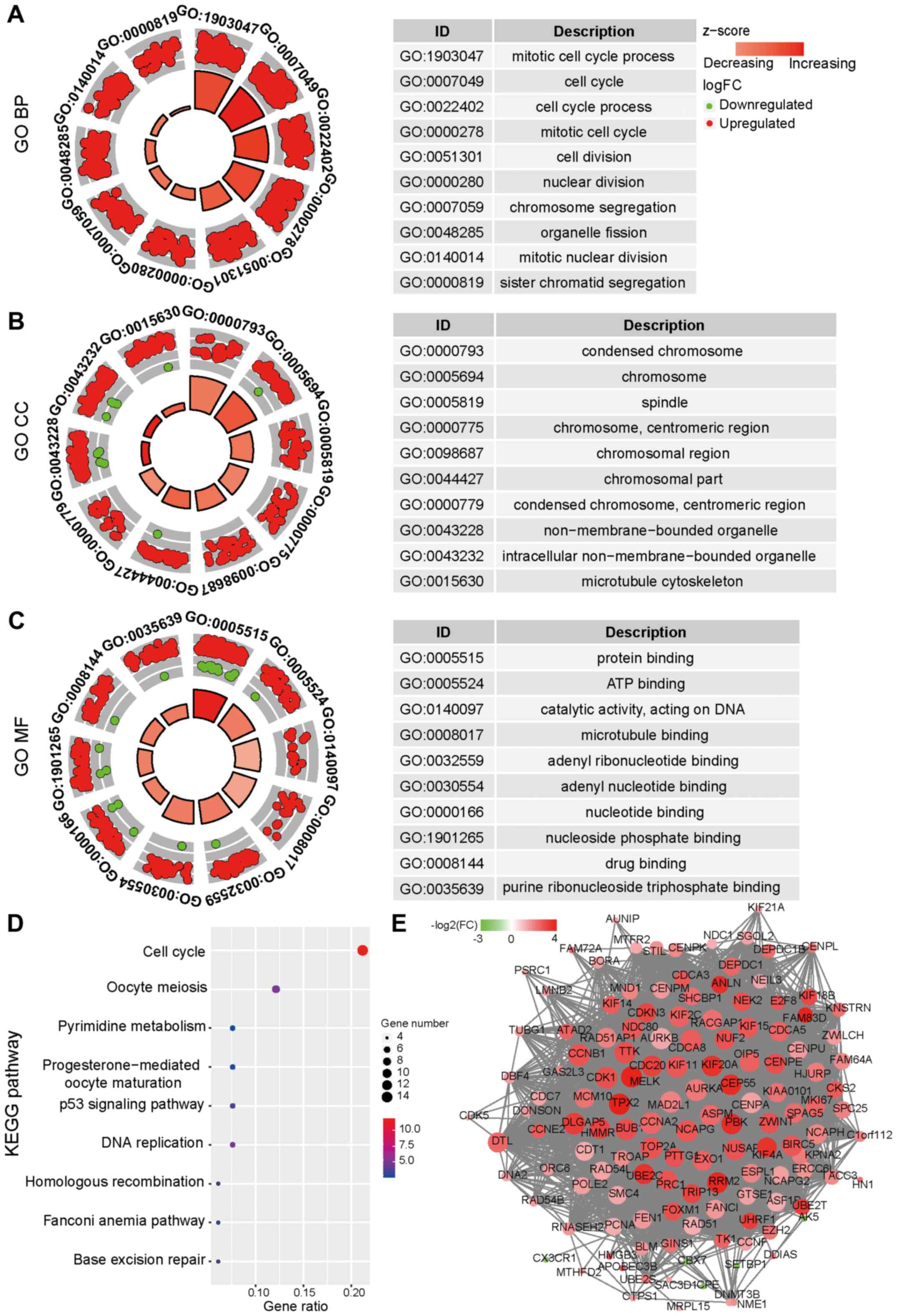 | Figure 2.Enrichment analysis and PPI network
of the genes in the turquoise module. (A) Results of the GO BP
analysis, (B) the GO CC analysis and (C) the GO MF analysis of the
turquoise module. The circles indicate the gene expression
distribution in each term, and the Z-score value indicates the
difference in the number of upregulated versus downregulated genes
divided by the square root of the total count. (D) Results of the
KEGG pathway analysis of the turquoise module. The colors indicate
the significance [-log10(P-value)], and the size of the circles
represents the number of genes enriched in the corresponding
annotation. (E) The PPI network of the genes in the turquoise
module. The size represents the degree of connectivity, and the
color represents FC (red, upregulated genes; green, downregulated
genes). GO, Gene Ontology; BP, biological process; CC, cellular
component; MF, molecular function; PPI, protein-protein
interaction; KEGG, Kyoto Encyclopedia of Genes and Genomes; FC,
fold-change. |
Potential independent prognosis
factors in breast cancer
The association of the turquoise module with tumor
prognosis was analyzed based on TCGA data. Using univariate Cox
analysis, several genes (SHCBP1, RAD54B, KIF21A and C8orf76)
associated with prognosis were identified (Table I). The results of the Kaplan-Meier
survival analysis identified RAD54B, KIF21A and C8orf76 as
potential independent prognosis factors, which were negatively
associated with overall patient survival time (Fig. 3A-C) and upregulated in late clinical
tumor stages (III and IV) compared with early stages (I and II;
Fig. S1).
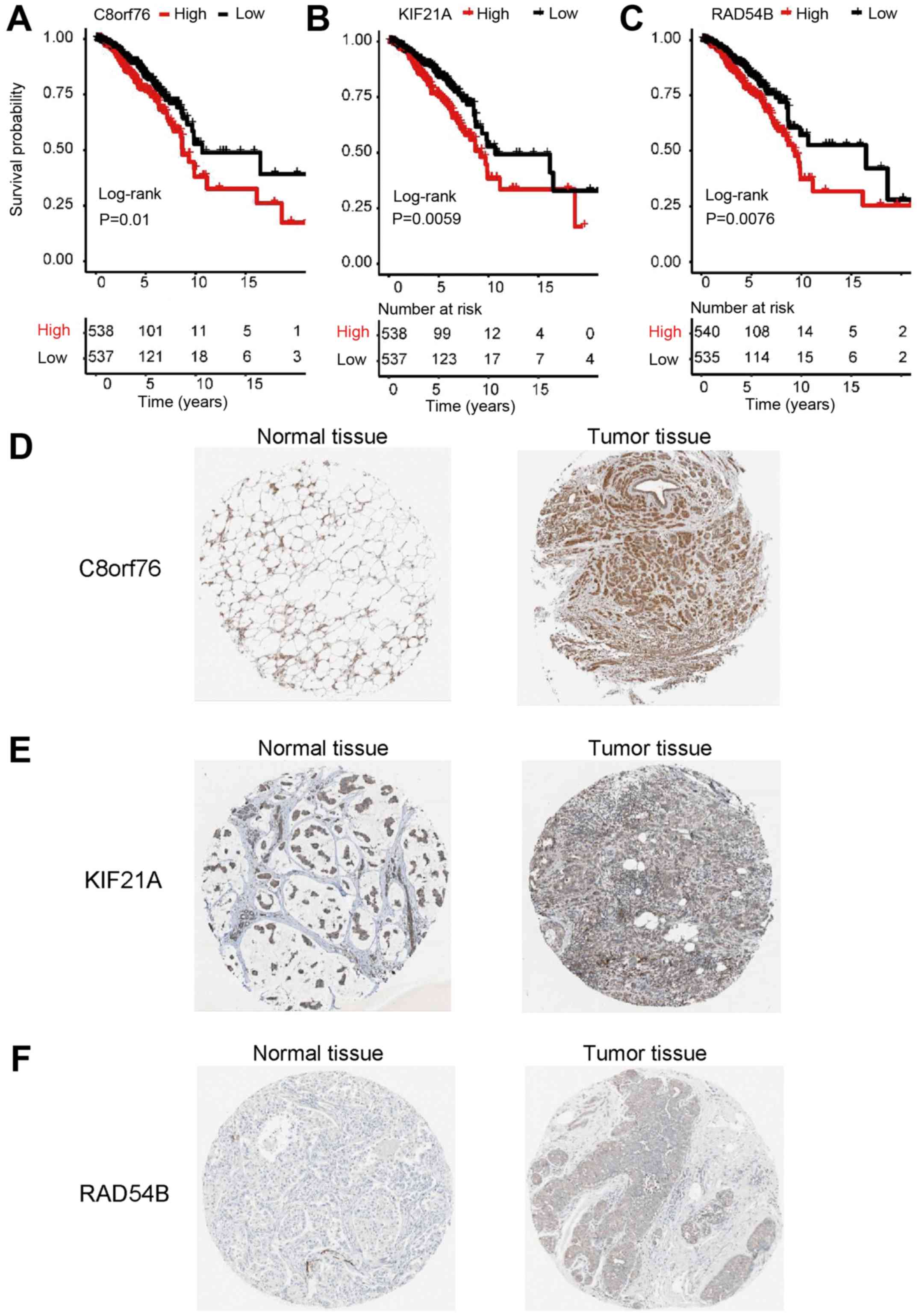 | Figure 3.Overall survival and
immunohistochemical analysis of the three genes identified in
breast cancer. (A-C) Survival analysis based on Kaplan-Meier
plotter; P-values were obtained from the log-rank test. Based on
the median expression, the patients were classed into the
high-level or the low-level group for (A) C8orf76, (B) KIF21A and
(C) RAD54B. (D) The protein levels of C8orf76 in normal tissue
(left: Staining, medium; intensity, moderate; quantity >75%) or
tumor tissue (right: Staining, medium; intensity, moderate;
quantity >75%). (E) The protein levels of KIF21A in normal
tissue (left: Staining, low; intensity, weak; quantity >75%) or
tumor tissue (right: Staining, medium; intensity, moderate;
quantity >75%). (F) The protein levels of RAD54B in normal
tissue (left: Staining, not detected; intensity, not detected;
quantity, not detected) or tumor tissue (right: Staining, low;
intensity, weak; quantity >75%). Images (D-F) were obtained from
the Human Protein Atlas (http://www.proteinatlas.org/). RAD54B, RAD54 homolog
B; KIF21A, kinesin family member 21A; C8orf76, chromosome 8 open
reading frame 76. |
 | Table I.Univariate Cox analysis and
Kaplan-Meier survival analysis based on the data of patients with
breast cancer from TCGA dataset. |
Table I.
Univariate Cox analysis and
Kaplan-Meier survival analysis based on the data of patients with
breast cancer from TCGA dataset.
|
| Univariate
analysis | Kaplan-Meier
survival analysis |
|---|
|
|
|
|
|---|
| Factor | HR (95% CI) | P-value | Log-rank test
P-value |
|---|
| Pathological
stage | 2.31
(1.62–3.31) |
<0.001c |
|
| Lymph nodes | 2.31
(1.6–3.33) |
<0.001c |
|
| SHCBP1 | 1.18
(1.03–1.35) | 0.019a | >0.05 |
| KIF21A | 1.19
(1.01–1.39) | 0.034a | 0.006b |
| C8orf76 | 1.27
(1.03–1.58) | 0.027a | 0.010a |
| RAD54B | 1.2
(1.01–1.41) | 0.035a | 0.008b |
The Human Protein Atlas database, the data of which
are open access to all researchers, contains a systems-based
analysis of protein expression in 17 types of cancer using data
from 8,000 patients, as well as immunohistochemistry images
directly demonstrating the indicated protein expression in tumor
tissues (37,38). The database was searched to analyze
the expression of RAD54B, KIF21A and C8orf76 in normal and breast
cancer tissues. With the exception of C8orf76, the protein
expression of RAD54B and KIF21A was increased in breast cancer
tissues compared with that in normal tissues (Fig. 3D-F).
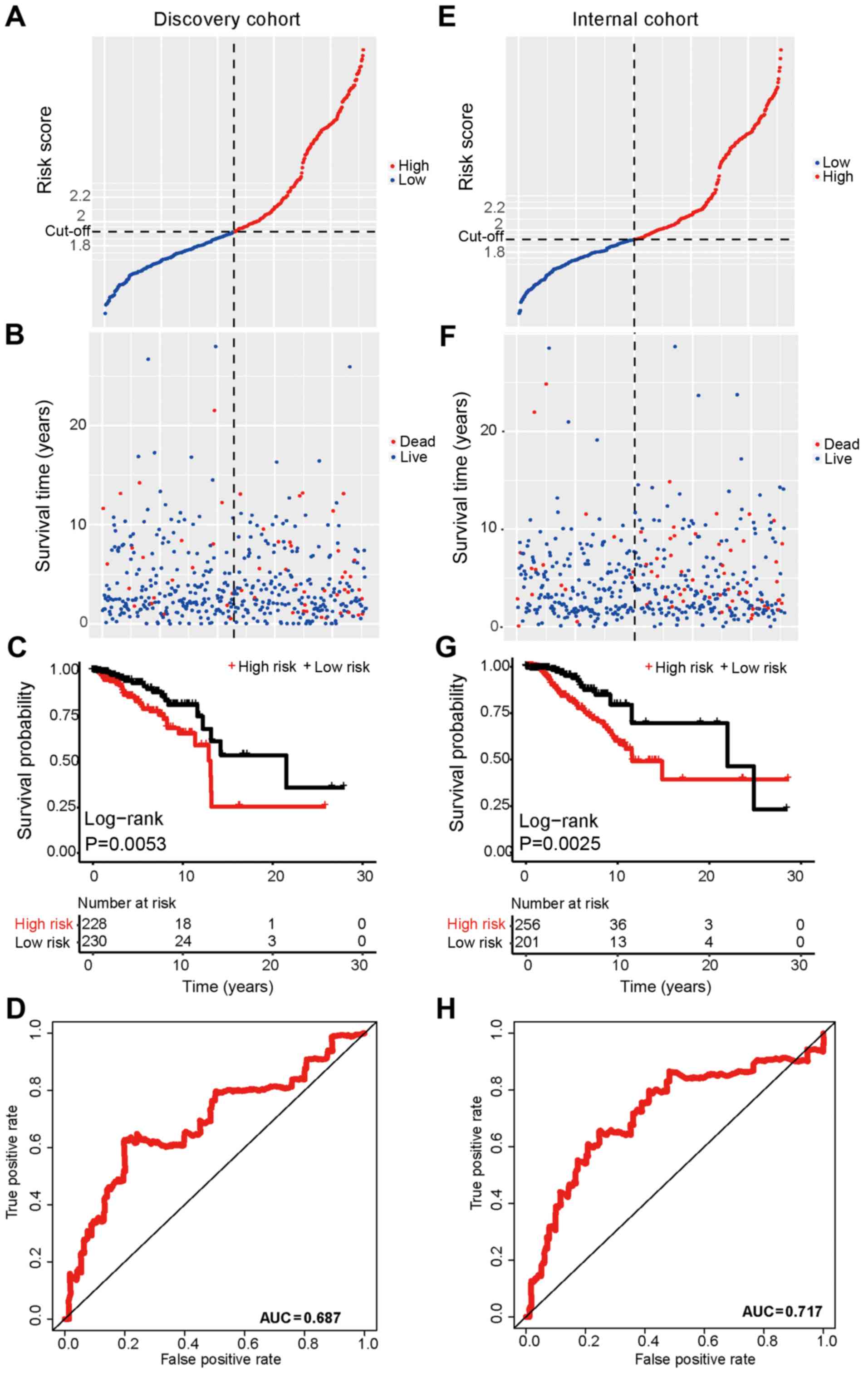
RAD54B combined with TNM stage may
predict the survival of patients with breast cancer
Since RAD54B, KIF21A and C8orf76 were significantly
associated with the overall survival of patients with breast
cancer, the patients from TCGA Project database were randomly
divided into two groups: The discovery cohort and the internal
cohort (Table SII). Combined with
the clinicopathological characteristics in the datasets, a
prognostic module was developed using forward conditional stepwise
regression with multivariate Cox analysis in the discovery cohort.
From the AIC values, a prognostic signature containing one gene
(RAD54B) and one clinical trait (TNM stage) was identified. Based
on this signature, the risk score was calculated for each sample in
the discovery cohort using the following formula: Risk score=RAD54B
expression × 0.236 + TNM stage (I/II=0 or III/IV=1) × 1.025. The
samples were ranked by score and divided into high-risk and
low-risk groups based on the median of the risk scores (median,
1.915), which was set as the cut-off point (Fig. 4A). The sample with the median value
was assigned to the high-risk group. The patients in the high-risk
group exhibited shorter overall survival times compared with the
patients in the low-risk group (Fig. 4B
and C). In addition, the ROC curve analysis revealed that the
AUC was 0.687 based on the risk scores (Fig. 4D).
The risk score model was further evaluated using the
internal cohort. Using the risk-score formula and cut-off point
derived from the discovery cohort, the patients were divided into
high-risk (n=256) and low-risk (n=201) groups. The overall survival
of the patients in the internal cohort revealed a similar trend to
that in the discovery cohort (Fig.
4E-H). In addition, the nomogram based on the signature and the
decision curve analysis demonstrated that this prognostic signature
was effective for evaluating the prognosis of patients with breast
cancer (Fig. S2).
Japonicone A may inhibit MCF-7 cell
proliferation by targeting RAD54B
As RAD54B may serve an important role in breast
cancer and may be a potential therapeutic target, the data from
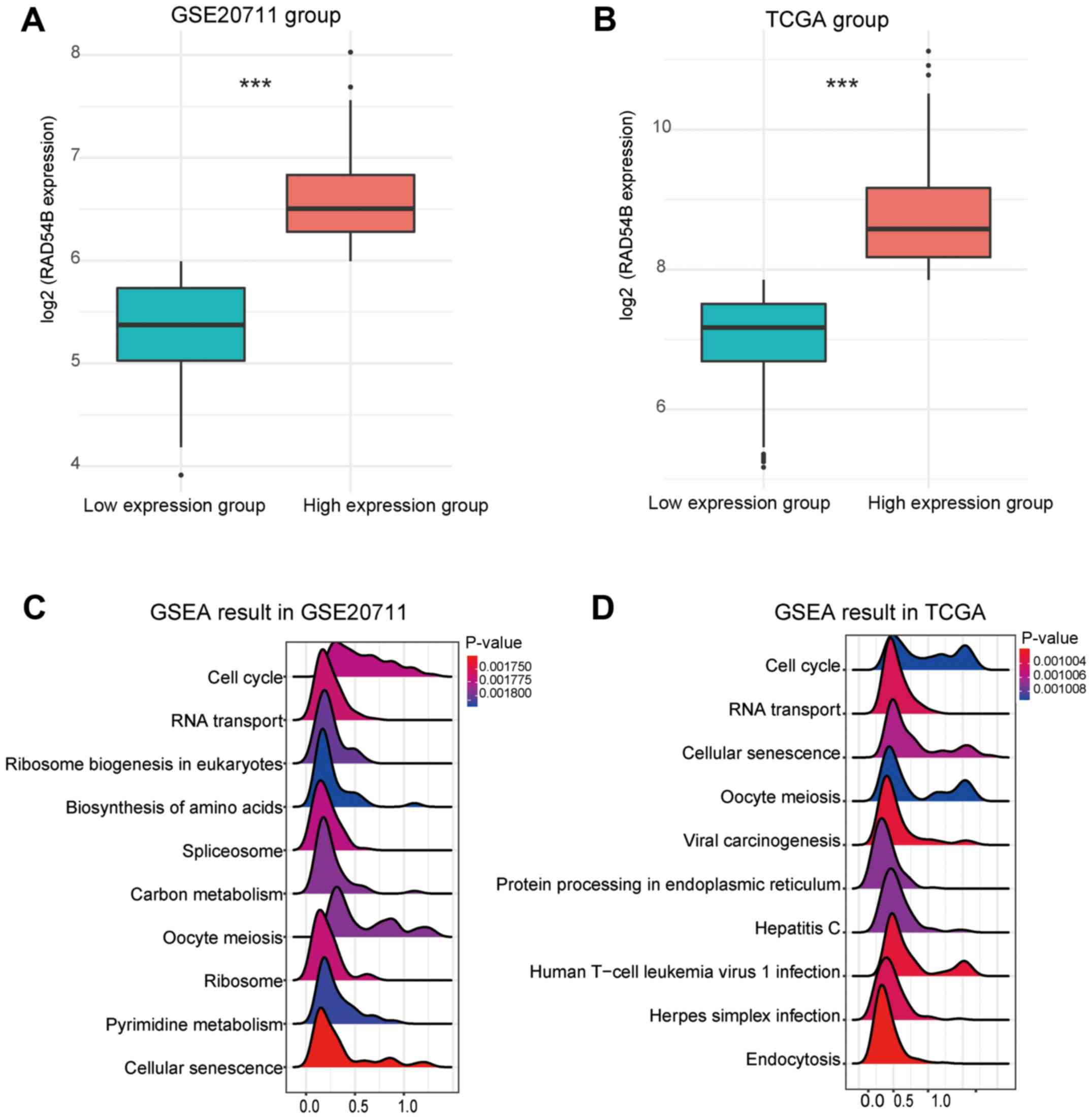
TCGA and GSE20711 were divided according to the expression of
RAD54B into high and low expression groups (Fig. 5A and B). GSEA analysis demonstrated
that the cell cycle function exhibited the strongest association
with the expression of RAD54B in the two datasets (Fig. 5C and D). These results suggested that
RAD54B may regulate the proliferation of breast cancer cells.
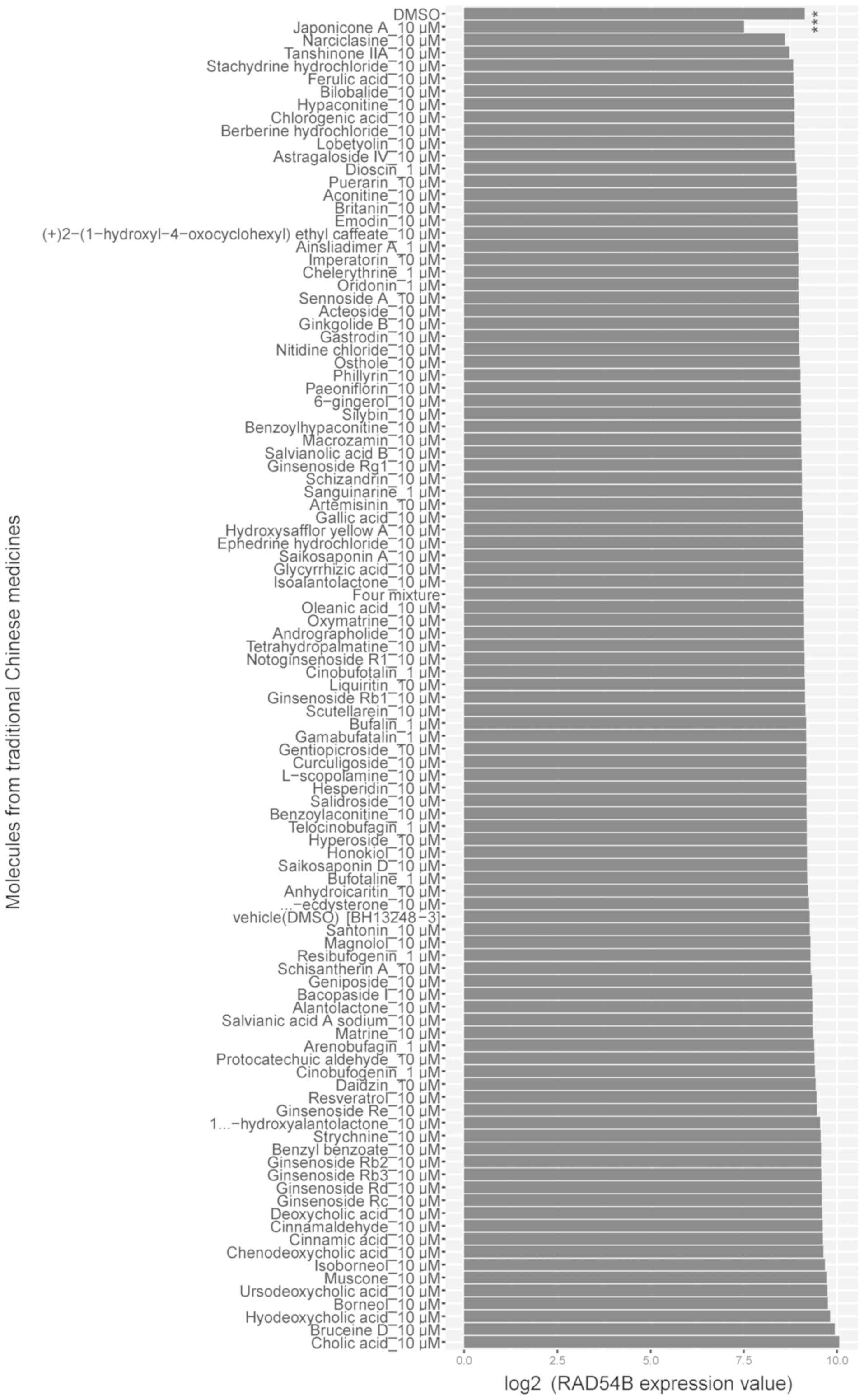
Based on the GSE85871 dataset, the expression of
RAD54B in breast cancer MCF-7 cells treated with 102 different
molecules used in traditional Chinese medicine was analyzed. The
results demonstrated that treatment with a compound known as
Japonicone A resulted in downregulated RAD54B expression in these
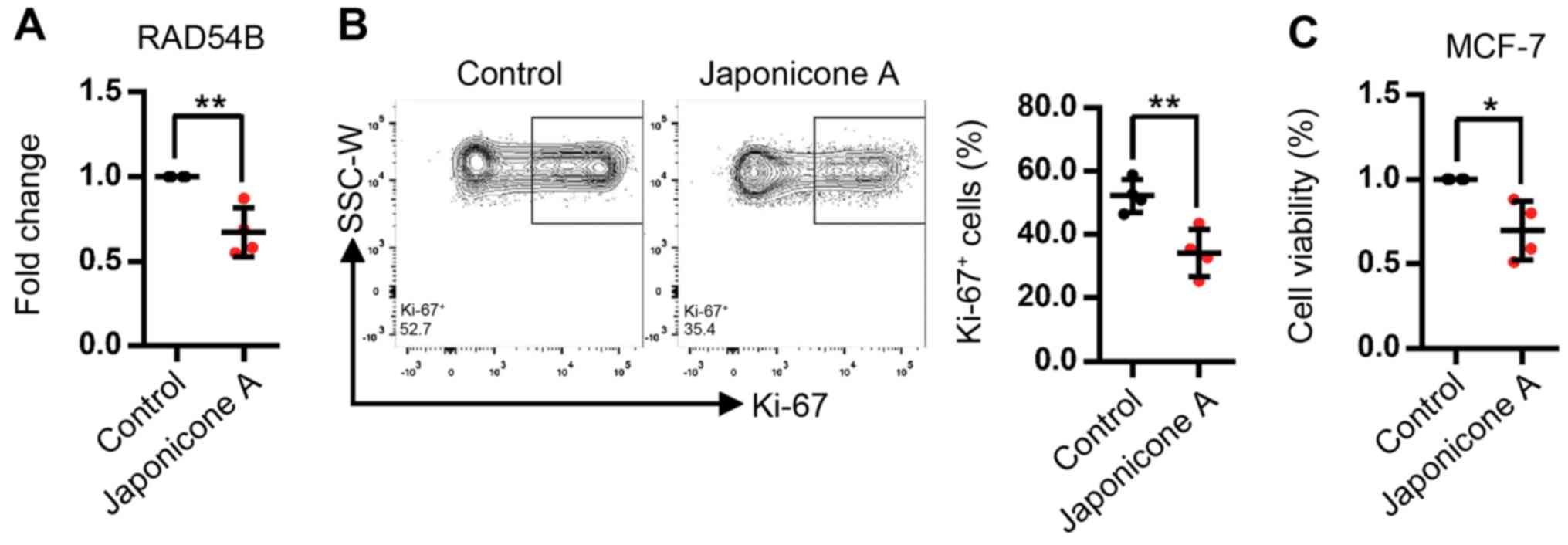
cells (Fig. 6). To confirm this
result, an inhibitory assay with Japonicone A on MCF-7 cells was
performed. RT-qPCR analysis revealed that the expression of RAD54B
was inhibited in MCF-7 cells treated with 10 µM Japonicone A for 24
h (Fig. 7A). In addition, the
proportion of Ki-67+ MCF-7 cells, which was the
indicator for cell proliferation used in the present study, was
reduced (Fig. 7B). The viability of
MCF-7 cells was determined by MTT assay (39); the results demonstrated that cell
viability was decreased following treatment with10 µM Japonicone A
(Fig. 7C).
Discussion
Breast cancer is a major cause of mortality in
females. Despite research on breast cancer treatment in the past
decades, the currently available therapeutic strategies are still
inadequate, especially for aggressive breast cancer (40). This is partly due to the lack of
knowledge about the molecular pathogenesis of the disease.
Therefore, exploring the molecular mechanisms and identifying
biomarkers for breast cancer may provide more effective
target-specific or personalized therapeutic strategies. In the
current study, based on the data from the GSE20711 dataset, a
sequential bioinformatics analysis was performed, and three genes
that may have impact on the overall survival of patients with
breast cancer were identified. One of the three genes, RAD54B, may
be a useful prognostic factor for breast cancer.
RAD54B belongs to the Snf2 superfamily and maps to
human chromosome 8q21.3-q22 (41).
RAD54B was initially identified as a homolog of RAD54, which serves
a central role in homologous recombination and the DNA repair
process (41–43). RAD54B also functions as a scaffold
for p53 degradation in response to DNA damage, thus regulating the
cell fate between cell cycle arrest and progression (37). As a result, constitutive upregulation
of RAD54B promotes genomic instability; this has been observed in
tumors, such as hepatic carcinoma (44). A recent study has demonstrated that
inhibition of RAD54B significantly reduced the proliferation and
colony formation of hepatoma cells (45). In the present study, RAD54B was
upregulated in breast cancer cells, and its expression level was
associated with the poor overall survival of patients with breast
cancer. In addition, when combining RAD54B with patient TNM stage
to construct a model, the model could be used to predict the
prognosis of patients more accurately than TNM stage alone, which
may be beneficial to the development of personalized treatments for
patients. The results of the GO, KEGG and GSEA analyses
demonstrated that RAD54B was primarily involved in the cell cycle
regulation of breast cancer cells, and the in vitro
inhibitory assay demonstrated that reduced expression of RAD54B
significantly inhibited the proliferation of MCF-7 cells.
Therefore, RAD54B may be used as a potential therapeutic target for
breast cancer treatment.
Traditional Chinese medicine has been used for the
prevention and treatment of diseases for centuries. Following the
development of modern pharmacognosy, various biologically active
natural compounds in traditional Chinese medicine, such as
berberine and artemisinin, have been identified to exhibit
therapeutic efficacy with minimal adverse effects, which provides
new sources and platforms for developing first-line drugs (46–48).
Thus, in combination with the aforementioned bioinformatics
analysis, the data from the GSE85871 dataset, which includes the
gene expression profiles of MCF-7 cells following treatment with
102 molecules from traditional Chinese medicine, were analyzed. The
results demonstrated that Japonicone A effectively inhibited RAD54B
expression in MCF-7 cells. Japonicone A is a component in the
aerial parts of Inula japonica, which was traditionally used
to treat bronchitis, digestive disorders, diabetes and general
inflammation (49). Previous studies
have demonstrated that Japonicone A may suppress the growth of
Burkitt lymphoma cells via the NF-κB pathway (31) and the growth of non-small cell lung
cancer cells via mitochondria-mediated pathways (50). In the present study, the
bioinformatics analysis and the in vitro inhibitory assay
revealed that Japonicone A may inhibit the expression of RAD54B in
breast cancer cells, resulting in the inhibition of cell
proliferation. In vivo experiments are required to provide
stronger evidence; these will be performed in our future studies to
explore the curative effect of Japonicone A on breast cancer.
In conclusion, the present study identified RAD54B
as a prognostic factor and a potential therapeutic target for
breast cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The current study was supported by the Special
Research Fund of Chongqing Medical and Pharmaceutical College
(grant no. ygz 2016103).
Availability of data and materials
The datasets analyzed during the current study are
available in the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo). All other data
generated or analyzed during this study are included in this
published article.
Authors' contributions
JF and JH performed the experiments and analyzed the
data. YX conceived and designed the experiments, supervised the
study and wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
WGCNA
|
weighted gene co-expression network
analysis
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
BP
|
biological process
|
|
TCGA
|
The Cancer Genome Atlas
|
|
MF
|
molecular function
|
|
CC
|
cellular component
|
|
DEGs
|
differentially expressed genes
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harbeck N and Gnant M: Breast cancer.
Lancet. 389:1134–1150. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Redig AJ and McAllister SS: Breast cancer
as a systemic disease: A view of metastasis. J Intern Med.
274:113–126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stratton MR, Campbell PJ and Futreal PA:
The cancer genome. Nature. 458:719–724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Balmain A, Gray J and Ponder B: The
genetics and genomics of cancer. Nat Genet. 33 (Suppl):S238–S244.
2003. View
Article : Google Scholar
|
|
6
|
Nguyen DX and Massague J: Genetic
determinants of cancer metastasis. Nat Rev Genet. 8:341–352. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Irish JM, Kotecha N and Nolan GP: Mapping
normal and cancer cell signalling networks: Towards single-cell
proteomics. Nat Rev Cancer. 6:146–155. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Papaleo E, Gromova I and Gromov P: Gaining
insights into cancer biology through exploration of the cancer
secretome using proteomic and bioinformatic tools. Expert Rev
Proteomics. 14:1021–1035. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aftab A, Shahzad S, Hussain HMJ, Khan R,
Irum S and Tabassum S: CDKN2A/P16INK4A variants association with
breast cancer and their in-silico analysis. Breast Cancer.
26:11–28. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klahan S, Wong HS, Tu SH, Chou WH, Zhang
YF, Ho TF, Liu CY, Yih SY, Lu HF, Chen SC, et al: Identification of
genes and pathways related to lymphovascular invasion in breast
cancer patients: A bioinformatics analysis of gene expression
profiles. Tumour Biol. 39:10104283177055732017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang J, Kong D, Cui Q, Wang K, Zhang D,
Gong Y and Wu G: Prognostic genes of breast cancer identified by
gene co-expression network analysis. Front Oncol. 8:3742018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheng D, He H and Liang B: A
three-microRNA signature predicts clinical outcome in breast cancer
patients. Eur Rev Med Pharmacol Sci. 22:6386–6395. 2018.PubMed/NCBI
|
|
13
|
Dedeurwaerder S, Desmedt C, Calonne E,
Singhal SK, Haibe-Kains B, Defrance M, Michiels S, Volkmar M,
Deplus R, Luciani J, et al: DNA methylation profiling reveals a
predominant immune component in breast cancers. EMBO Mol Med.
3:726–741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lv C, Wu X, Wang X, Su J, Zeng H, Zhao J,
Lin S, Liu R, Li H, Li X and Zhang W: The gene expression profiles
in response to 102 traditional Chinese medicine (TCM) components: A
general template for research on TCMs. Sci Rep. 7:3522017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mounir M, Lucchetta M, Silva TC, Olsen C,
Bontempi G, Chen X, Noushmehr H, Colaprico A and Papaleo E: New
functionalities in the TCGAbiolinks package for the study and
integration of cancer data from GDC and GTEx. PLoS Comput Biol.
15:e1006701. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wen Q, Yang Y, Chen XH, Pan XD, Han Q,
Wang D, Dang Y, Li XH, Yan J and Zhou JH: Competing endogenous RNA
screening based on long noncoding RNA-messenger RNA co-expression
profile in Hepatitis B virus-associated hepatocarcinogenesis. J
Tradit Chin Med. 37:510–521. 2017. View Article : Google Scholar
|
|
18
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan L, Zeng G, Chen L, Wang G and Wang X,
Cao X, Lu M, Liu X, Qian G, Xiao Y and Wang X: Identification of
key genes and pathways in human clear cell renal cell carcinoma
(ccRCC) by co-expression analysis. Int J Biol Sci. 14:266–279.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang T, Wu B, Zhang X, Zhang M, Zhang S,
Huang W, Liu T, Yu W, Li J and Yu X: Identification of gene
coexpression modules, hub genes, and pathways related to spinal
cord injury using integrated bioinformatics methods. J Cell
Biochem. Jan 17–2019.(Epub ahead of print). doi:
10.1002/jcb.27908.
|
|
21
|
Yu G, Wang LG, Han Y and He QY:
ClusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Walter W, Sánchez-Cabo F and Ricote M:
GOplot: An R package for visually combining expression data with
functional analysis. Bioinformatics. 31:2912–2914. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mo XG, Liu W, Yang Y, Imani S, Lu S, Dan
G, Nie X, Yan J, Zhan R, Li X, et al: NCF2, MYO1F, S1PR4, and FCN1
as potential noninvasive diagnostic biomarkers in patients with
obstructive coronary artery: A weighted gene co-expression network
analysis. J Cell Biochem. Jan 17–2019.(Epub ahead of print).
View Article : Google Scholar
|
|
24
|
Yang Y, Lu Q, Shao X, Mo B, Nie X, Liu W,
Chen X, Tang Y, Deng Y and Yan J: Development of a three-gene
prognostic signature for hepatitis B virus associated
hepatocellular carcinoma based on integrated transcriptomic
analysis. J Cancer. 9:1989–2002. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang N, Guo H, Dong Z, Chen Q, Zhang X,
Shen W, Bao Y and Wang X: Establishment and validation of a
7-microRNA prognostic signature for non-small cell lung cancer.
Cancer Manag Res. 10:3463–3471. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Colwill K; Renewable Protein Binder
Working Group, ; Gräslund S: A roadmap to generate renewable
protein binders to the human proteome. Nat Methods. 8:551–558.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu H, Zhang Y, Qi L, Ding L, Jiang H and
Yu H: NFIX Circular RNA promotes glioma progression by regulating
miR-34a-5p via notch signaling pathway. Front Mol Neurosci.
11:2252018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu Z, Qin J, Zhang H, Wang D, Hua Y, Ding
J, Shan L, Jin H, Zhang J and Zhang W: Japonicone A antagonizes the
activity of TNF-α by directly targeting this cytokine and
selectively disrupting its interaction with TNF receptor-1. Biochem
Pharmacol. 84:1482–1491. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qin JJ, Jin HZ, Fu JJ, Hu XJ, Wang Y, Yan
SK and Zhang WD: Japonicones A-D, bioactive dimeric sesquiterpenes
from Inula japonica Thunb. Bioorg Med Chem Lett. 19:710–713.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li X, Yang X, Liu Y, Gong N, Yao W, Chen
P, Qin J, Jin H, Li J, Chu R, et al: Japonicone A suppresses growth
of Burkitt lymphoma cells through its effect on NF-κB. Clin Cancer
Res. 19:2917–2928. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Du Y, Gong J, Tian X, Yan X, Guo T, Huang
M, Zhang B, Hu X, Liu H, Wang Y, et al: Japonicone A inhibits the
growth of non-small cell lung cancer cells via
mitochondria-mediated pathways. Tumour Biol. 36:7473–7482. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Patrick J: survivalROC: Time-dependent ROC
curve estimation from censored survival data. https://cran.r-project.org/web/packages/survivalROC/index.htmlMay.
2019
|
|
35
|
Vickers AJ and Elkin EB: Decision curve
analysis: A novel method for evaluating prediction models. Med
Decis Making. 26:565–574. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Marshall B: rmda: Risk model decision
analysis. https://cran.r-project.org/web/packages/rmda/index.htmlMarch
20–2018
|
|
37
|
Uhlen M, Zhang C, Lee S, Sjöstedt E,
Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et
al: A pathology atlas of the human cancer transcriptome. Science.
357(pii): eaan25072017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pontén F, Jirström K and Uhlen M: The
human protein atlas-a tool for pathology. J Pathol. 216:387–393.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming
QL, Huang C, Li P and Gao N: A novel autophagy/mitophagy inhibitor
liensinine sensitizes breast cancer cells to chemotherapy through
DNM1L-mediated mitochondrial fission. Autophagy. 11:1259–1279.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nagini S: Breast cancer: Current molecular
therapeutic targets and new players. Anticancer Agents Med Chem.
17:152–163. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hiramoto T, Nakanishi T, Sumiyoshi T,
Fukuda T, Matsuura S, Tauchi H, Komatsu K, Shibasaki Y, Inui H,
Watatani M, et al: Mutations of a novel human RAD54 homologue,
RAD54B, in primary cancer. Oncogene. 18:3422–3426. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Miyagawa K, Tsuruga T, Kinomura A, Usui K,
Katsura M, Tashiro S, Mishima H and Tanaka K: A role for RAD54B in
homologous recombination in human cells. EMBO J. 21:175–180. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wesoly J, Agarwal S, Sigurdsson S, Bussen
W, Van Komen S, Qin J, van Steeg H, van Benthem J, Wassenaar E,
Baarends WM, et al: Differential contributions of mammalian Rad54
paralogs to recombination, DNA damage repair, and meiosis. Mol Cell
Biol. 26:976–989. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yasuhara T, Suzuki T, Katsura M and
Miyagawa K: Rad54B serves as a scaffold in the DNA damage response
that limits checkpoint strength. Nat Commun. 5:54262014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang R, Li Y, Chen Y and Wang L, Wu Q, Guo
Y, Li Y, Liu J and Wang L: Inhibition of RAD54B suppresses
proliferation and promotes apoptosis in hepatoma cells. Oncol Rep.
40:1233–1242. 2018.PubMed/NCBI
|
|
46
|
Mathur S and Hoskins C: Drug development:
Lessons from nature. Biomed Rep. 6:612–614. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Clardy J and Walsh C: Lessons from natural
molecules. Nature. 432:829–837. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ertl P and Schuffenhauer A:
Cheminformatics analysis of natural products: Lessons from nature
inspiring the design of new drugs. Prog Drug Res. 66:217, 219–235.
2008.
|
|
49
|
Qin JJ, Jin HZ, Zhu JX, Fu JJ, Hu XJ, Liu
XH, Zhu Y, Yan SK and Zhang WD: Japonicones E-L, dimeric
sesquiterpene lactones from Inula japonica Thunb. Planta
Med. 76:278–283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
West AP, Khoury-Hanold W, Staron M, Tal
MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff
DA, et al: Mitochondrial DNA stress primes the antiviral innate
immune response. Nature. 520:553–557. 2015. View Article : Google Scholar : PubMed/NCBI
|