Introduction
Breast cancer is one of the most common malignancies
in women in both developed and developing countries, and each year
>1,300,000 cases of breast cancer are reported globally
(1). For decades,
clinicopathological features have been utilized to evaluate the
prognosis of patients with breast cancer, including tumor size,
clinical stage, intrinsic subtype and lymph node status (2–5).
However, these factors are limited in their prognostic capability
and are only useful in a number of patients (6). It is widely accepted that the
underlying molecular mechanisms of breast cancer are complex and
may involve the alterations of specific genomic regions, as well as
epigenetic modifications in mammary epithelial cells (7,8). To the
best of our knowledge, patients with similar disease
characteristics, who have received similar treatments may present
with markedly different clinical outcomes. Therefore, accurately
predicting patient outcome and subsequently selecting the
appropriate treatment, reducing morbidity and prolonging survival
time are of great clinical importance.
Long non-coding RNAs (lncRNAs) are a class of RNAs
with >200 nucleotides and no known protein coding capability
(9,10). lncRNAs have been implicated in a wide
range of biological processes, including tumor-suppressor
modulation, RNA-RNA interactions, and epigenetic and
post-transcriptional regulation (11–14). As
additional biological functions of lncRNAs are identified, they
have become the focus of an increasing number of studies. These
studies have revealed that lncRNAs serve a role in carcinogenesis
and possess specific expression patterns in cancer (15–17). To
date, several lncRNAs have been regarded as diagnostic and/or
prognostic biomarkers for specific malignancies, such as lncRNA HOX
transcript antisense RNA (HOTAIR), which is overexpressed in
breast cancer, thus promoting cancer cell invasion and metastasis
by altering the methylation and gene expression of histone H3K27
via polycomb repressive complex 2 (18). Another lncRNA, metastasis-associated
lung adenocarcinoma transcript 1, was first identified in non-small
cell lung cancer (NSCLC); its high level of expression was strongly
correlated with an increased risk of NSCLC, and was associated with
metastasis and poor patient outcome (19).
However, the predictive ability of single lncRNAs is
still unsatisfactory, resulting in high numbers of both false
positive and negative results (20).
Therefore, the present study aimed to identify a four-lncRNA
signature able to predict the overall survival (OS) rate of
patients with breast cancer, and to validate the prognostic value
of the identified lncRNAs using high-throughput sequencing data
from The Cancer Genome Atlas (TCGA) database.
Materials and methods
Breast cancer gene expression data
from TCGA and Gene Expression Omnibus (GEO) databases
Breast cancer gene expression data, including coding
and non-coding RNA sequence data, were acquired from TCGA
(https://cancergenome.nih.gov/) together
with the corresponding clinical information. Until 2017, 1,098
breast cancer samples were available from TCGA, though in the
present study only those including patient survival status were
selected (n=768); this enabled the determination of any association
between the expression of lncRNAs of the lncRNA-expression
signature and the corresponding OS time for breast cancer. These
768 breast cancer samples were divided equally into a training set
(to identify the gene expression signature) and a validation set
(to validate the gene expression signature). To confirm the
expression levels of the differentially expressed genes, the gene
expression dataset GSE5764 (21),
containing 10 breast cancer tissue samples and 20 non-cancerous
samples, was downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/; Affymetrix GPL570
platform, Affymetrix Human Genome U133 Plus 2.0 Array; Affymetrix;
Thermo Fisher Scientific, Inc.).
Identification of lncRNAs
RNA genes downloaded from TCGA were compared with
published lncRNAs from the MiTranscriptome database (http://mitranscriptome.org/). Potential lncRNAs were
identified as transcriptome sequences that were mapped to
corresponding lncRNAs, rather than any protein-coding region, and
were not identified as protein-coding genes in the National Center
for Biotechnology Information database (https://www.ncbi.nlm.nih.gov/).
Gene expression data analysis
Raw read counts of the transcriptomic data from TCGA
were normalized using the quartile normalization method and
logarithmically transformed to a normal distribution. The
Bioconductor package DESeq2 (https://www.bioconductor.org/packages/release/bioc/html/DESeq2.html,
version 1.24.0) was used to perform the normalization and identify
differentially expressed genes in breast cancer samples compared
with adjacent normal tissues, with an adjusted P<0.05 and an
absolute log2-based fold-change >0.5. For gene expression data
from GEO, the R package limma (https://www.bioconductor.org/packages/release/bioc/html/limma.html,
version 3.40.6) was used to conduct differential expression
analysis.
Establishment of a prognostic
signature
To establish a prognostic signature for breast
cancer, a two-step method was employed using the R package SIS
(https://CRAN.R-project.org/package=SIS, version 0.8–6)
for sure independence screening. Firstly, univariate Cox regression
analysis was performed to identify survival-associated genes.
Secondly, SIS (based on the least absolute shrinkage and selection
operator, Cox-penalized regression model) was used to identify
important variables and construct multi-gene-based prognostic
signatures for OS rate prediction.
Guilt by association analysis
To identify genes that correlated with the four
lncRNAs of the prognostic signature, data from TCGA were used to
evaluate the pairwise Pearson's correlation between the expression
levels of the target lncRNAs. Only associated genes with an
absolute r≥0.3 and a significant correlation (P<0.05) were
retained. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG; http://www.genome.jp/kegg/) pathway analyses were
performed using the Database for Annotation, Visualization and
Integrated Discovery (DAVID; http://david.ncifcrf.gov/).
Statistical analysis
Kaplan-Meier analysis was used to estimate the
performance of the prognostic signatures, and log-rank test was
performed to evaluate statistical significance. A risk score was
calculated for each patient according to the formula of the
four-lncRNA signature, and patients were divided into high- and
low-risk groups using the median score as a cut-off. Receiver
operating characteristic (ROC) analysis was used to evaluate the
sensitivity and specificity of the four-lncRNA signature and other
biomarkers, including TP53, MKI67, ESR1, PGR, ERBB2 and HOTAIR. All
statistical analyses were conducted using R 3.5.2 (https://www.r-project.org/), and P<0.05 was
considered to indicate a statistically significant difference.
Results
Patient characteristics
All 768 patients were diagnosed with breast cancer
based on clinicopathological evaluation. The clinical stage and
histological subtype were determined using the
Tumor-Node-Metastasis staging (22)
and immunohistochemical molecular typing methods, respectively.
According to the data, the estrogen receptor (ER), progesterone
receptor (PR) and human epidermal growth factor receptor 2 (HER2)
status of each patient was indicated as positive, negative or
indeterminate. In addition, the ranges of the OS and relapse-free
survival times were 1–8,605 and 1–8,556 days, respectively. Patient
characteristics are displayed in Table
I.
 | Table I.Demographic characteristics of the
768 patients with breast cancer included in the present study. |
Table I.
Demographic characteristics of the
768 patients with breast cancer included in the present study.
|
Characteristics | Training set
(n=384) | Validation set
(n=384) | Total set (n=768),
% |
|---|
| Sex |
|
|
|
|
Male | 4 | 3 | 7 (0.91) |
|
Female | 380 | 381 | 761 (99.09) |
| TNM stage (22) |
|
|
|
| Stage
I | 66 | 67 | 133 (17.32) |
| Stage
II | 226 | 223 | 449 (58.46) |
| Stage
III | 85 | 86 | 171 (22.27) |
| Stage
IV | 7 | 8 | 15 (1.95) |
| ER status |
|
|
|
|
Negative | 90 | 92 | 182 (23.70) |
|
Positive | 291 | 290 | 581 (75.65) |
|
Indeterminate | 3 | 2 | 5 (0.65) |
| PR status |
|
|
|
|
Negative | 125 | 128 | 253 (32.94) |
|
Positive | 256 | 254 | 510 (66.41) |
|
Indeterminate | 3 | 2 | 5 (0.65) |
| HER2 status |
|
|
|
|
Negative | 228 | 231 | 459 (59.77) |
|
Positive | 92 | 87 | 179 (23.31) |
|
Indeterminate | 64 | 66 | 130 (16.93) |
| Vital status |
|
|
|
|
Alive | 326 | 326 | 652 (84.90) |
|
Deceased | 58 | 58 | 116 (15.10) |
| OS time
(range), days | 7-8,556 | 1-8,605 | 1-8,605 |
| RFS status |
|
|
|
|
Relapsed | 295 | 288 | 583 (75.91) |
|
Relapse-free | 89 | 96 | 185 (24.09) |
| RFS
time (range), days | 7-8,556 | 1-8,391 | 1-8,556 |
Differential expression analysis and
determination of the four-lncRNA signature in the training set
Differential expression analysis was employed to
select differentially expressed RNAs in normal and cancerous
tissues; a total of 8,854 upregulated and 5,939 downregulated RNAs
were identified. Subsequently, all possible combinations of four
lncRNAs were analyzed and compared using the two-step Cox
regression method. A total of 7 models consisting of four lncRNAs
were identified (Table SI); among
these candidates, one model was identified as the most suitable for
predicting the OS of patients with breast cancer. Patients were
divided into high- and low-risk groups using the median risk score
as a cut-off, with the risk score calculated as follows: Risk
score=(−0.015× expression value of PVT1) + (−0.193×
expression value of MAPT-AS1) + (−0.116× expression value of
LINC00667) + (0.098× expression value of LINC00938).
The coefficients of this formula were derived from multivariate Cox
regression analysis (Table SI).
Kaplan-Meier analysis was also performed to determine the
association between the expression levels of the four-lncRNA
signature and patient OS. Compared with those of the low-risk
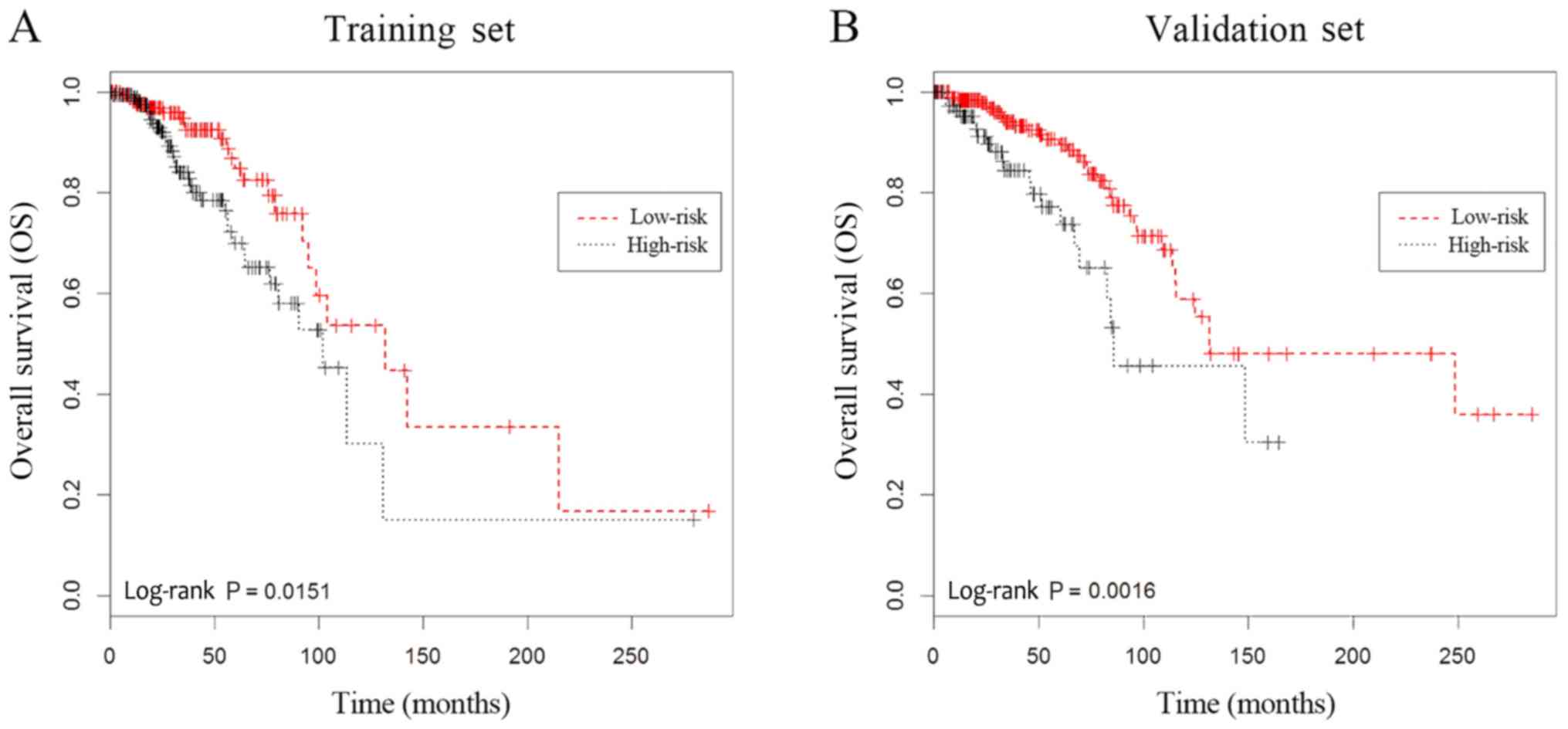
group, high-risk patients exhibited significantly poorer OS rate
(log-rank P=0.0151; Fig. 1A).
Validation of the four-lncRNA
signature in the validation set
To confirm the predictive capacity of the
four-lncRNA signature identified in the training set, the
equivalent analyses were also performed in the validation set.
Patients were divided into low- and high-risk groups, and the
differences between patient OS rates were compared using
Kaplan-Meier analysis. Patients in the high-risk group possessed
significantly lower OS rate than those of patients in the low-risk
group (log-rank P=0.0016; Fig. 1B),
which was consistent with the findings from the training set.
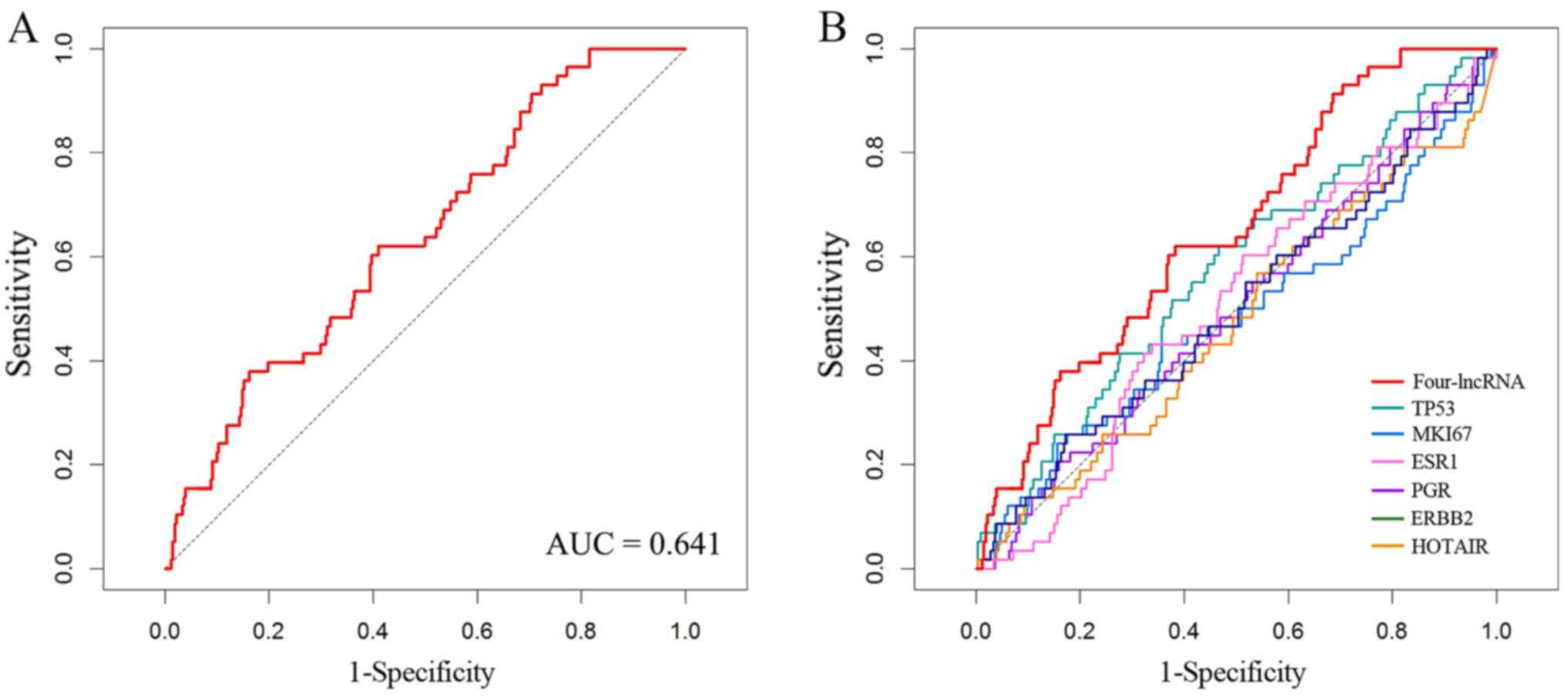
Furthermore, ROC analysis was performed to evaluate the sensitivity
and specificity of survival prediction; the area under the curve
(AUC) was 0.641 (Fig. 2A),
indicating that the four-lncRNA signature was able to accurately
predict the survival of patients with breast cancer.
Four-lncRNA signature in different
clinical stages and molecular subtypes
ROC analyses were performed to investigate whether
the four-lncRNA signature was applicable to different breast cancer
stages and molecular subtypes. In stages I–IV, the AUC values were
0.595, 0.687, 0.634 and 0.645, respectively (Fig. S1), indicating that the four-lncRNA
signature was able to predict the survival of patients at different
clinical stages of breast cancer. Regarding subtype, the AUC values
in the four molecular subgroups were 0.637, 0.654, 0.688 and 0.613,
respectively (Fig. S2), suggesting
that the four-lncRNA signature served as a prognostic indicator for
patients with different breast cancer subtypes.
Performance of the four-lncRNA
signature compared with that of known biomarkers and individual
lncRNAs
For further clarification, the performance of the
four-lncRNA signature was compared with that of several known
breast cancer biomarkers, including TP53, MKI67, ESR1, PGR,
ERBB2 and HOTAIR, using ROC and Kaplan-Meier analyses.
The sensitivity and specificity of these six known biomarkers are
displayed in Fig. 2B. The AUC values
of TP53, MKI67, ESR1, PGR, ERBB2 and HOTAIR were
0.574, 0.483, 0.510, 0.501, 0.501 and 0.473 (data not shown),
respectively, while the AUC value (0.641) of the four-lncRNA
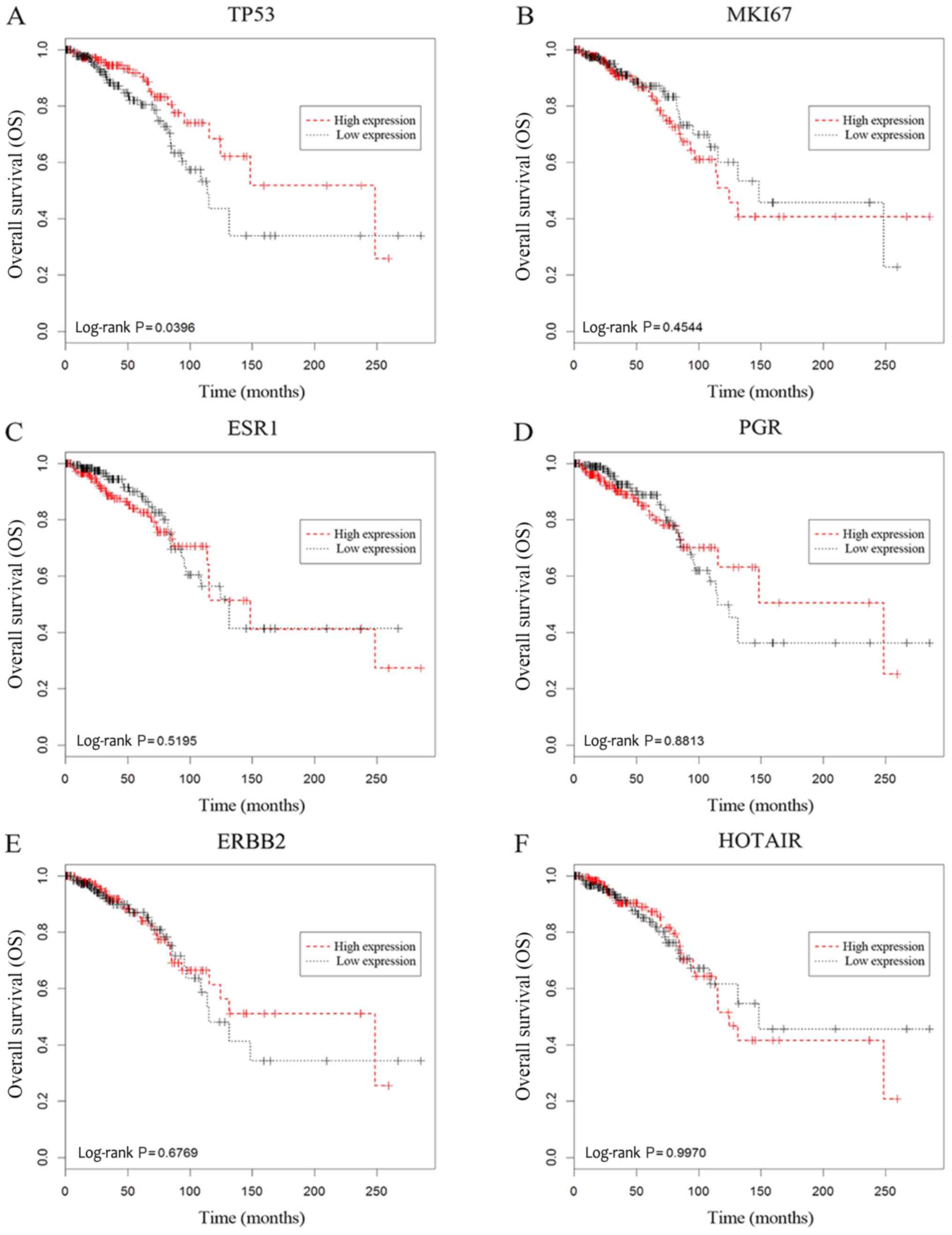
signature was greater. Kaplan-Meier analysis revealed that only
TP53 was significantly associated with patient OS (Fig. 3A; log-rank P=0.0396), while the other
selected biomarkers were not (Fig.
3B-F). Moreover, the four lncRNAs from the identified model
were also evaluated. ROC curve analysis generated AUC values for
PVT1, MAPT-AS1, LINC00667 and LINC00938 as 0.532,
0.553, 0.550 and 0.480, respectively (Fig. S3), which were smaller than that of
the four-lncRNA signature. In addition, the results of Kaplan-Meier
analysis indicated that the differential expression of these
lncRNAs was not significantly associated with the OS rate of
patients with breast cancer (log-rank P>0.05; Fig. S4).
Relative expression levels and
potential biological functions of the four lncRNAs of the lncRNA
signature
To further investigate the potential functions of
the four lncRNAs of the signature, gene expression data from the
GSE5764 dataset were downloaded from the GEO database, and
differential expression analysis was performed. The fold-change
values of PVT1, MAPT-AS1, LINC00667 and LINC00938
were 2.031, 3.057, 1.579, 0.455, respectively. This result
indicated that PVT1, MAPT-AS1 and LINC00667 were
upregulated in breast cancer tissues, and that LINC00938 was
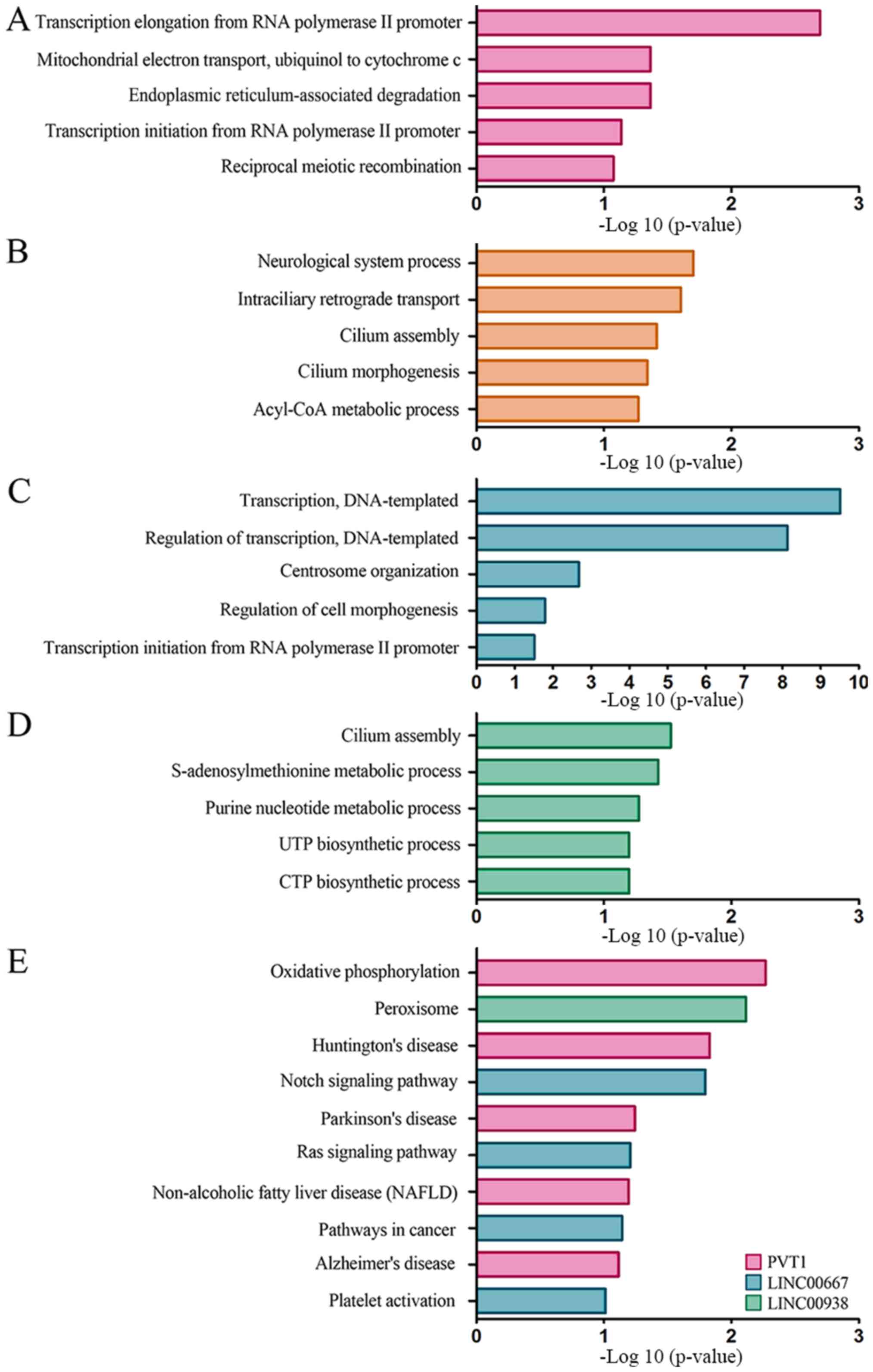
downregulated. GO enrichment and KEGG pathway analyses were
conducted. According to the results of GO analysis, the primary
PVT1-associated functions were ‘transcription elongation’,
‘mitochondrial electron transport’ and ‘endoplasmic
reticulum-associated degradation’ (Fig.
4A). MAPT-AS1 was associated with ‘cilium morphogenesis’
and ‘cilium assembly’, ‘intraciliary retrograde transport’ and
‘neurological system process’ (Fig.
4B). LINC00667 was associated with ‘regulation of
transcription, DNA-templated’, ‘centrosome organization’ and
‘regulation of cell morphogenesis’ (Fig.
4C), and LINC00983 was associated with biological
processes involved in ‘cilium assembly’ and ‘S-adenosylmethionine
metabolic process’ (Fig. 4D).
Moreover, KEGG analysis also indicated several biological processes
and pathways that potentially associated with the signature
lncRNAs, including ‘Notch signaling pathway’, ‘oxidative
phosphorylation’ and ‘Huntington's disease’ (Fig. 4E).
Discussion
In the present study, the RNA expression data of 768
patients from TCGA were analyzed, and 14,793 RNAs that were
differently expressed in normal vs. cancerous tissues were
selected. Following multivariate Cox regression analysis (data not
shown), a model consisting of four lncRNAs (PVT1, MAPT-AS1,
LINC00667 and LINC00938) was determined to predict the
survival of patients with breast cancer, while Kaplan-Meier and ROC
analyses confirmed that this model was able to predict OS to an
acceptable degree of specificity and sensitivity.
In previous years, an increasing number of lncRNAs
have been identified and are widely considered to be a novel class
of gene regulators in various types of cancer (11,23).
With the development of high-throughput sequencing, a gradually
increasing number of sequencing data have been used to study
cancer-associated lncRNAs. Using transcriptome sequencing, Zou
et al (24) identified two
novel lncRNAs, LCE5A-1 and KCTD6-3, that are associated with head
and neck carcinogenesis, and Ylipää et al (25) established prostate cancer associated
transcript-1 as a novel oncogenic lncRNA, confirming its
association with castration-resistant prostate cancers. To date,
aberrant lncRNA expression levels have been observed in numerous
cancer types, making these lncRNAs novel and reliable biomarkers
for cancer diagnosis and prognosis (26,27). The
present study profiled the expression of lncRNAs associated with
breast cancer prognosis using next-generation sequencing data, and
determined a set of four lncRNAs that, when combined, may be used
as a potential biomarker for the prognosis of breast cancer.
Previous studies have identified various biomarkers
with prognostic value in breast cancer. TP53 is a recognized
tumor-suppressor gene, the encoded protein of which responds to a
diverse range of cellular stimuli to regulate the expression of
target genes. Mutations in these genes are associated with a
variety of human cancers, such as gastric cancer, colorectal cancer
and breast cancer (28).
MKI67 is a nucleoprotein-coding gene; its expression
product, Ki-67, has been identified as a biomarker of cell
proliferation, which is regarded as a predictor of patient outcome
(29). Additionally, lncRNA
HOTAIR has been confirmed to promote breast cancer invasion
and metastasis (30). The roles of
ESR1, PGR and ERBB2, also known as ER, PR and
HER2, respectively, have been widely recognized for breast
cancer molecular typing and prognostics (31). In the present study, the prognostic
value of several commonly used clinical prognostic molecular
indicators was evaluated using ROC analysis and was compared with
that of the four-lncRNA signature. ESR1, PGR, ERBB2, MKI67
and TP53 were all included in the analysis, and the result
showed that the AUC value of the four-lncRNA signature was greater
than that of the aforementioned biomarkers, which confirmed the
four lncRNA signature as a potentially superior prognostic
predictor. Additionally, according to the results of Kaplan-Meier
analysis, the four individual lncRNAs of the signature were not
adequate as independent prognostic predictors. This showed that,
although a particular lncRNA may be associated with breast cancer,
it may not reliably predict patient survival. However, the
combination of these four lncRNAs was able to predict the outcomes
of patients with breast cancer with satisfactory sensitivity and
specificity.
lncRNAs are expressed at numerous cellular locations
and fulfill a wide variety of regulatory roles at almost all stages
of gene expression (32). Although
specific lncRNAs have been implicated in a number of biological
processes, the majority of their functions are not fully
understood. Of the four lncRNAs of the signature, PVT1 is
located on chromosome 8q24.21; a previous study has demonstrated
that supernumerary copies of this chromosomal region are associated
with various types of cancer, including breast and ovarian cancer,
acute myeloid leukemia and Hodgkin's lymphoma (33). MAPT-AS1 is an 840-bp lncRNA
transcribed from the anti-sense strand of the MAPT promoter,
and has been identified as a potential epigenetic regulator of
MAPT expression in Parkinson's disease (34). However, to the best of our knowledge,
there have been no reports of the association between
MAPT-AS1 and tumorigenesis to date. Furthermore, the
biological functions of LINC00667 and LINC00938
remain to be elucidated. To date, to the best of our knowledge,
there is no indication as to why the four-lncRNA signature may
serve as a prognostic marker. lncRNAs function in complex ways, and
the potential association between these molecules are crucial to
understanding their underlying mechanisms of action.
In the present study, differential expression
analysis was performed on gene expression data from TCGA and GEO
databases. Compared with non-cancerous samples, PVT1,
MAPT-AS1 and LINC00667 were upregulated, while
LINC00938 was downregulated in breast cancer tissues.
Therefore, PVT1, MAPT-AS1 and LINC00667 were
considered to be candidate oncogenes, while LINC00938 may
serve as a cancer-suppressor gene. Moreover, GO and KEGG analyses
were employed to investigate the potential functions of the four
lncRNAs. The results showed that PVT1 and LINC00667
were associated with transcription regulation, while MAPT-AS1,
LINC00667 and LINC00938 were associated with cellular
mitosis, and PVT1 was associated with mitochondrial energy
metabolism. These fundamental biological processes are essentially
involved in tumorigenesis and cancer progression (35,36).
Additionally, KEGG analysis indicated that LINC00667 was
associated with the ‘Notch signaling pathway’, and previous study
has demonstrated that dysregulated Notch signaling is oncogenic,
inhibits apoptosis and promotes cell survival (37).
In conclusion, the present study identified a
four-lncRNA signature with predictive value for breast cancer
prognosis, which may be used as a novel biomarker for the prognosis
of patients with breast cancer. Although the signature may
contribute to the prognostic evaluation of breast cancer, one of
the limitations of the present study is that it was a
bioinformatics analysis, and therefore further studies are required
using clinical samples, in order to evaluate the identified
four-lncRNA signature, in addition to determining the functional
mechanisms of these lncRNAs.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The breast cancer gene expression data, together
with the corresponding clinical information are available from TCGA
(https://cancergenome.nih.gov/). The
dataset GSE5764 was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The data of
associated genes and pathways for GO and KEGG analyses are
available in the DAVID database (https://david.ncifcrf.gov/).
Authors' contributions
JW and MZ designed the study and conducted
bioinformatic analysis. QL, HH, DP, CS and MZ sorted the data and
participated in the statistical analysis. MZ drafted the
manuscript. DP and CS participated in drafting the manuscript and
providing research guidance. JW reviewed and edited the manuscript.
All authors read and approved the final manuscript. All authors
agreed to be accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
lncRNA
|
long non-coding RNA
|
|
NSCLC
|
non-small cell lung cancer
|
|
OS
|
overall survival
|
|
ROC
|
receiver operating characteristic
|
|
SIS
|
sure independence screening
|
|
TCGA
|
The Cancer Genome Atlas
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Michaelson JS, Silverstein M, Wyatt J,
Weber G, Moore R, Halpern E, Kopans DB and Hughes K: Predicting the
survival of patients with breast carcinoma using tumor size.
Cancer. 95:713–723. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rakha EA, El-Sayed ME, Menon S, Green AR,
Lee AH and Ellis IO: Histologic grading is an independent
prognostic factor in invasive lobular carcinoma of the breast.
Breast Cancer Res Treat. 111:121–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shokouh TZ, Ezatollah A and Barand P:
Interrelationships Between Ki67, HER2/neu, p53, ER, and PR Status
and their associations with tumor grade and lymph node involvement
in breast carcinoma subtypes: Retrospective-observational
analytical study. Medicine (Baltimore). 94:e13592015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma J, Luo DX, Huang C, Shen Y, Bu Y,
Markwell S, Gao J, Liu J, Zu X, Cao Z, et al: AKR1B10
overexpression in breast cancer: Association with tumor size, lymph
node metastasis and patient survival and its potential as a novel
serum marker. Int J Cancer. 131:E862–E871. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Crabb SJ, Bajdik CD, Leung S, Speers CH,
Kennecke H, Huntsman DG and Gelmon KA: Can clinically relevant
prognostic subsets of breast cancer patients with four or more
involved axillary lymph nodes be identified through
immunohistochemical biomarkers? A tissue microarray feasibility
study. Breast Cancer Res. 10:R62008. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
M Braden A, V Stankowski R, M Engel J and
A Onitilo A: Breast cancer biomarkers: Risk assessment, diagnosis,
prognosis, prediction of treatment efficacy and toxicity, and
recurrence. Curr Pharm Des. 20:4879–4898. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nagini S: Breast Cancer: Current molecular
therapeutic targets and new players. Anticancer Agents Med Chem.
17:152–163. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
ENCODE Project Consortium, ; Birney E,
Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH,
Weng Z, Snyder M, Dermitzakis ET, et al: Identification and
analysis of functional elements in 1% of the human genome by the
ENCODE pilot project. Nature. 447:799–816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Clark MB, Johnston RL, Inostroza-Ponta M,
Fox AH, Fortini E, Moscato P, Dinger ME and Mattick JS: Genome-wide
analysis of long noncoding RNA stability. Genome Res. 22:885–898.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Prensner JR and Chinnaiyan AM: The
emergence of lncRNAs in cancer biology. Cancer Discov. 1:391–407.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rinn JL and Chang HY: Genome regulation by
long noncoding RNAs. Annu Rev Biochem. 81:145–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nagano T and Fraser P: No-nonsense
functions for long noncoding RNAs. Cell. 145:178–181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoon JH, Abdelmohsen K, Srikantan S, Yang
X, Martindale JL, De S, Huarte M, Zhan M, Becker KG and Gorospe M:
LincRNA-p21 suppresses target mRNA translation. Mol Cell.
47:648–655. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Peng WX, Koirala P and Mo YY:
LncRNA-mediated regulation of cell signaling in cancer. Oncogene.
36:5661–5667. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bhan A, Soleimani M and Mandal SS: Long
noncoding RNA and cancer: A new paradigm. Cancer Res. 77:3965–3981.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sanchez Calle A, Kawamura Y, Yamamoto Y,
Takeshita F and Ochiya T: Emerging roles of long non-coding RNA in
cancer. Cancer Sci. 109:2093–2100. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote cancer
metastasis. Nature. 464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gutschner T, Hammerle M and Diederichs S:
MALAT1-a paradigm for long noncoding RNA function in cancer. J Mol
Med (Berl). 91:791–801. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chandra Gupta S and Nandan Tripathi Y:
Potential of long non-coding RNAs in cancer patients: From
biomarkers to therapeutic targets. Int J Cancer. 140:1955–1967.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Turashvili G, Bouchal J, Baumforth K, Wei
W, Dziechciarkova M, Ehrmann J, Klein J, Fridman E, Skarda J,
Srovnal J, et al: Novel markers for differentiation of lobular and
ductal invasive breast carcinomas by laser microdissection and
microarray analysis. BMC Cancer. 7:552007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hortobagyi GN, Edge SB and Giuliano A: New
and Important Changes in the TNM staging system for breast cancer.
Am Soc Clin Oncol Educ Book. 38:457–467. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen
X, Zhang Q, Yan G and Cui Q: LncRNA Disease: A database for
long-non-coding RNA-associated diseases. Nucleic Acids Res.
41((Database Issue)): D983–D986. 2013.PubMed/NCBI
|
|
24
|
Zou AE, Ku J, Honda TK, Yu V, Kuo SZ,
Zheng H, Xuan Y, Saad MA, Hinton A, Brumund KT, et al:
Transcriptome sequencing uncovers novel long noncoding and small
nucleolar RNAs dysregulated in head and neck squamous cell
carcinoma. RNA. 21:1122–1134. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ylipää A, Kivinummi K, Kohvakka A, Annala
M, Latonen L, Scaravilli M, Kartasalo K, Leppänen SP, Karakurt S,
Seppälä J, et al: Transcriptome sequencing reveals PCAT5 as a novel
ERG-regulated long noncoding RNA in prostate cancer. Cancer Res.
75:4026–4031. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gutschner T and Diederichs S: The
hallmarks of cancer: A long non-coding RNA point of view. RNA Biol.
9:703–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gibb EA, Brown CJ and Lam WL: The
functional role of long non-coding RNA in human carcinomas. Mol
Cancer. 10:382011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song CV, Teo SH, Taib NA and Yip CH:
Surgery for BRCA, TP53 and PALB2: A literature review.
Ecancermedicalscience. 12:8632018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sundara Rajan S, Hanby AM, Horgan K,
Thygesen HH and Speirs V: The potential utility of geminin as a
predictive biomarker in breast cancer. Breast Cancer Res Treat.
143:91–98. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sørensen KP, Thomassen M, Tan Q, Bak M,
Cold S, Burton M, Larsen MJ and Kruse TA: Long non-coding RNA
HOTAIR is an independent prognostic marker of metastasis in
estrogen receptor-positive primary breast cancer. Breast Cancer Res
Treat. 142:529–536. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang YF, Liao YY, Yang M, Peng NF, Xie SR
and Xie YF: Discordances in ER, PR and HER2 receptors between
primary and recurrent/metastatic lesions and their impact on
survival in breast cancer patients. Med Oncol. 31:2142014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tseng YY, Moriarity BS, Gong W, Akiyama R,
Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnell
TC, et al: PVT1 dependence in cancer with MYC copy-number increase.
Nature. 512:82–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coupland KG, Kim WS, Halliday GM, Hallupp
M, Dobson-Stone C and Kwok JB: Role of the long non-coding RNA
MAPT-AS1 in regulation of microtubule associated protein tau (MAPT)
expression in Parkinson's disease. PLoS One. 11:e01579242016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bradner JE, Hnisz D and Young RA:
Transcriptional Addiction in Cancer. Cell. 168:629–643. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Valcarcel-Jimenez L, Gaude E, Torrano V,
Frezza C and Carracedo A: Mitochondrial Metabolism: Yin and yang
for tumor progression. Trends Endocrinol Metab. 28:748–757. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Aithal MG and Rajeswari N: Role of Notch
signalling pathway in cancer and its association with DNA
methylation. J Genet. 92:667–675. 2013. View Article : Google Scholar : PubMed/NCBI
|