Introduction
Lung cancer is one of the most common malignancies
worldwide and is one of the most lethal types of cancer (1). Non-small cell lung cancer (NSCLC)
accounts for ~85% of lung cancer cases (2). Early-stage lung cancer is characterised
by an insufficiency of emblematic clinical manifestations, and for
the majority of patients, tumours have advanced to later stages
when diagnosed. Surgery is the first choice of treatment for
patients with resectable NSCLC; however, even if pathological grade
II or III tumours are completely removed, there remains a high risk
of recurrence within 5 years, and their respective 5-year overall
survival (OS) rates are only 56.9 and 23.6% (3). At present, prolongation of the OS of
patients with advanced lung cancer mainly relies on multiple
courses of chemotherapy. The chemotherapy regimen based on
cisplatin (CDDP) has gradually become a standard adjuvant therapy
for patients with advanced NSCLC. However, CDDP chemotherapy does
not effectively improve OS due to apparent CDDP resistance
(4). Therefore, improving the
efficacy of CDDP chemotherapy and the subsequent treatment effects
for CDDP-resistant patients are important to improving OS.
CDDP mainly destroys tumour cells by inducing
apoptosis. However, multiple solid tumour cells, including NSCLC
cells, exhibit an evident resistance to apoptosis (5). Cells can be induced to undergo various
modes of programmed cell death, and apoptosis, which was identified
early and is frequently reported on in research, is not the only
mode. Ferroptosis is a widespread and promising mode of
non-apoptotic programmed cell death. Ferroptosis is known for its
distinct iron-dependence, which is induced by intracellular lipid
reactive oxygen species (ROS) accumulation in the cytoplasm
(6). A previous study indicated that
ferroptosis is fundamentally different from apoptosis; therefore,
the anti-apoptotic pathway of tumour cells cannot protect them from
cell death mediated by ferroptosis; therefore, inducing ferroptosis
may be an ideal solution for overcoming apoptosis resistance
(7). Nevertheless, studies
concerning ferroptosis in NSCLC are exceedingly scarce.
The classical mechanism underlying CDDP-induced
apoptosis is associated with formation of a CDDP-DNA complex, which
can alter the structure of nucleotides and DNA, influencing DNA
replication and homeostasis (8).
However, CDDP can also induce cell death through increasing
intracellular ROS levels, disrupting intracellular homeostasis and
triggering oxidative stress (9).
Since generated oxidative stress serves a crucial role in
CDDP-mediated cytotoxicity, the activation of genes driven by
transcription factor NF-E2 related factor 2 (Nrf2)/antioxidant
response element (ARE) is considered a predominant reason for CDDP
failure (10). In previous years,
numerous Nrf2 downstream target genes that participate in ROS
regulation, including NAD(P)H quinone dehydrogenase 1 (NQO1)
(11), heme oxygenase 1
(HO-1) (12) and light chain
of System xc− (xCT) (13), have been demonstrated to be involved
in modulating the sensitivity of tumour cells to CDDP. Activation
of the transmembrane transport protein xCT directly exerts its
functions on the most important antioxidative stress molecule,
glutathione (GSH), at the synthesis level (13). A number of studies have indicated
that xCT significantly contributes to antioxidative stress of
malignant tumour cells (13,14). However, how the Nrf2-xCT pathway and
the activation of other downstream genes of Nrf2 regulate the CDDP
sensitivity of NSCLC cells, and how to effectively eliminate
CDDP-resistant NSCLC cells remains unknown. Notably, with further
study of ferroptosis mechanisms, small molecular inducers of
ferroptosis may be identified and these may promote intracellular
lipid ROS, and eventually induce ferroptosis. Among these small
molecular inducers, the classic inducers erastin and sorafenib,
which are used to treat advanced renal and liver cancer, possess
clinical development and application prospects. These two molecules
exert their function by inhibiting the cystine import activity of
xCT, resulting in a reduction of GSH synthetic materials and
accumulation of lipid ROS, thus ultimately inducing ferroptosis
(6,15). However, to the best of our knowledge,
the effects of the aforementioned ferroptosis inducers on NSCLC,
particularly NSCLC with CDDP treatment failure, have not been
studied. Yet it is reasonable to hypothesize that the application
of ferroptosis inducers is an ideal way of solving CDDP resistance
in NSCLC.
In the present study, CDDP induced activation of the
Nrf2/xCT pathway in different NSCLC cell lines, and its activation
level was associated with the extent of CDDP resistance.
Additionally, the expression levels of Nrf2/xCT in CDDP-resistant
NSCLC cells were markedly increased, and the classical ferroptosis
inducers erastin and sorafenib clearly induced ferroptosis in
CDDP-resistant NSCLC cells. When a low dose of CDDP was used in
combination with erastin/sorafenib, it effectively eliminated
CDDP-resistant NSCLC cells. Finally, the present study demonstrated
that erastin/sorafenib may inhibit CDDP-resistant NSCLC cell growth
in vivo. This study indicated that ferroptosis induced by
erastin/sorafenib may provide a novel perspective for the treatment
of patients with NSCLC following failed CDDP treatment, and may
improve the OS of patients.
Materials and methods
Cell lines and cell culture
The A549 and NCI-H1299 human NSCLC cell lines were
purchased from Shanghai Institutes for Biological Sciences, Chinese
Academy of Cell Resource Center. N2 and N5 cells were surgically
obtained from patients (N2: Male, 61 years old; N5: male, 58 years
old) at the Shengjing Hospital of China Medical University from May
to July of 2014, once written informed consent had been obtained.
The study was approved by the Ethics Committee of China Medical
University. Primary NSCLC cell isolation and expansion were
performed as previously described (16). The N5 CDDP-resistant (N5CP) variant
cell line was established by continuous culturing of N5 cells with
increasing concentrations of CDDP (1–10 µg/ml; cat. no. C2210000;
Sigma-Aldrich; Merck KGaA) for 6 months. The culture medium of N5CP
cells contained 10 µg/ml CDDP to maintain drug resistance. All
cells were cultured in RPMI-1640 medium (cat. no. 11875093; Gibco;
Thermo Fisher Scientific, Inc.) containing 10% foetal bovine serum
(cat. no. 10100147; Gibco; Thermo Fisher Scientific, Inc.), 100
U/ml penicillin and 100 µg/ml streptomycin (cat. no. 15140122;
Gibco; Thermo Fisher Scientific, Inc.) in a humidified atmosphere
at 37°C with 5% CO2.
Cell survival rate analysis
Cell survival rate was assessed using the Cell
Counting kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.),
according to the manufacturer's protocol. Cells (2×104
cells/well) were seeded in 96-well plates and; after 24 h, the
cells were subjected to with CDDP, erastin (E7781; Sigma-Aldrich;
Merck KGaA) or sorafenib (284461-73-0; Sigma-Aldrich; Merck KGaA)
with or without deferoxamine (DFO; 138-14-7, Sigma-Aldrich; Merck
KGaA) or Vitamin E (Vit-E; 10191-41-0, Sigma-Aldrich; Merck KGaA)
for an additional 48 h. After refreshing the culture media, 10
µl/well CCK-8 solution was added and the plates were incubated for
an additional 45 min in a 37°C incubator. The absorbance of cells
at 450 nm was detected using a microplate reader (iMark™ Microplate
Absorbance Reader; Bio-Rad Laboratories, Inc.).
Propidium iodide (PI) staining
The PI Flow Cytometry kit (cat. no. ab139418; Abcam)
was used to determine cell death. Briefly, cells (2×105
cells/well) were seeded in 6-well plates and incubated for 24 h at
37°C. Subsequently, the cells were treated with erastin (10 µM) or
sorafenib (20 µM). After 48 h, the cells were harvested, washed
twice with PBS and subsequently re-suspended in PI staining
solution at room temperature for 15 min in the dark, according to
the manufacturer's protocol. Samples were assessed by flow
cytometry (Muse; Sigma-Aldrich; Merck KGaA) and the data were
analysed using FlowJo software version 9 (FlowJo LLC).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA of A549, N5 and N5CP cells was extracted
using TRIzol® Reagent (cat. no. 15596026; Invitrogen;
Thermo Fisher Scientific, Inc.). Extracted RNA was quantified and
RNA purity was confirmed by measuring 260/230 and 260/280 nm
absorbance ratios (NanoDrop 2000 Spectrophotometer; NanoDrop
Technologies; Thermo Fisher Scientific, Inc.). Subsequently, 1 µg
total RNA was used for cDNA synthesis, using an iScript™ cDNA
Synthesis kit (cat. no. 1708891; Bio-Rad Laboratories, Inc.),
according to the manufacturer's protocols. SYBR Premix Ex Taq (cat.
no. RR420A; Takara Bio, Inc., Otsu, Japan) was used to measure
individual gene expression on the CFX96 Real-Time PCR Detection
system (Bio-Rad Laboratories, Inc.). The primer sequences used for
RT-qPCR were as follows: Human Nrf2, forward,
5′-AGTCCTGGTCATCGGAAAAC-3′, reverse 5′-ATGGAGAGCTTTTGCCCTAA-3′;
human xCT, forward 5′-CCATGAACGGTGGTGTGTT-3′, reverse
5′-GACCCTCTCGAGACGCAAC-3′); human HO-1, forward
5′-CTCTTGGCTGGCTTCCTTAC-3′, reverse 5′-TCCTTCCTCCTTTCCAGAGA-3′);
human NQO1, forward 5′-TGGCTCCATGTACTCTCTGC-3′, reverse
5′-CAGAAATGCAGAATGCCACT-3′); and human GAPDH, forward
5′-AGTCAGCCGCATCTTCTTTT-3′ and reverse 5′-CAATACGACCAAATCCGTTG-3′.
Gene expression was normalised to GAPDH. Each sample was
analysed in triplicate. The PCR reaction was performed under the
following conditions: One cycle at 95°C for 2 min; followed by 45
cycles at 95°C for 5 sec and extension at 62°C for 30 sec; melting
curves were recorded for 5 sec per temperature step during a
temperature gradient from 65.0°C to 95.0°C with an increment of
0.5°C (17). All expression levels
were calculated using the 2−ΔΔCq method (17).
Luciferase reporter assay
A549 and N5 cells were seeded in 12-well plates at a
density of 1×105 cells/well. The following day, cells
were co-transfected with the ARE luciferase reporter vector and
Renilla luciferase vector (ARE Reporter kit; cat. no. 60514;
BPS Bioscience, Inc.) with Lipofectamine® LTX Reagent
(cat. no. 15338100; Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturers' protocols. Renilla
luciferase activity was used as an internal control. A total of 24
h post-transfection, the culture media were changed and 20 µg/ml
CDDP or DMSO (cat. no. M81802; Sigma-Aldrich, Merck KGaA) were
added. After 12 h, the cells were collected and luciferase activity
was detected using a Dual-Luciferase Reporter Assay system (cat.
no. E1910; Promega Corporation). Mean values from triplicate
analysis were presented.
Western blotting
The cells treated with CDDP, siRNA, overexpression
plasmids, erastin or sorafenib were washed twice with ice-cold PBS
at the end of the experiment. Whole cell protein lysates were
prepared by dissolving the cell pellets in lysis buffer [62.5 mM
Tris-HCl (pH 6.8), 2% SDS and 10% glycerol]. Protein concentrations
were measured with a Pierce™ Bicinchoninic Acid Protein Assay kit
(cat. no. 23225; Thermo Fisher Scientific, Inc.). Total proteins
(20 µg/lane) were separated by 8–10% SDS-PAGE. Subsequently,
proteins were transferred to polyvinylidene difluoride (PVDF)
membranes (cat. no. IPVH09120; EMD Millipore) and the membranes
were blocked with 1% skimmed milk for 1 h at room temperature.
After three washes with Tris-buffered saline with 0.1% Tween-20
(TBST), the PVDF membranes were incubated with anti-human Nrf2
(1:1,000 dilution; cat. no. ab31163; Abcam), xCT (1:1,000 dilution;
cat. no. ab175186; Abcam) and GAPDH (1:1,000 dilution; cat. no.
ab8245; Abcam) antibodies diluted in TBST at room temperature for 1
h. After incubating with a goat anti-rabbit IgG H&L for
detecting Nrf2 and xCT (1:10,000 dilution; cat. no. ab97051; Abcam)
or a goat anti-mouse IgG H&L for detecting GAPDH (1:10,000
dilution; cat. no. ab6708; Abcam) at room temperature for 1 h, the
membranes were visualised using Pierce™ Enhanced Chemiluminescence
Western Blotting Substrate (cat. no. 32106; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
ROS determination
ROS generation was determined using
6-carboxy-2′,7′-dichlorofluorescein diacetate dye
(H2DCFDA; cat. no. D399; Thermo Fisher Scientific,
Inc.). The medium was refreshed following treatment with CDDP,
erastin, sorafenib or DMSO, and 20 µl/well H2DCFDA was
added to the medium 30 min prior to the end of the experiment at
37°C. Subsequently, the cells were washed twice with ice-cold PBS
and digested with trypsin. ROS production was analysed using a flow
cytometer (Muse; Sigma-Aldrich; Merck KGaA) and FlowJo v.9
software.
Knockdown and overexpression
experiment
For the knockdown experiment, A549 cells were seeded
in 12-well plates at a density of 1.5×105 cells/well.
The following day, the cells were transfected with a final
concentration of 20 nM anti-human Nrf2 small interfering RNA
(siRNA; cat. no. 107966; Thermo Fisher Scientific, Inc.),
anti-human xCT siRNA (cat. no. 108517; Thermo Fisher Scientific,
Inc.) or scrambled siRNA (cat. no. AM4611; Thermo Fisher
Scientific, Inc.) using Lipofectamine® RNAiMax reagent
(cat. no. 13778150; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Subsequently, 24 h post-transfection,
the medium was replaced with fresh medium containing 20 µg/ml CDDP
and the cells were incubated for an additional 48 h. For the
overexpression experiment, N5 cells were seeded as aforementioned
and were then transfected with a final concentration of 0.5 ng/µl
pcDNA3-human Nrf2, pcDNA3-human xCT or pcDNA3 vector using
Lipofectamine® LTX Reagent. The plasmids of pcDNA3-hNrf2
and pcDNA3-hxCT were constructed as described previously (14). After 24 h, the medium was replaced
with fresh medium containing 40 µg/ml CDDP. After 48 h, cell
survival rate measurements were performed.
Xenograft assay
A total of 60 BALB/c-nu/nu nude mice (male; age, 4–6
weeks; weight, 16–22 g) were obtained from the Shanghai Laboratory
Animal Co., Ltd. Mice were housed under pathogen-free conditions in
barrier facilities under a 12-h dark/light cycle. The room
temperature was maintained at 23°C with a humidity of 50–60%; food
and water were ad libitum. All animal research was performed
in accordance with the approved animal protocols and the guidelines
of the Institutional Animal Care and Use Committee of China Medical
University. The present animal study was approved by the
Institutional Animal Care and Use Committee of China Medical
University. N5CP cells (5×106) were suspended in 200 µl
DMEM and Matrigel (cat. no. 354234; BD Biosciences) mixture at a
ratio of 1:1. Subsequently, the mixture was injected subcutaneously
into the upper right flank of 20 nude mice. After 10 days, the mice
were randomly divided into four groups and were treated with CDDP
(5 mg/kg/2 days), erastin (10 mg/kg/2 days), sorafenib (10 mg/kg/2
days) or PBS by intraperitoneal injection. Two days after the third
injection, the mice were sacrificed and tumours were carefully
removed. For the combination experiment, CDDP (1 mg/kg) and erastin
(5 mg/kg) or sorafenib (3 mg/kg) were also injected three times.
Subsequently, tumour weight was measured. The mice were euthanised
when a humane endpoint was reached (when the mice were distressed
by the tumour burden or tumour volume was >2,000
mm3).
Statistical analysis
Statistical analysis was performed using SPSS v.20.0
software (IBM Corp.). All values are expressed as the mean ±
standard error and are representative of at least three independent
experiments. The data were analysed using one-way ANOVA. When the
results were significant, post hoc testing of differences among
groups was performed using a Bonferroni test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Sensitivity of NSCLC cells to CDDP is
negatively associated with Nrf2 pathway activation
It has been demonstrated that Nrf2 expression is
negatively associated with CDDP cytotoxicity; however, to the best
of our knowledge, there are no detailed reports on either the key
Nrf2 downstream target genes or the mechanisms of regulation
(18). To further investigate the
association between CDDP sensitivity and Nrf2, as well as to
identify the downstream target genes, the cytotoxicity of CDDP in
four NSCLC cell lines was compared. NCI-H1299 and A549 cells are
commonly used in the laboratory, and N2 and N5 cells were primary
cultured cells from patients with NSCLC obtained during surgical
resection. As shown in Fig. 1A, CDDP
exerted different cytotoxic effects on these cells. A549 cells were
most resistant to CDDP, whereas N5 cell were most sensitive to
CDDP. To investigate the differences in more detail, in a follow-up
experiment, the two representative cell lines A549 and N5 were
selected and the effects of CDDP on the Nrf2 pathway were compared.
As shown in Fig. 1B and C, in A549
and N5 cell lines, CDDP markedly upregulated the expression of
Nrf2 and its downstream target gene xCT at the mRNA
level, but no significant alterations in the expression of
HO-1 and NQO1 were observed. In A549 cells, the
expression of Nrf2 was only slightly increased by CDDP,
whereas xCT expression was increased 8.6-fold. By comparing
the influence of CDDP on overall activation levels of the Nrf2
pathway in the two cell lines, the activation level of the
CDDP-mediated Nrf2 pathway was revealed to be higher in A549 cells
than in N5 cells (Fig. 1B and C).
Western blot analysis demonstrated that Nrf2 and xCT protein
expression were markedly increased in A549 cells in the presence of
CDDP, which was consistent with the mRNA expression analysis
results (Fig. 1D). In particular,
Nrf2 protein was detected in unstimulated A549 cells, which may be
caused by a Keap1 mutation, which hydrolyses Nrf2 by ubiquitination
(19). Keap1 mutation leads to the
sustained accumulation of Nrf2, as well as its downstream target
genes. This may be why CDDP did not cause further significant
upregulation of Nrf2 downstream target genes in A549 cells. The
present study demonstrated that N5 cells with lower levels of Nrf2
activation were more sensitive to CDDP treatment than A549 cells
with higher activation levels of Nrf2. To further explore the
regulation of Nrf2/ARE-driven downstream gene transcription by
CDDP, A549 and N5 cells were transiently transfected with a
luciferase reporter plasmid containing ARE. CDDP increased the
luciferase activity of reporter genes and had a clearer effect on
Nrf2 transcription activity in A549 cells than in N5 cells
(Fig. 1E). These results indicated
that CDDP induced activation of the Nrf2/xCT pathway in NSCLC
cells, and the activation level was negatively associated with the
sensitivity of cells to CDDP.
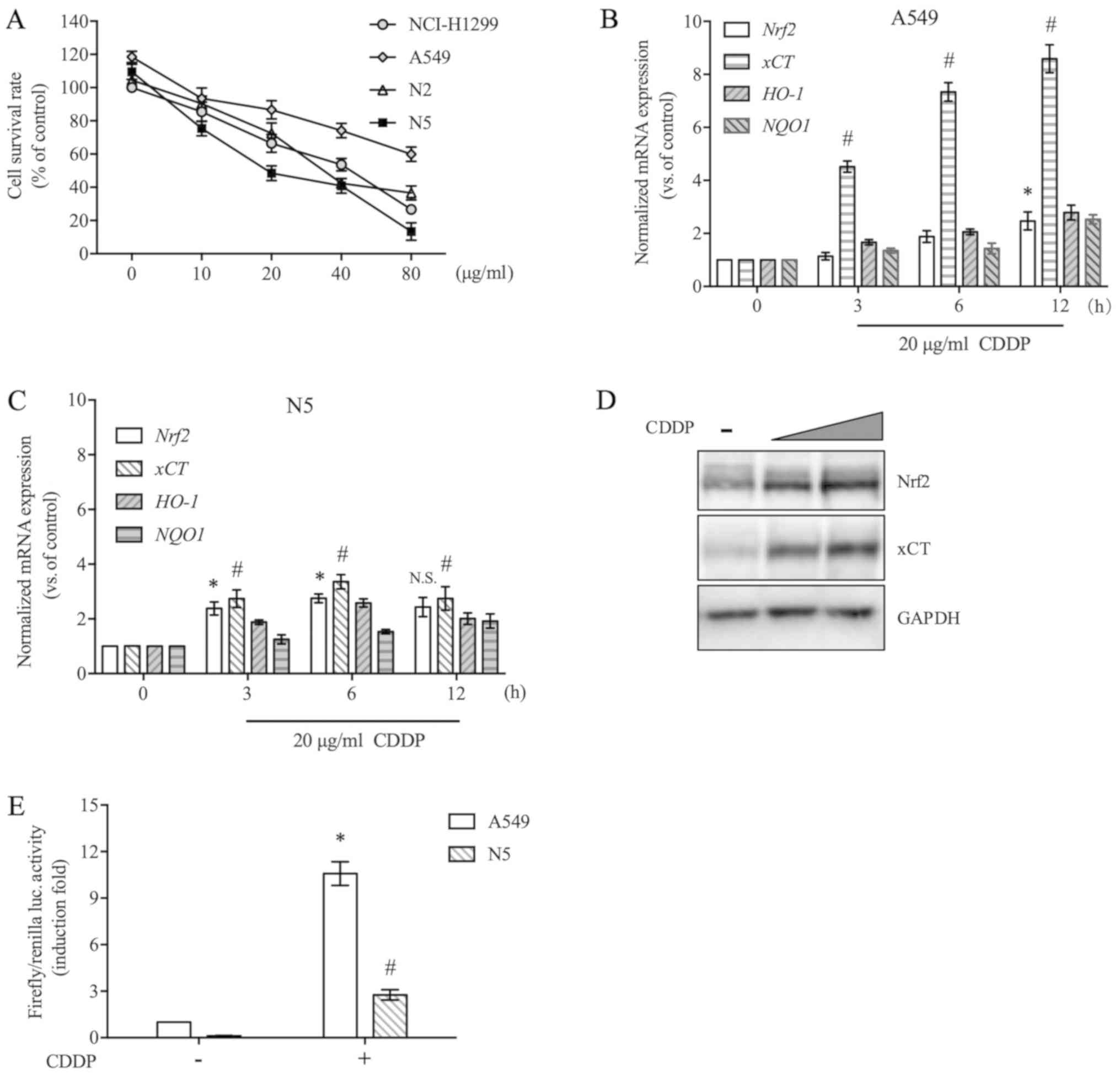 | Figure 1.Nrf2 pathway is activated by CDDP and
negatively regulates CDDP toxicity. (A) NCI-H1299, A549, N2 and N5
cells were treated with a series of CDDP doses (10–80 µg/ml) for 48
h, and the cell survival rate was analysed using a Cell Counting
kit-8 assay. (B and C) A549 and N5 cells were exposed to 20 µg/ml
CDDP for 3–12 h. The mRNA expression levels of Nrf2, xCT,
HO-1 and NQO1 were evaluated by reverse
transcription-quantitative PCR and normalised to GAPDH. (D)
A549 cells were exposed to 20 or 40 µg/ml CDDP treatment for 12 h.
Nrf2 and xCT protein expression was detected by western blotting.
GAPDH was used as an internal control. (E) A549 and N5 cells were
transfected with an antioxidant response element luciferase
reporter vector and Renilla luciferase vector. After 24 h of
transfection, the cells were incubated in the presence of 20 µg/ml
CDDP for an additional 12 h and were then subjected to a luciferase
assay. Luciferase activity was normalised to Renilla
luciferase activity and then induction fold were compared with A549
cells without CDDP treatment. Data are presented as the mean ±
standard error from at least three independent experiments.
Differences among groups were assessed by one-way ANOVA with a
Bonferroni post hoc test. *P<0.05 vs. Nrf2 in (B) and (C), vs.
A549 in (E); #P<0.05 vs. xCT in (B) and (C), vs. N5
in (E). CDDP, cisplatin; HO-1, heme oxygenase 1; luc, luciferase;
NQO1, NAD(P)H quinone dehydrogenase 1; Nrf2, NF-E2 related factor
2; N.S., not significant; xCT, light chain of System
xc−. |
Nrf2/xCT expression determines the
sensitivity of cells to CDDP
The Nrf2/xCT pathway can resist oxidative stress and
downregulate intracellular ROS levels by regulating glutathione
synthesis (14). In the present
study, the ROS levels of A549 and N5 cells following CDDP treatment
were measured. As shown in Fig. 2A,
CDDP significantly increased ROS accumulation in A549 and N5 cells,
but the effect was greater in N5 cells (Fig. 2A). To further explain the
contribution of Nrf2/xCT to CDDP sensitivity in NSCLC cells, A549
cells resistant to CDDP were transfected with siRNA to knockdown
Nrf2 or xCT. The knockdown efficiency of individual siRNAs was
confirmed through western blot analysis (Fig. 2C). As shown in Fig. 2B, when the expression of Nrf2 or xCT
was decreased, the sensitivity of A549 cells to CDDP was markedly
enhanced. Additionally, the effect of Nrf2 knockdown on CDDP
cytotoxicity was stronger than that of xCT knockdown. Conversely,
Nrf2 or xCT were transiently overexpressed in N5 cells that were
relatively sensitive to CDDP (Fig.
2E). With increasing Nrf2 or xCT expression levels, CDDP
cytotoxicity to N5 cells was significantly decreased (Fig. 2D). Compared with the results of
Fig. 2A, overexpression or knockdown
of xCT expression reversed the regulatory effect of CDDP on ROS
levels in N5 and A549 cells (Fig.
2F).
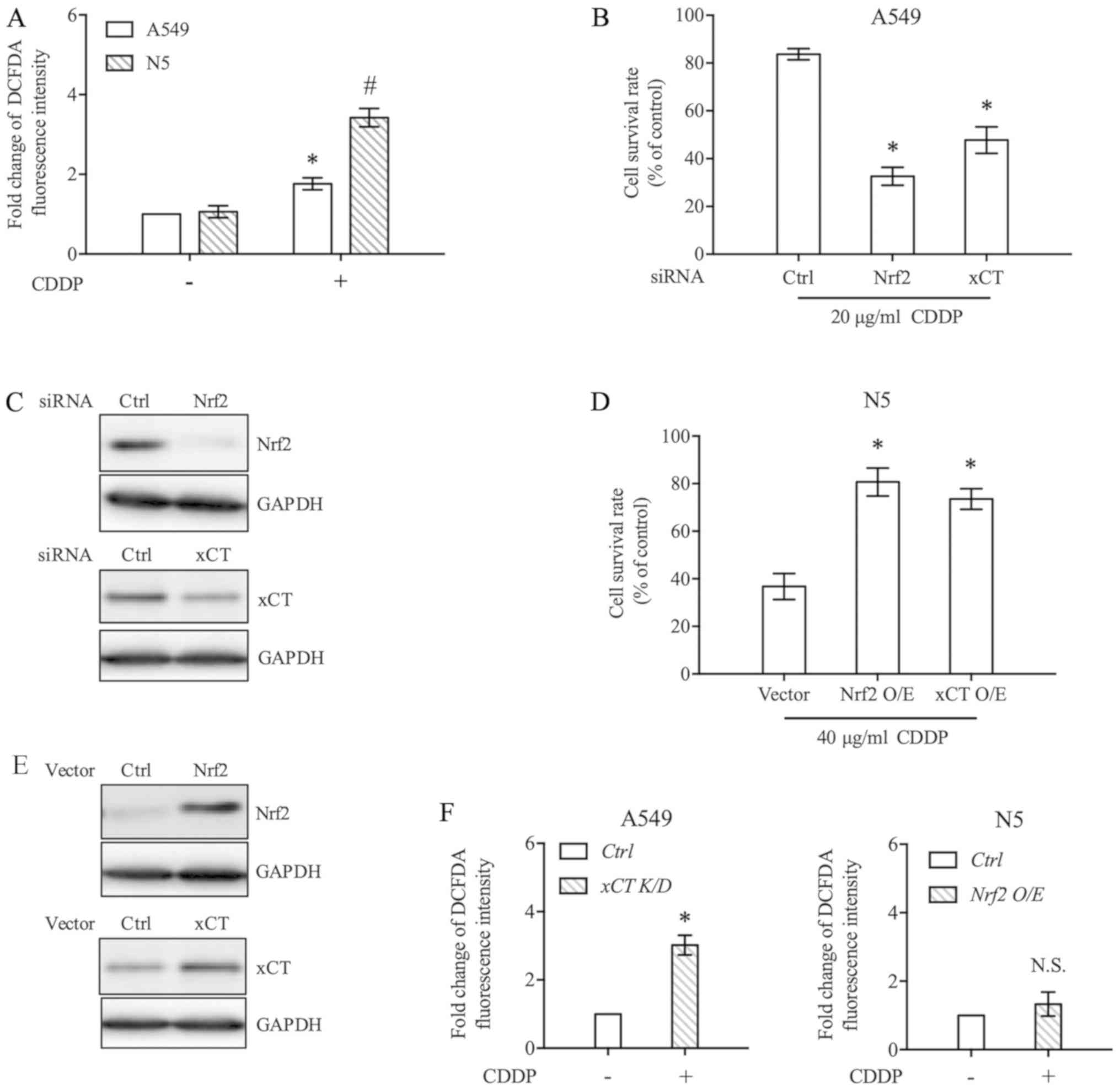 | Figure 2.Nrf2/xCT expression determines CDDP
sensitivity. (A) A549 and N5 cells were exposed to 20 µg/ml CDDP or
DMSO for 6 h. DCFDA fluorescence, indicative of intracellular ROS
level, was analysed by flow cytometry. Data were presented relative
to the DMSO-treated control. (B) A549 cells were transfected with
Nrf2 or xCT siRNA. After 24 h, the cells were treated with 20 µg/ml
CDDP for 48 h, and cell survival rate was determined. (C) Nrf2 and
xCT expression in A549 cells was inhibited by specific siRNAs. At
24 h post-transfection, whole cell lysate was extracted, and
western blot analysis was performed to detect Nrf2 and xCT
expression. GAPDH was used as a loading control. (D) N5 cells were
transfected with Nrf2 or xCT expression vector. The pcDNA3 vector
was used as a control. After 24 h of incubation, the cells were
treated with 40 µg/ml CDDP for 48 h and subjected to cell survival
rate analysis. (E) Western blot analysis was performed to detect
the protein expression levels of Nrf2 and xCT following
transfection with individual overexpression plasmids. (F) The
expression of xCT was downregulated by siRNA in A549 cells and
upregulated by overexpression plasmid in N5 cells. Cells were then
treated with 20 µg/ml CDDP for 6 h, and ROS levels were detected by
flow cytometry. Data are presented as the mean ± standard error
from at least three independent experiments. Differences among
groups were assessed by one-way ANOVA with a Bonferroni post hoc
test. *P<0.05 vs. (A) A549, vs. (B) Ctrl, vs. (D) Vector.
#P<0.05 vs. N5 (A). Ctrl, control; CDDP, cisplatin;
DCFDA, 6-carboxy-2′,7′-dichlorofluorescein diacetate dye; K/D,
knockdown; N.S., not significant; Nrf2, NF-E2 related factor 2;
O/E, overexpression; ROS, reactive oxygen species; siRNA, short
interference RNA; xCT, light chain of System
xc−. |
Erastin and sorafenib effectively
induce N5CP cell ferroptosis by modulating the Nrf2/xCT
pathway
According to the aforementioned results, activation
of the Nrf2/xCT pathway may be a major mechanism underlying NSCLC
cell CDDP resistance, and inhibiting the Nrf2/xCT pathway
effectively reversed resistance to CDDP in NSCLC cells. One
induction mechanism of ferroptosis is to inhibit cystine import via
xCT (6). Therefore, the induction of
ferroptosis may be a feasible option to eliminate CDDP-resistant
NSCLC cells. To obtain more clinically meaningful CDDP-resistant
NSCLC cells, N5 cells that were relatively sensitive to CDDP were
screened and cultured with gradually increasing concentrations of
CDDP for 6 months, and the resulting cells were termed N5CP cells.
Cell survival rate experiments demonstrated that N5CP cells
exhibited marked CDDP resistance compared with N5 cells (Fig. 3A). The mRNA expression levels of
Nrf2 and xCT in N5 and N5CP cells were compared by
RT-qPCR, and were revealed to be significantly higher in N5CP cells
than in N5 cells (Fig. 3B). Erastin
and sorafenib are traditional ferroptosis inducers that mainly
inhibit the transport function of xCT. In the present study,
erastin and sorafenib significantly reduced the survival rate of
N5CP cells (Fig. 3C and D).
Furthermore, erastin and sorafenib-induced cell death was markedly
suppressed by the specific inhibitors of ferroptosis deferoxamine
mesylate (DFO) and Vitamin E (Vit-E), thus verifying that the type
of cell death induced was ferroptosis (Fig. 3C and D). Additionally, PI staining
was employed to detect cell death induced by erastin and sorafenib,
in order to validate the results of viability experiments
previously conducted (20,21). Erastin and sorafenib markedly
increased the number of PI-positive N5CP cells (Fig. 3E and F). In addition, the expression
of Nrf2 and xCT at the mRNA and protein levels were measured
following erastin and sorafenib treatment. Erastin and sorafenib
did not significantly alter Nrf2 expression at the mRNA level, but
markedly reduced Nrf2 protein expression (Fig. 3G and H). This may be due to Nrf2
degradation being increased by erastin and sorafenib. As a target
gene of Nrf2, xCT expression was significantly decreased at the
mRNA and protein level. These findings indicated that erastin and
sorafenib efficiently eliminated CDDP-resistant NSCLC cells by
modulating the Nrf2/xCT pathway.
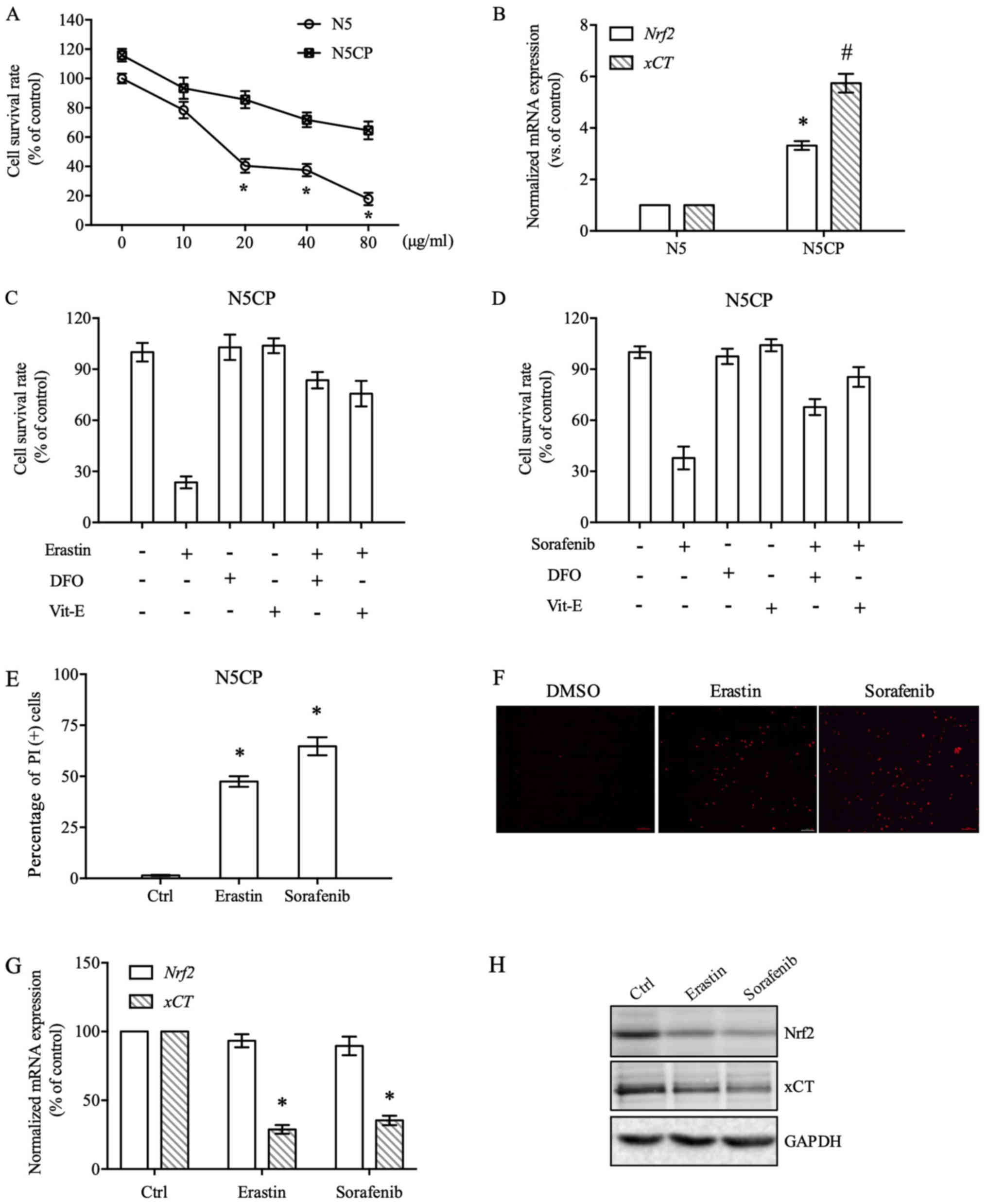 | Figure 3.Erastin and sorafenib induce N5CP
cell ferroptosis. (A) N5 and N5CP cells were exposed to different
doses of CDDP, as indicated, for 48 h and were then subjected to
cell survival rate determination. (B) The expression of Nrf2
and xCT mRNA in N5 and N5CP cells was measured using
RT-qPCR. The data were normalised to GAPDH. (C and D) N5CP
cells were treated with erastin (10 µM), sorafenib (20 µM), DFO (50
µM) and Vit-E (100 µM) individually or in combination, as
indicated, for 48 h, and then cell survival rate detection was
performed. (E) N5CP cells were exposed to erastin (10 µM),
sorafenib (20 µM) or DMSO for 48 h and subjected to PI staining
analysis. Representative images are shown in (F). (G and H) N5CP
cells were treated with erastin (10 µM), sorafenib (20 µM) or DMSO
for 12 h, and the expression of Nrf2 and xCT was determined by
RT-qPCR and western blotting. Data are presented as the mean ±
standard error, and differences among groups were analysed by
one-way ANOVA with a Bonferroni post hoc test. *P<0.05 vs. N5CP
(A), vs. Nrf2 (B), vs. DMSO (C-E), vs. xCT (G);
#P<0.05 vs. xCT (B). CDDP, cisplatin; Ctrl, control;
DFO, deferoxamine mesylate; Nrf2, NF-E2 related factor 2; PI,
propidium iodide; RT-qPCR, reverse transcription-quantitative PCR;
Vit-E, vitamin E; xCT, light chain of System
xc−. |
CDDP combined with erastin/sorafenib
effectively induces N5CP cell ferroptosis
N5CP cells were treated with low doses of CDDP
and/or erastin/sorafenib, as indicated, in order to explore the
combined effect of low dose drugs, to allow for comparisons despite
the inconsistent doses used across different experiments.
Individual treatment with CDDP, erastin or sorafenib did not
markedly affect cell survival rates. When cells were cultured in
CDDP combined with erastin or sorafenib, the cell survival rate was
significantly decreased (Fig. 4A),
whereas ROS accumulation was increased at the same time (Fig. 4B). Furthermore, the cell death caused
by cooperation of CDDP and erastin/sorafenib could be partially
reversed by DFO, indicating that the type of cell death induced was
ferroptosis (Fig. 4A).
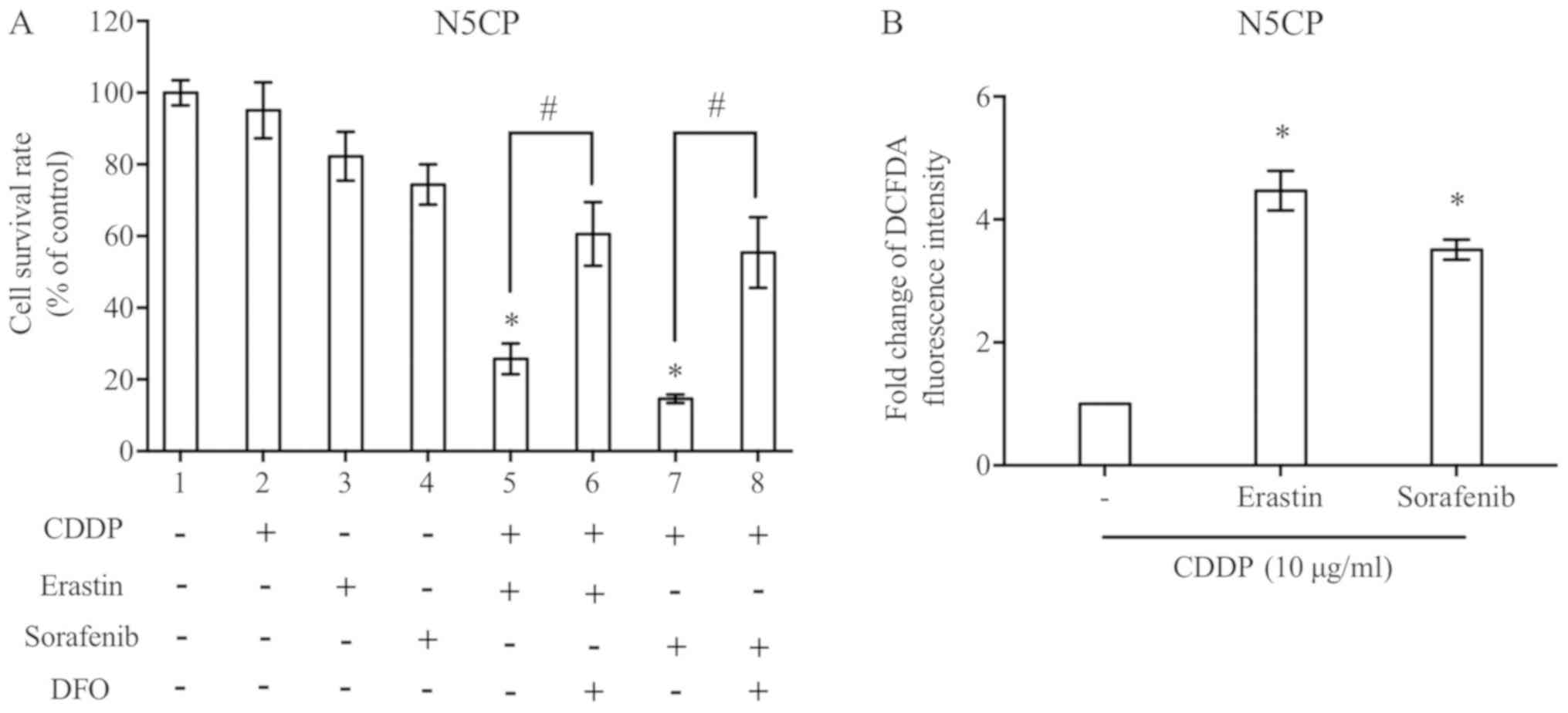 | Figure 4.CDDP combined with erastin/sorafenib
effectively induces N5CP cell ferroptosis. (A) N5CP cells were
treated as indicated for 48 h. A Cell Counting kit-8 assay was used
to detect the cell survival rate. The concentrations of CDDP,
erastin, sorafenib and DFO were 10, 5, 10 and 50 µM, respectively.
(B) N5CP cells were exposed to CDDP combined with erastin,
sorafenib or control at the same concentrations for 12 h, and the
reactive oxygen species level was detected using a DFCDA probe and
flow cytometry. Data are presented as the mean ± standard error
from at least three independent experiments. Differences among
groups were assessed by one-way ANOVA with a Bonferroni post hoc
test. *P<0.05 vs. (A) DMSO, vs. (B) CDDP only;
#P<0.05. CDDP, cisplatin; DCFDA,
6-carboxy-2′,7′-dichlorofluorescein diacetate dye; DFO,
deferoxamine mesylate. |
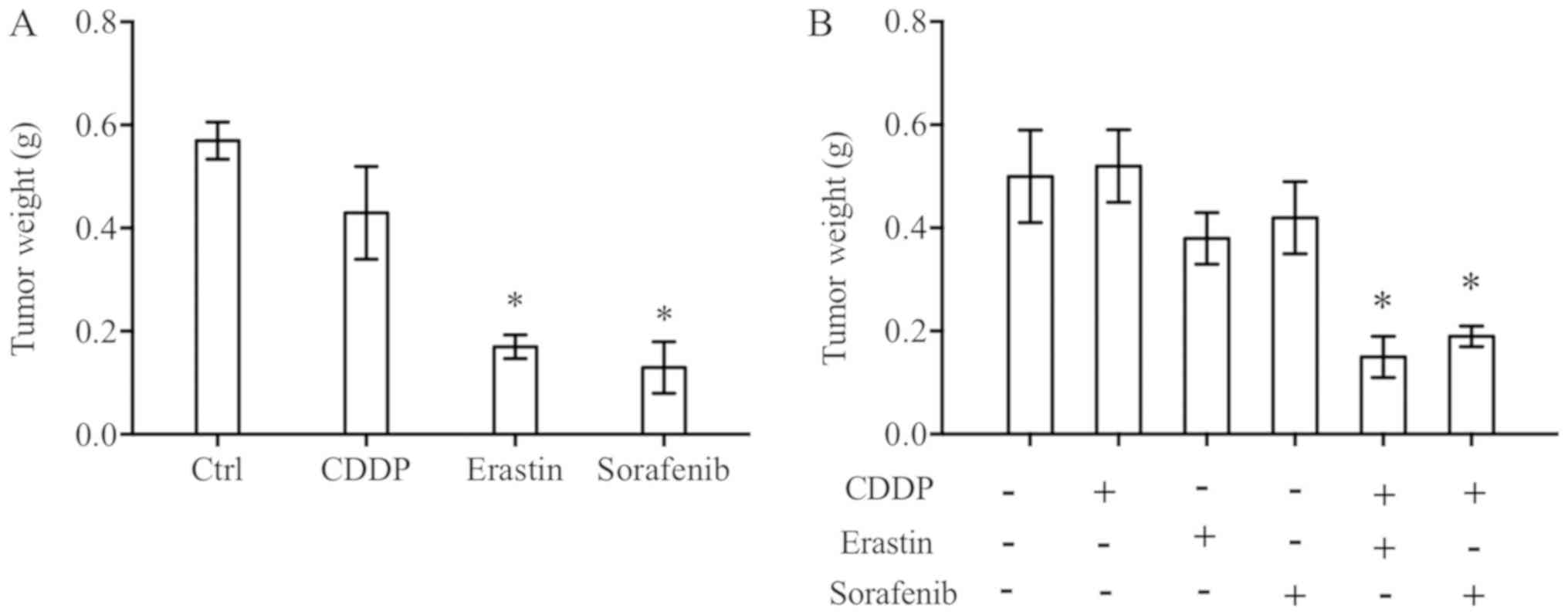
Erastin/sorafenib restrains in vivo
tumour growth in nude mice xenograft models
N5CP cells were seeded under nude mice skin; ~10
days later, when the tumour volume reached ~600 mm3, the
drugs CDDP (3 mg/kg), erastin (20 mg/kg) or sorafenib (10 mg/kg)
were intraperitoneally injected into the nude mice every other day,
and the tumour was resected and weighed after three injections
(22). The results demonstrated that
erastin/sorafenib inhibited tumour growth in vivo (Fig. 5A). Additionally, the present study
demonstrated that low doses of CDDP (1 mg/kg), erastin (5 mg/kg)
and sorafenib (3 mg/kg) did not exhibit clear inhibitory effects on
tumour growth effects when used alone; however, when used in
combination, tumour growth was significantly inhibited (Fig. 5B). Based on the aforementioned
results, it may be hypothesised that apparent reductions in tumour
size due to treatment with erastin/sorafenib alone were possibly
due to the dosages being high enough. It was evident that the
effective concentrations of in vivo and in vitro
experiments were inconsistent. It is a common phenomenon that the
effects of drugs are not consistent. Evidence suggests that the
effect of drugs is associated with numerous factors apart from
concentration, including the route of administration and
liposoluble activity (23).
Discussion
Resistance to drugs can usually be divided into two
subtypes: Primary resistance, which is associated with
chemoresistance prior to chemotherapy; and acquired resistance,
which is mainly involved in drug resistance following chemotherapy
(24). A variety of solid tumour
cells, represented by NSCLC, exhibit inherent disorders of CDDP
transport and metabolism, accompanied by high expression levels of
multidrug resistance genes. Additionally, the anti-apoptotic
pathway can be activated by CDDP; therefore, the coexistence of
these two drug-resistance subtypes clearly restricts the curative
effect and application of CDDP (25,26). In
addition, although carboplatin (CBP) is widely used in NSCLC
treatment as the second generation of platinum medicine and has
excellent properties, including lower ototoxicity and
nephrotoxicity, there is a cross-resistance to CBP and CDDP, such
that CBP neither eliminates CDDP resistance nor markedly improves
the prognosis of patients with NSCLC (27). In previous years, progress in NSCLC
treatment research has been made regarding targeted drugs in
addition to the exploration of platinum. In particular, the
introduction of drugs that specifically target ALK receptor
tyrosine kinase, epidermal growth factor receptor and programmed
cell death 1/programmed death-ligand 1 substantially improves the
therapeutic outcomes of patients with certain gene mutations
(28,29). However, the effect of these targeted
drugs is unsatisfactory in patients without these mutations and
gene expression characteristics. In the present study, a
CDDP-resistant NSCLC cell model was used to reveal the CDDP
sensitivity of tumour cells, which was reduced by the Nrf2/xCT
pathway. Additionally, the results indicated that inducing
ferroptosis could efficiently eliminate CDDP-resistant NSCLC
cells.
Ferroptosis was fortuitously identified during the
process of screening compounds that selectively produce lethal
effects on RAS mutation-containing tumour cells. Two small
molecules, erastin and Ras selective lethal (RSL) 3 compound
(RSL3), of entirely different chemical structures are known as RSL
compounds. The two can inactivate the intracellular GSH-dependent
antioxidant defence and induce ferroptosis by different mechanisms
(6). Erastin mainly inhibits the
import of cystine through xCT, whereas RSL3 directly binds and
restrains glutathione peroxidase 4 (Gpx4). Since Gpx4 is
indispensable for the physiological function of cells, RSL3
exhibits cytotoxicity against normal cells, which limits the
application of Gpx4 inhibitors (6,30).
Similar to erastin, the small molecules sulfasalazine, glutamate,
phosphatidylethanolamines (PE) and sorafenib mainly trigger
ferroptosis by affecting xCT (31,32).
Glutamate is rarely used clinically due to its neurotoxicity, and a
study regarding the mechanism of ferroptosis induced by PE is
currently in its infancy (33). The
present study investigated whether erastin/sorafenib used
individually or in combination with a small dose of CDDP could
effectively induce ferroptosis in CDDP-resistant NSCLC cells.
Sulfasalazine is a clinical anti-inflammatory drug used to treat
inflammatory bowel diseases and rheumatoid arthritis (34). Although a previous report claimed
that sulfasalazine can increase the sensitivity of colon cancer
cells to CDDP in a GSH-dependent manner (35), in Phase I clinical trials of patients
with gastric cancer who were positive for splice variant isoforms
of CD44, which can stabilize xCT, co-application of sulfasalazine
and CDDP to CDDP-resistant advanced gastric cancer exhibited
insufficient effectiveness (36).
Consistent with the aforementioned studies, sulfasalazine did not
exhibit clear cytotoxic effects with CDDP in the present
CDDP-resistant NSCLC cell model (data not shown). These
contradictory results may be explained by the specificity of tumour
cells, xCT blocking the efficacy of the drug or another unknown
reason. In the present study, only the role of erastin/sorafenib in
one type of CDDP-resistant NSCLC cells was observed, so there are
limitations to this analysis. In future studies, we will continue
to explore the function of erastin/sorafenib in other
CDDP-resistant NSCLC cell lines, as well as other pathological
types of cells. Besides blocking xCT, sorafenib is also a
multi-target tyrosine kinase inhibitor possessing
anti-proliferative and anti-angiogenic effects (37). In the present study, the lethality of
sorafenib, whether used individually or in combination with CDDP,
on N5CP cells was clearer than that of erastin; however, it was
inconsistent with the mediated intracellular lipid ROS accumulation
levels. Additionally, DFO and Vit-E failed to completely inhibit
the cytotoxicity of sorafenib. These results suggested that the
cell death mechanism, induced by sorafenib alone or in combination
with CDDP, may be mainly ferroptosis in combination with multiple
other cell death mechanisms.
The transcription factor Nrf2 is the master
regulator of cell antioxidants for oxidative or electrophilic
stress. ROS can induce Nrf2 accumulation in the nucleus and then
combine with basic leucine zipper proteins, including small MAF
bZIP transcription factor, to eventually form a transactivation
complex to bind to AREs, which regulate downstream gene
transcription (38). The ARE
sequence is mostly present in the promoter or enhancer regions of
genes encoding phase II detoxification enzymes (39). These enzymes driven by Nrf2/ARE are
principally involved in the regulation of intracellular ROS levels,
and their expression level directly affects the occurrence of
ferroptosis. HO-1 mediates ferroptosis induced by BAY11-7085
(40). NQO1, HO-1 and ferritin heavy
chain 1 provide resistance to erastin- and sorafenib-induced
hepatocellular carcinoma cell ferroptosis by regulating iron
metabolism and lipid peroxidation (22). Metallothionein-1 has recently been
revealed to be the downstream target gene of Nrf2, which is the
biomarker of tumour cell ferroptosis induced by sorafenib (41). Therefore, Nrf2 influences ferroptosis
through multiple downstream target genes and is considered the
central regulatory factor of ferroptosis. The present study also
revealed that the regulatory effect of knockdown or overexpression
of Nrf2 on CDDP sensitivity was significantly higher than that of
xCT, which may be associated with the simultaneous regulation of
additional Nrf2 target genes. Therefore, the ferroptosis induction
efficiency of Nrf2 inhibitors may be higher than that of downstream
target gene inhibitors. Although a variety of small molecules have
been reported to specifically inhibit Nrf2, their effects on
ferroptosis induction have rarely been reported on (42).
In conclusion, the present study revealed that Nrf2
pathway activation is one of the predominant regulatory mechanisms
underlying NSCLC cell resistance to CDDP. Erastin and sorafenib
could efficiently induce CDDP-resistant NSCLC cell ferroptosis. In
addition, a low concentration of CDDP in combination with
erastin/sorafenib effectively induced N5CP cell ferroptosis; the
potential mechanism by which sorafenib and erastin induced
ferroptosis in CDDP-resistant NSCLC cells may be associated with
inhibition of the expression of the Nrf2 downstream target gene
xCT. In a nude mouse xenograft model, erastin and sorafenib
markedly restrained N5CP cell growth. Ferroptosis inducers,
represented by erastin and sorafenib, may benefit the OS of
patients with advanced NSCLC or even following CDDP treatment
failure, which provides a novel perspective for the treatment of
NSCLC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL and LZ designed the research and wrote the paper.
YL, HYY, XMX and HBL implemented the experiments and data
collection. CW performed the statistical analyses and
interpretation. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
China Medical University, and written informed consent was obtained
from the patients. The animal study was approved by the
Institutional Animal Care and Use Committee of China Medical
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ARE
|
antioxidant response element
|
|
CDDP
|
cisplatin
|
|
DFO
|
deferoxamine mesylate
|
|
Gpx4
|
glutathione peroxidase 4
|
|
GSH
|
glutathione
|
|
HO-1
|
heme oxygenase 1
|
|
NSCLC
|
non-small cell lung cancer
|
|
NQO1
|
NAD(P)H quinone dehydrogenase 1
|
|
Nrf2
|
NF-E2 related factor 2
|
|
OS
|
overall survival
|
|
ROS
|
reactive oxygen species
|
|
RSL3
|
Ras selective lethal 3 compound
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Casiraghi M, Maisonneuve P, Piperno G,
Bellini R, Brambilla D, Petrella F, Marinis F and Spaggiari L:
Salvage surgery after definitive chemoradiotherapy for non-small
cell lung cancer. Semin Thorac Cardiovasc Surg. 29:233–241. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferlay J, Steliarova-Foucher E,
Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D and
Bray F: Cancer incidence and mortality patterns in Europe:
Estimates for 40 countries in 2012. Eur J Cancer. 49:1374–1403.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tiseo M, Franciosi V, Grossi F and
Ardizzoni A: Adjuvant chemotherapy for non-small cell lung cancer:
Ready for clinical practice? Eur J Cancer. 42:8–16. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roh JL, Kim EH, Jang HJ, Park JY and Shin
D: Induction of ferroptotic cell death for overcoming cisplatin
resistance of head and neck cancer. Cancer Lett. 381:96–103. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaminskyy VO, Piskunova T, Zborovskaya IB,
Tchevkina EM and Zhivotovsky B: Suppression of basal autophagy
reduces lung cancer cell proliferation and enhances
caspase-dependent and -independent apoptosis by stimulating ROS
formation. Autophagy. 8:1032–1044. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tian Y, Wu K, Liu Q, Han N, Zhang L, Chu Q
and Chen Y: Modification of platinum sensitivity by KEAP1/NRF2
signals in non-small cell lung cancer. J Hematol Oncol. 9:832016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hou X, Bai X, Gou X, Zeng H, Xia C, Zhuang
W, Chen X, Zhao Z, Huang M and Jin J:
3′,4′,5′,5,7-pentamethoxyflavone sensitizes Cisplatin-resistant
A549 cells to Cisplatin by inhibition of Nrf2 pathway. Mol Cells.
38:396–401. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ansari MA: Sinapic acid modulates
Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in
rats. Biomed Pharmacother. 93:646–653. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang P, Wang W, Wei Z, Xu LI, Yang X and
Du Y: xCT expression modulates cisplatin resistance in Tca8113
tongue carcinoma cells. Oncol Lett. 12:307–314. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ye P, Mimura J, Okada T, Sato H, Liu T,
Maruyama A, Ohyama C and Itoh K: Nrf2- and ATF4-dependent
upregulation of xCT modulates the sensitivity of T24 bladder
carcinoma cells to proteasome inhibition. Mol Cell Biol.
34:3421–3434. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lachaier E, Louandre C, Godin C, Saidak Z,
Baert M, Diouf M, Chauffert B and Galmiche A: Sorafenib induces
ferroptosis in human cancer cell lines originating from different
solid tumors. Anticancer Res. 34:6417–6422. 2014.PubMed/NCBI
|
|
16
|
Pillai MM, Elakkiya V, Gopinathan J,
Sabarinath C, Shanthakumari S, Sahanand KS, Dinakar Rai BK,
Bhattacharyya A and Selvakumar R: A combination of biomolecules
enhances expression of E-cadherin and peroxisome
proliferator-activated receptor gene leading to increased cell
proliferation in primary human meniscal cells: An in vitro study.
Cytotechnology. 68:1747–1761. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Homma S, Ishii Y, Morishima Y, Yamadori T,
Matsuno Y, Haraguchi N, Kikuchi N, Satoh H, Sakamoto T, Hizawa N,
et al: Nrf2 enhances cell proliferation and resistance to
anticancer drugs in human lung cancer. Clin Cancer Res.
15:3423–3432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Namani A, Cui QQ, Wu Y, Wang H, Wang XJ
and Tang X: NRF2-regulated metabolic gene signature as a prognostic
biomarker in non-small cell lung cancer. Oncotarget. 8:69847–69862.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen D, Fan Z, Rauh M, Buchfelder M,
Eyupoglu IY and Savaskan N: ATF4 promotes angiogenesis and neuronal
cell death and confers ferroptosis in a xCT-dependent manner.
Oncogene. 36:5593–5608. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hamilton G and Rath B: A short update on
cancer chemoresistance. Wien Med Wochenschr. 164:456–460. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Friedman R: Drug resistance in cancer:
Molecular evolution and compensatory proliferation. Oncotarget.
7:11746–11755. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Batra A, Thakar A and Bakhshi S:
Ototoxicity in retinoblastoma survivors treated with carboplatin
based chemotherapy: A cross-sectional study of 116 patients.
Pediatr Blood Cancer. 62:20602015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nishio M, Kim DW, Wu YL, Nakagawa K,
Solomon BJ, Shaw AT, Hashigaki S, Ohki E, Usari T, Paolini J, et
al: Crizotinib versus chemotherapy in asian patients with
ALK-positive advanced non-small cell lung cancer. Cancer Res Treat.
50:691–700. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reck M, Rodriguez-Abreu D, Robinson AG,
Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe
S, et al: Pembrolizumab versus chemotherapy for PD-L1-positive
non-small-cell lung cancer. N Engl J Med. 375:1823–1833. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Latunde-Dada GO: Ferroptosis: Role of
lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta
Gen Subj. 1861:1893–1900. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Macrez R, Stys PK, Vivien D, Lipton SA and
Docagne F: Mechanisms of glutamate toxicity in multiple sclerosis:
Biomarker and therapeutic opportunities. Lancet Neurol.
15:1089–1102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zenlea T and Peppercorn MA:
Immunosuppressive therapies for inflammatory bowel disease. World J
Gastroenterol. 20:3146–3152. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma MZ, Chen G, Wang P, Lu WH, Zhu CF, Song
M, Yang J, Wen S, Xu RH, Hu Y and Huang P: Xc- inhibitor
sulfasalazine sensitizes colorectal cancer to cisplatin by a
GSH-dependent mechanism. Cancer Lett. 368:88–96. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shitara K, Doi T, Nagano O, Fukutani M,
Hasegawa H, Nomura S, Sato A, Kuwata T, Asai K, Einaga Y, et al:
Phase 1 study of sulfasalazine and cisplatin for patients with
CD44v-positive gastric cancer refractory to cisplatin (EPOC1407).
Gastric Cancer. 20:1004–1009. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wilhelm SM, Adnane L, Newell P, Villanueva
A, Llovet JM and Lynch M: Preclinical overview of sorafenib, a
multikinase inhibitor that targets both Raf and VEGF and PDGF
receptor tyrosine kinase signaling. Mol Cancer Ther. 7:3129–3140.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rushmore TH, Morton MR and Pickett CB: The
antioxidant responsive element. Activation by oxidative stress and
identification of the DNA consensus sequence required for
functional activity. J Biol Chem. 266:11632–11639. 1991.PubMed/NCBI
|
|
39
|
Hayes JD and McMahon M: Molecular basis
for the contribution of the antioxidant responsive element to
cancer chemoprevention. Cancer Lett. 174:103–113. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chang LC, Chiang SK, Chen SE, Yu YL, Chou
RH and Chang WC: Heme oxygenase-1 mediates BAY 11–7085 induced
ferroptosis. Cancer Lett. 416:124–137. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Houessinon A, Francois C, Sauzay C,
Louandre C, Mongelard G, Godin C, Bodeau S, Takahashi S, Saidak Z,
Gutierrez L, et al: Metallothionein-1 as a biomarker of altered
redox metabolism in hepatocellular carcinoma cells exposed to
sorafenib. Mol Cancer. 15:382016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sun H, Zhu J, Lin H, Gu K and Feng F:
Recent progress in the development of small molecule Nrf2
modulators: A patent review (2012–2016). Expert Opin Ther Pat.
27:763–785. 2017. View Article : Google Scholar : PubMed/NCBI
|