Introduction
Esophageal cancer (EC) is one of the most common
malignancies that leads to high mortality and poor prognosis
worldwide (1). Approximately 572,034
new diagnosed cases and 508,585 deaths occurred in 2018 worldwide
(2). In China, esophageal squamous
cell carcinoma (ESCC), which is capable of direct invasion and
early metastasis, is the most prevalent pathological type of EC;
ESCC accounted for >90% of EC cases in 2011 in China (3,4). Despite
advances in diagnostic technologies and treatment, the overall
survival of patients with ESCC remains poor (5–7). Thus,
identifying the mechanistic basis of ESCC progression and
developing novel therapeutic targets for patients with ESCC is
urgently needed.
Cyclin-dependent kinase inhibitor 3 (CDKN3) serves
crucial roles in the cell cycle and proliferation (8–10). CDKN3
performs its functions by binding to cyclin proteins, which results
in dephosphorylation of CDK1 and CDK2 proteins and inhibition of
cell cycle progression (11–13). The dynamic expression of CDKN3 and
its oncogenic role have been widely explored in various types of
cancer (14–16). A previous study has reported that
CDKN3 is upregulated in cervical cancer and associated with reduced
survival time, indicating that it may be a potential biomarker for
patients with cervical cancer (15).
Zang et al (16) have
demonstrated that CDKN3 is expressed at high levels in lung
adenocarcinoma and is associated with poor survival outcomes.
Silencing CDKN3 suppresses cell proliferation and tumorigenesis in
nasopharyngeal carcinoma by regulating the expression of p27
(17). Deng et al (18) demonstrated that CDKN3 exhibits a high
expression in breast cancer cell lines, thus promoting apoptosis
and inhibiting cell migration. Xu et al (19) used pathway analysis to explore the
differentially expressed genes in ESCC; the results indicated that
CDKN3 is upregulated in ESCC and functions as a key gene in signal
transduction networks (including PI3K-Akt signaling pathway, and
cell cycle). Although a number of studies have demonstrated that
CDKN3 expression is upregulated in ESCC, limited information is
available regarding the function and mechanism of CDKN3 in ESCC
development.
In the present study, database search was used to
determine the levels of CDKN3 expression in ESCC tissues and cells.
Functional experiments were also employed to explore the functions
of CDKN3 in ESCC cells.
Materials and methods
Cell lines
ESCC cell lines EC-1 (cat. no. BNCC339894, l), EC-7
(named KYSE510; cat. no. BNCC342111), Eca-109 (cat. no. BNCC337687)
and TE-1 (cat. no. BNCC100151) were obtained from the BeNa Culture
Collection. An epithelial cell line Het1A (cat. no. ATCC CRL-2692)
was obtained from the American Type Culture Collection. ESCC cells
were cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% fetal bovine serum at 37°C with 5%
CO2.
Cell transfection
Small interfering (si)RNAs si-CDKN3 and si-NC were
obtained from Guangzhou RiboBio Co., Ltd. The sequences were as
follows: CDKN3 siRNA 1, 5′-GTGGAATTATCACCCATCA-3′; CDKN3 siRNA 2,
5′-CTGCTTGTCTCCTACTATA-3′; si-NC, 5′-GGCUCUAGAAAAGCCUAUGC-3′. For
transfection, Eca-109 and TE-1 cells were cultured in 6-well plates
(1.5×105 cells/well) and transfected with 2 µg si-CDKN3
or 2 µg si-NC using Lipofectamine® iMAX (Invitrogen;
Thermo Fisher Scientific, Inc.). Following transfection, Eca-109
and TE-1 cells were cultured at 37°C with 5% CO2 for 48
h prior to subsequent experiments. Reverse
transcription-quantitative PCR (RT-qPCR) and western blotting were
used to determine transfection efficiency.
RT-qPCR assay
RNA was extracted from Eca-109 and TE-1 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). cDNA was reverse-transcribed from RNA using the PrimeScript™
High Fidelity RT-PCR kit (Takara Biotechnology Co., Ltd.) for mRNA
expression analysis. The reaction conditions for reverse
transcription were 37°C for 15 min and 85°C for 5 sec. RT-qPCR was
conducted using SYBR®-Green (Applied Biosystems; Thermo
Fisher Scientific, Inc.) on an ABI7500 real-time PCR instrument
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The
thermocycling conditions were as follows: 95°C for 30 sec, followed
by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The specific
primers for CDKN3 and β-actin used were: CDKN3 forward,
5′-GTCCCAAACCTTCTGGATCTCTAC-3′ and reverse,
5′-AGCTCTTCCATTATTTCACAGCAG-3′; β-actin forward,
5′-GGACTTCGAGCAAGAGATGG-3′ and reverse, 5′-AGCACTGTGTTGGCGTACAG-3′.
The relative expression of CDKN3 was calculated using the
2−ΔΔCq method (20).
Cell proliferation assays
At 48 h post-transfection, 3×103
transfected cells/well (Eca-109 and TE-1 cells) were inoculated
into 96-well plates and incubated at 37°C with 5% CO2.
Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc.) solution
(10 µl) was added to each well for 1 h at 37°C and absorbance was
recorded at 450 nm on days 1, 2, 3 and 4. For colony formation
assay, transfected ESCC cells (100 cells/well) were seeded in a
6-well plate and incubated at 37°C with 5% CO2 for 10
days. The colonies were fixed with 4% formaldehyde for 30 min and
stained with 0.2% crystal violet for 2 h at room at room
temperature. Colonies containing >50 cells were counted using a
light microscope (magnification, ×200) using the ImageJ software
(National Institutes of Health).
Wound-healing assay
Wound-healing assay was used to detect the migration
of Eca-109 and TE-1 cells. ESCC cells were transfected with siCDKN3
or si-NC in six-well plates and cultured for 48 h until 90%
confluency was reached. A 200-µl pipette tip was used to create an
artificial wound. The cells were washed with PBS and cultured in
serum-free RPMI-1640 medium. Wound closure was visualized at 0 and
24 h using an inverted light microscope (magnification, ×200). The
distance between the edges of the wound was calculated as
previously described.
Invasion assay
Matrigel assay was used to determine cell
invasiveness. Transfected Eca-109 and TE-1 cells
(~2×104) were harvested, resuspended in 200-µl
serum-free RPMI-1640 medium and added into the upper Transwell
chamber pre-coated with Matrigel. The lower part of the chamber was
filled with medium containing 20% FBS. Following 12 or 24 h of
incubation, the cells on the top surface of the filters were
removed from the upper chamber using a cotton swab. The cells that
invaded through the Matrigel membrane were fixed with 4%
paraformaldehyde and stained with 0.5% crystal violet at 37°C for 2
h at room temperature. Cell images from five randomly fields were
selected and counted using a light microscope with ×200
magnification.
Flow cytometric analysis
Following transfection, 1×105 cells
(Eca-109 and TE-1 cells) were seeded onto 6-well plates and
cultured for 24 h at 37°C. The cells were harvested and fixed with
70% ethanol for 24 h at −20°C. The processed cells were stained
with propidium iodide (PI) staining solution (BD Pharmingen) at
37°C for 10 min. Cell cycle distribution was analyzed by
FACScalibur flow cytometer (BD Biosciences) and the ModFit LT
(version 3.2) software. All experiments were repeated three
times.
Western blot analysis
Transfected Eca-109 and TE-1 cells were suspended
and lysed with RIPA buffer (Beyotime Institute of Biotechnology).
Protein was extracted from the cells and quantified using a
Bicinchoninic Acid kit (Beyotime Institute of Biotechnology).
Protein (15 µg/lane) was separated using 10% SDS-PAGE and
transferred onto a PVDF membrane (EMD Millipore). The membrane was
subsequently blocked with 5% skimmed milk in Tris-buffered saline
containing 0.1% Tween-20 (TBST) for 2 h at 37°C and incubated at
4°C overnight with the following antibodies: Anti-CDKN3 (1:1,000;
cat. no. ab175393; Abcam), anti-AKT (1:1,000; cat. no. ab179463;
Abcam), anti-p-AKT (1:1,000; cat. no. ab38449; Abcam) and
anti-cyclinD1 (1:1,000; cat. no. 2922; Cell Signaling Technology,
Inc.). Anti-p27 (1:1,000; cat. no. 3686S; Cell Signaling
Technology, Inc.) and β-actin (1:2,000; cat. no. 4970; Cell
Signaling Technology, Inc.) were used as loading controls.
Following three washes with TBST, the membranes were incubated with
horseradish peroxidase-conjugated secondary goat anti-rabbit
immunoglobulin G antibody (1:1,000; cat. no. 7074; Cell Signaling
Technology, Inc.) for 1 h at 37°C. Immunoreactive bands were
visualized using ECL reagent (Beyotime Institute of
Biotechnology).
Bioinformatics analysis
The Cancer Genome Atlas (TCGA) analysis was
performed using the UALCAN website (http://ualcan.path.uab.edu/index.html) as previously
described (21). Microarray data of
mRNA expression from the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) data sets
GSE20347 and GSE70409 (22,23) were downloaded and analyzed by DESeq
packages in R (http://www.bioconductor.org/packages/release/bioc/html/DESeq.html).
The list of genes co-expressed with CDKN3 obtained from the UALCAN
database were annotated for Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analyses using DAVID (https://david.ncifcrf.gov/).
Statistical analysis
All experiments were repeated three times and data
are presented as the mean ± standard deviation. Data were processed
using SPSS 20.0 (IBM Corp.) and GraphPad Prism 7.0 (GraphPad
Software, Inc.). Student's t-test was used to compare the
differences between two groups. Comparisons among multiple groups
were analyzed using one-way ANOVA followed by Tukey's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
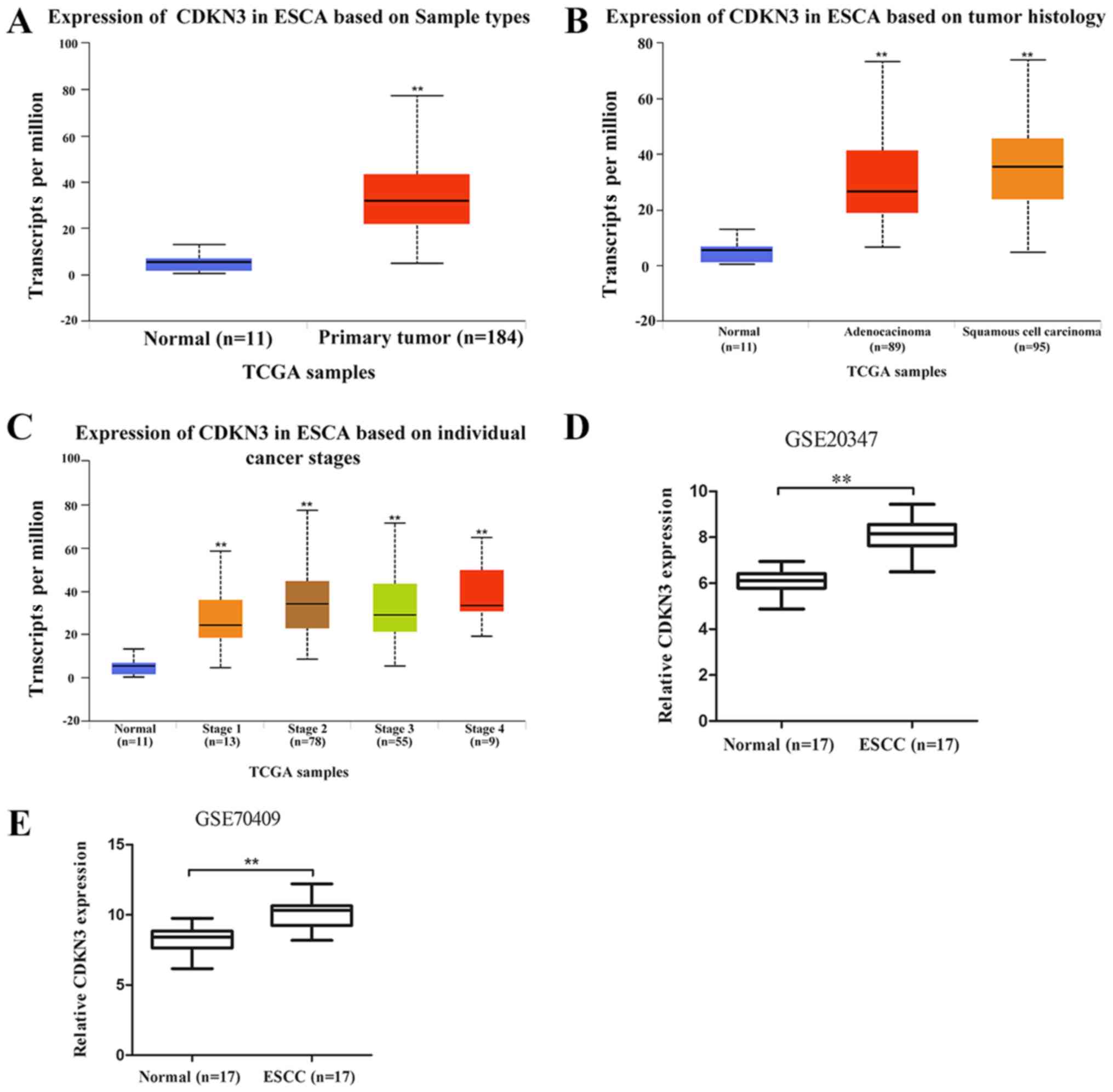
CDKN3 is upregulated in ESCC
To determine the role of CDKN3 in ESCC, an
interactive website UALCAN (http://ualcan.path.uab.edu), which is based on TCGA
gene expression data, was used to analyze CDKN3 expression in ESCC.
The results demonstrated that CDKN3 was significantly upregulated
in esophageal carcinoma (ESCA) tissues compared with normal samples
(Fig. 1A). The difference in CDKN3
expression between esophageal adenocarcinoma (EAC) and ESCC was not
significant (P=0.06); CDKN3 expression levels in EAC and ESCC were
higher compared with those in normal samples (Fig. 1B). The expression of CDKN3 in
patients with esophageal cancer, according to their stage, is shown
in Fig. 1C. The data from the GEO
database revealed higher CDKN3 expression in ESCC compared with
that in normal esophageal samples in the GSE20347 and GSE70409
datasets, which confirmed that CDKN3 was upregulated in ESCC.
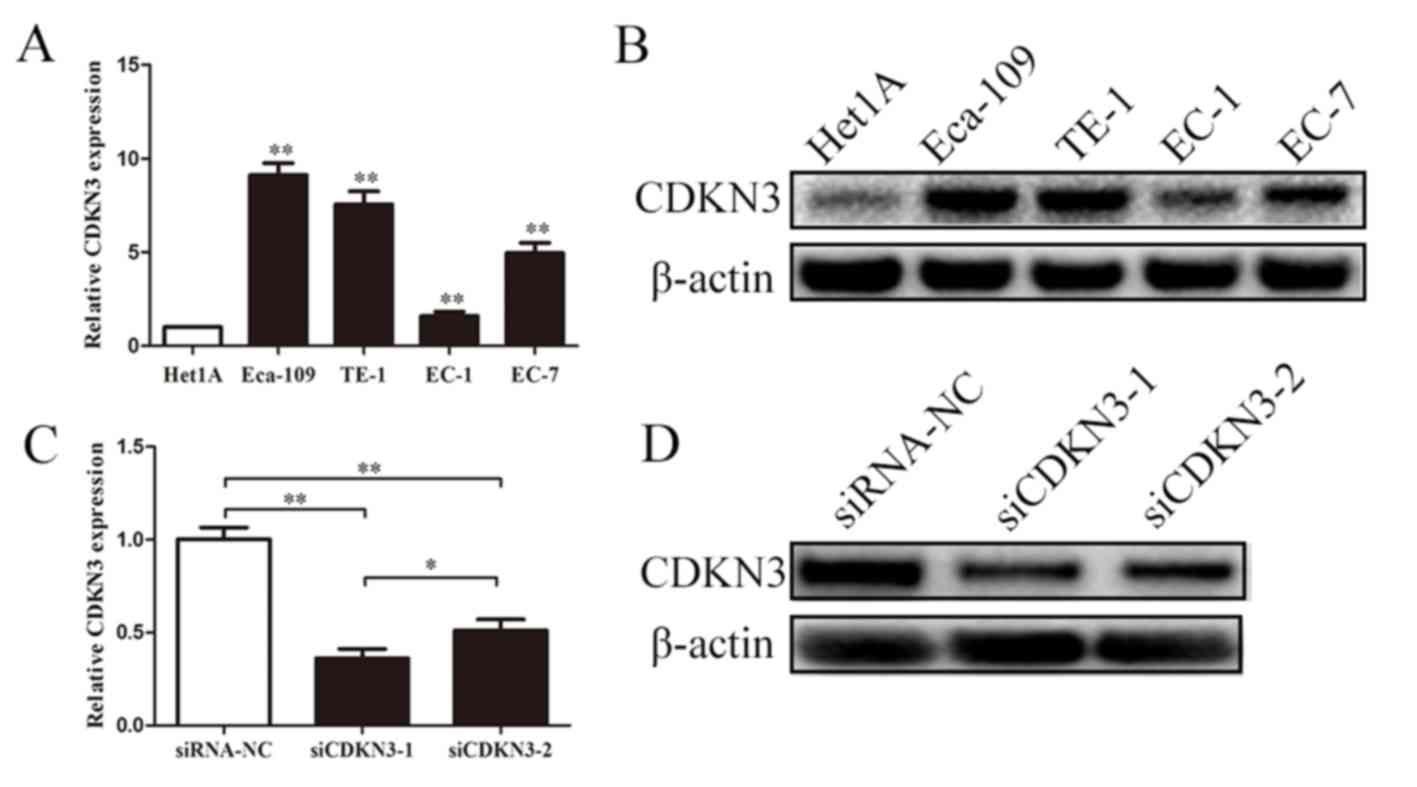
CDKN3 is upregulated in ESCC cell
lines and effectively knocked down by siRNA
CDKN3 expression in ESCC was examined using qPCR and
western blot analyses. The expression levels of CDKN3 were
significantly upregulated in EC-1, EC-7, Eca-109 and TE-1 cells
compared with those in normal esophageal epidermal cells (Het1A).
Subsequent experiments were performed to determine whether the two
siRNAs effectively knocked down CDKN3 expression in ESCC cells.
Transfection with siRNAs reduced the mRNA and protein expression
levels of CDKN3 compared with the negative control, and siCDKN3-1
exhibited a stronger inhibitory effect compared with siCDKN3-2
(Fig. 2C and D). These results
suggested that CDKN3 was upregulated in ESCC cells, and siCDKN3-1
was an effective approach for silencing CDKN3 expression.
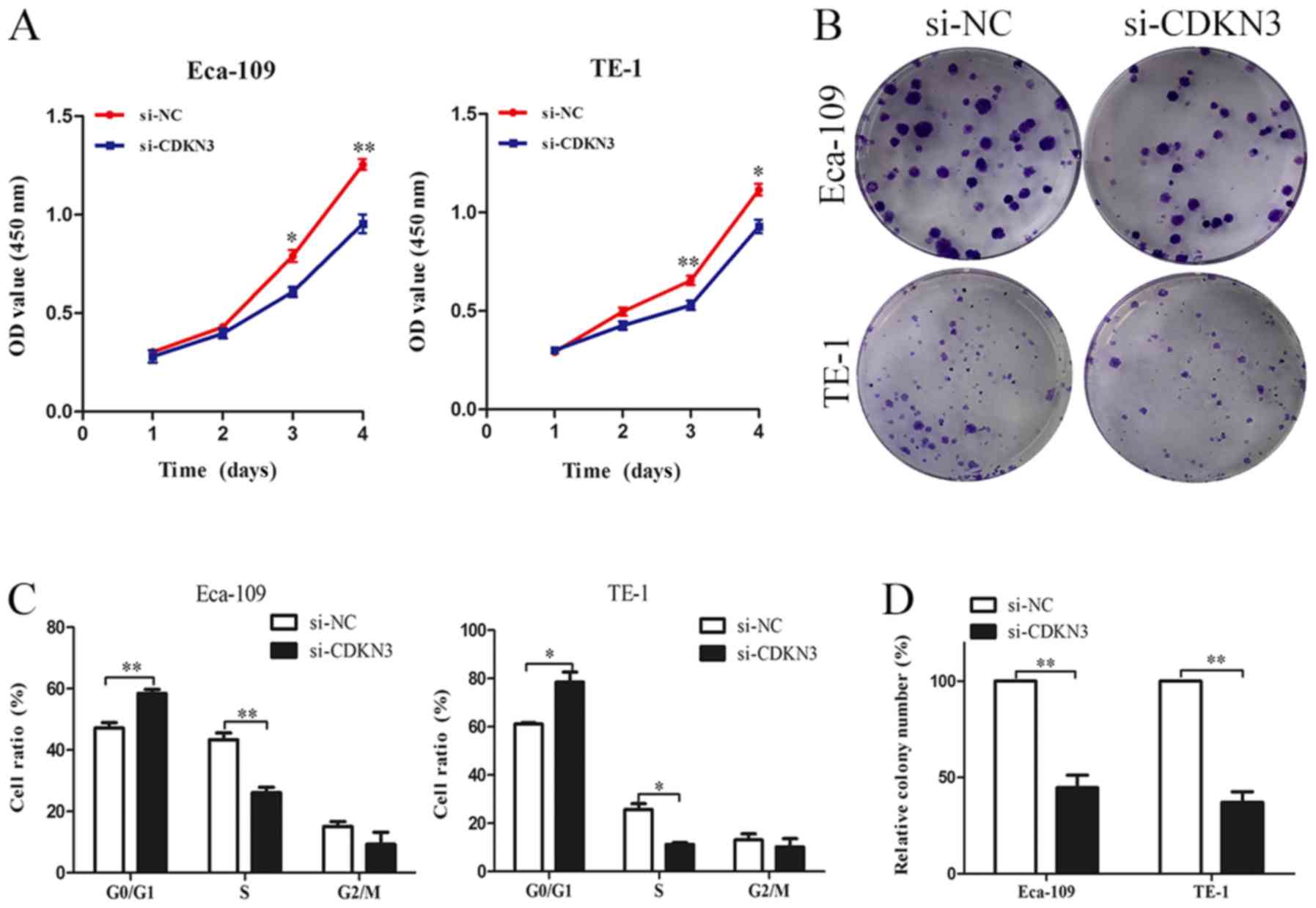
CDKN3 knockdown suppresses ESCC cell
proliferation, invasion and migration
The biological functions of CDKN3 in ESCC cell lines
were characterized. CCK-8 assay revealed that ESCC cells
transfected with si-CDKN3 exhibited lower cell viability compared
with the control groups (Fig. 3A).
The colony formation assay also demonstrated that CDKN3 knockdown
significantly reduced the proliferation of ESCC cells (Fig. 3B and D). The cell cycle analysis
results suggested that CDKN3 knockdown increased the proportion of
cells in the G0/G1 phase, which indicated that CDKN3 knockdown may
inhibit the proliferation of ESCC cells by suppressing the G1/S
transition (Fig. 3C). To determine
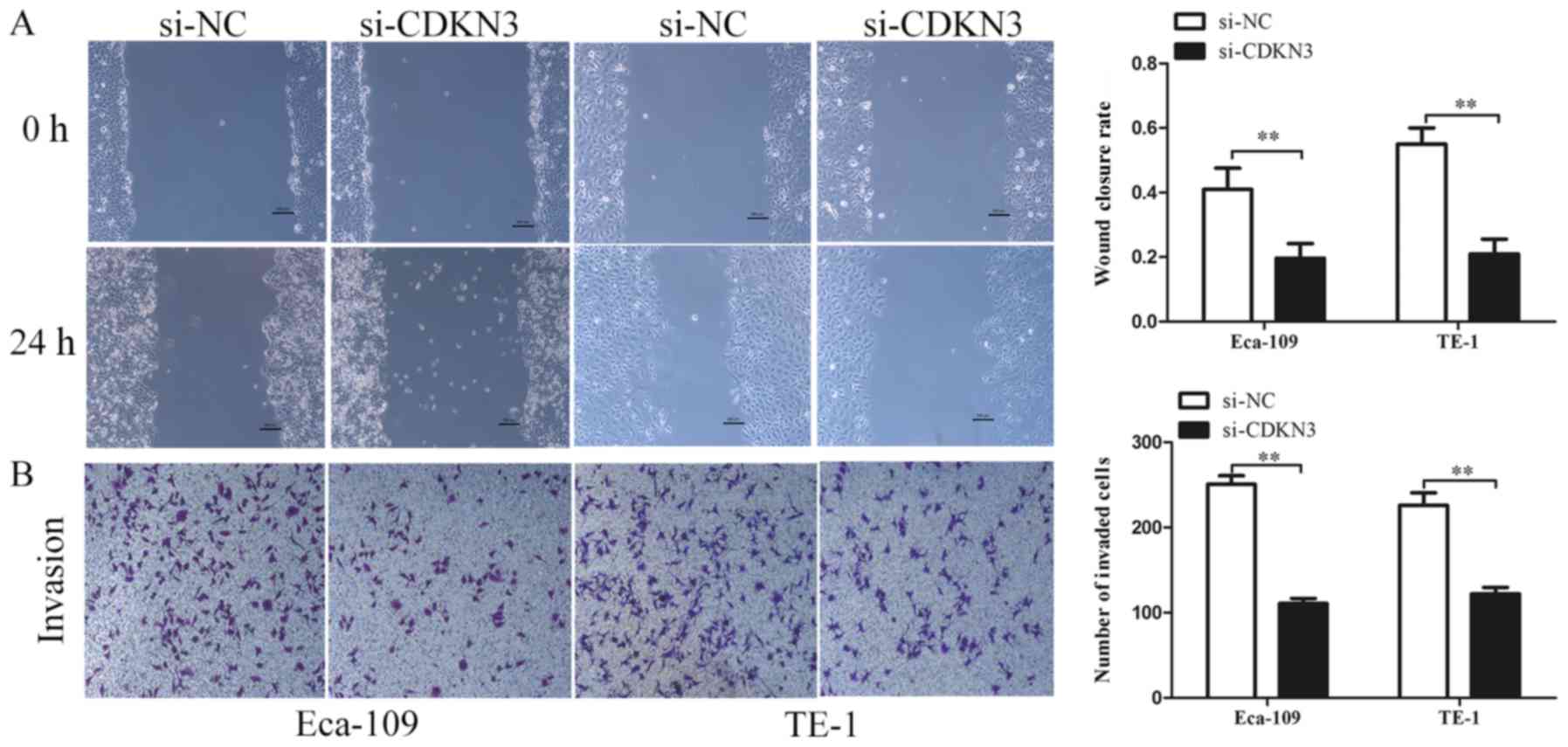
cell invasive and migratory abilities, wound-healing and Transwell
invasion assays were used. The wound closure rate of ESCC cells in
the si-CDKN3 groups was lower compared with that in the controls
(Fig. 4A). The Transwell invasion
assay revealed that silencing CDKN3 expression significantly
reduced the cell invasive capability compared with the control
groups (Fig. 4B). In conclusion, the
results of the functional assays suggested that CDKN3 enhanced the
viability, invasion and migration of ESCC cells.
CDKN3 knockdown reduces cell
proliferation and invasion by inhibiting the AKT signaling pathway
in ESCC
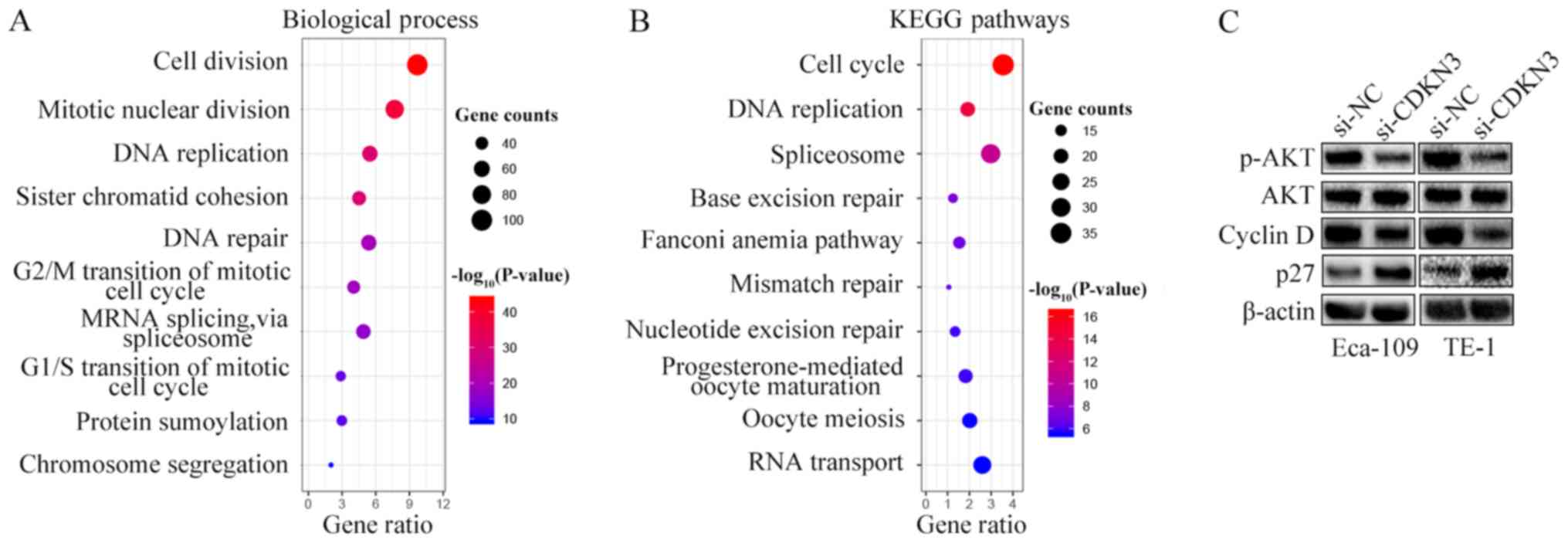
To explore the mechanisms underlying the cell
proliferation, invasion and migration enhancement by CDKN3, GO and
KEGG pathway enrichment analyses were performed by characterizing
the genes co-expressed with CDKN3 obtained from the UALCAN
database. Processes and pathways associated with the cell cycle
accounted for the highest enrichment for the co-expressed genes
(Fig. 5A and B). Since CDKN3
knockdown reduced ESCC cell proliferation, invasion and migration
and promoted the G0/G1 phase, the expression of cell cycle
signaling pathway-related proteins were detected by western blot
analysis. The results demonstrated that si-CDKN3 treatment
downregulated the levels of p-AKT and cyclin D1 and increased the
levels of p27 compared with si-NC treatment; however, no
differences were observed between the protein expression levels of
AKT in the two groups (Fig. 5C).
Thus, CDKN3 promoted cell proliferation and invasion by activating
the AKT signaling pathway.
Discussion
ESCC capable of direct invasion and early metastasis
is the fourth dominant cause of tumor-related mortality in China in
2015 (24). Identification of the
molecular basis underlying ESCC progression and the development of
novel diagnostic biomarkers is imperative. In the present study,
data from two databases, UALCAN and GEO, demonstrated that the
expression of CDKN3 was higher in ESCC compared with that in normal
esophageal tissues. The qPCR and western blot analyses also
revealed that CDKN3 was upregulated in ESCC cell lines. The
functional assays demonstrated that CDKN3 knockdown decreased the
capability of ESCC cells to proliferate, invade and migrate and
suppressed the G1/S transition. Further analysis indicated that
CDKN3 facilitated the promotion of ESCC cell proliferation,
invasion and migration by activating the AKT signaling pathway.
CDKN3 is a cyclin-dependent kinase (CDK) inhibitor
that serves important roles in regulating the cell cycle
progression (25). CDKN3 has been
reported to be involved in tumor development and progression
(14–17). Chang et al (26) have demonstrated that CDKN3 is
overexpressed in nasopharyngeal cancer (NPC) tissues and that high
expression of CDKN3 is associated with an advanced tumor stage and
poor survival; thus, CDKN3 may serve as a predictive target for
NPC. In cervical cancer, the expression of CDKN3 is associated with
poor survival, and silencing CDKN3 expression significantly reduces
cell proliferation (15). CDKN3
knockdown also inhibits cell invasion, proliferation and adhesion
and induces cell apoptosis; high CDKN3 expression predicts poor
outcomes, indicating that CDKN3 is involved in the oncogenic
transformation process (27). Xu
et al (19) used pathway
analysis to explore the differentially expressed genes in ESCC and
identified that CDKN3 is upregulated in ESCC and functions as a key
gene in signal transduction networks. The results of the present
study demonstrated that CDKN3 was upregulated in ESCC tissues and
cell lines compared with normal tissues and normal esophageal
epidermal cells, respectively. The CCK-8 and colony formation
assays indicated that si-CDKN3 treatment significantly reduced the
viability of ESCC cells, and flow cytometry demonstrated that
knockdown of CDKN3 promoted the G0/G1 phase compared with the
negative control groups. The wound-healing and Transwell invasion
assays confirmed that silencing CDKN3 expression significantly
reduced cell migratory and invasive capability compared with the
controls. However, a number of previous studies have reported that
CDNK3 causes opposite effects on tumor development. For example,
Dai et al (28) have
demonstrated that CDKN3 expression decreases in hepatocellular cell
carcinoma and is related to advanced tumor stage. Another study has
revealed that CDKN3 inhibition exaggerates cell survival by
activating the AKT pathway (24).
Studies explaining the deregulation of CDKN3 in different cancer
types are limited; additional experiments must be performed to
elucidate the role of CDKN3 in carcinogenesis.
A list of genes co-expressed with CDKN3 obtained
from the UALCAN database was analyzed in the present study to
explore the potential mechanism of CDKN3 in enhancing cell
proliferation, invasion and migration. The GO and KEGG results
demonstrated that the biological processes and pathways associated
with the cell cycle accounted for the highest enrichment of the
co-expressed genes. Wang et al (17) reported that CDKN3 performed its
biological functions by targeting p27 in NPC. By contrast, Dai
et al (28) have demonstrated
that silencing CDKN3 expression activates the AKT/p53/p21 axis. The
AKT signaling pathway serves important roles in inducing the cell
cycle, proliferation and invasion. Therefore, the protein levels of
the vital genes involved in the AKT signaling pathway (26–28) were
determined in the present study. The results demonstrated that
knockdown of CDKN3 suppressed the phosphorylation of AKT and the
expression of cyclin D1 and upregulated the expression of p27.
These results are inconsistent with those of Dai et al
(28). Further study is needed to
confirm the mechanism of CDKN3 in different types of cancer.
The experiments performed by Wang et al
(29) focused on the functions of
CDKN3 in ESCA progression and chemoresistance. ESCA includes EAC
and ESCC. In the present study, the role of CDKN3 in regulating
cell proliferation, invasion and migration was explored in ESCC
cells. Another study using bioinformatics analysis has identified
that CDKN3 is the key gene involved in signal transduction networks
(19); however, it did not explore
the functions and the underlying mechanism of CDKN3 in ESCC.
Neither of the two studies investigated the role of
CDKN3 in regulating ESCC cell proliferation, invasion and migration
(19,29). A recent study by Liu et al
(30) has reported that CDKN3
promoted the ESCC cell cycle, invasion and migration by modulating
the p-AKT-p53-p21-axis. The results of the present study were
consistent with their study and provided further evidence that
CDKN3 may regulate ESCC progression by activating the AKT signaling
pathway.
The present study is preliminary and there were
certain limitations. Only the expression levels of cyclin D1 and
p27 were detected, which was not conclusive. In future studies, the
expression levels of other cyclin-related proteins, such as cyclin
E, p21 and cyclin B, or other proteins in the AKT pathway will be
analyzed. In addition, the effects of CDKN3 on apoptosis and the
functions of CDKN3 in vivo need to be determined.
In conclusion, the results of the present study
demonstrated that CDKN3 was upregulated in ESCC tissues and cells
compared with the control groups, and CDKN3 knockdown significantly
reduced the proliferation, migration and invasion and suppressed
the G1/S transition of ESCC cell. Mechanistic analysis also
revealed that CDKN3 exerted its tumor-promoting effects partly by
inducing the expression of AKT signaling pathway-associated
proteins. These results supported the oncogenic role of CDKN3 in
the tumorigenesis of ESCC and provided a potential target for ESCC
treatment.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81672989), the Six Talent
Peaks Project in Jiangsu Province (grant no. LGY2016024) and the
Natural Science Foundation of Jiangsu Province (grant no.
H201614).
Availability of data and materials
The datasets used and/or analysed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HY and MD designed the study. HY and JY performed
the experiments. MD and JJY reviewed, analyzed and interpreted the
data. XH and LY developed the original concept, wrote and revised
the manuscript. All authors approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics. 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zeng H, Zheng R, Zhang S, Zuo T, Xia C,
Zou X and Chen W: Esophageal cancer statistics in China, 2011:
Estimates based on 177 cancer registries. Thorac Cancer. 7:232–237.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pennathur A, Gibson MK, Jobe BA and
Luketich JD: Oesophageal carcinoma. Lancet. 381:400–412. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang W, Li H, Chen X, Su M, Lin R and Zou
C: Phase II study of concurrent chemoradiotherapy with a modified
target volumes delineation method for inoperable oesophagealcancer
patients. Br J Radiol. 90:201703282017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lagergren J, Smyth E, Cunningham D and
Lagergren P: Oesophageal cancer. Lancet. 390:2383–2396. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cress WD, Yu P and Wu J: Expression and
alternative splicing of the cyclin-dependent kinase inhibitor-3
gene in human cancer. Int J Biochem Cell Biol. 91:98–101. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu D, Zhang J, Wu Y, Shi G, Yuan H, Lu Z,
Zhu Q, Wu P, Lu C, Guo F, et al: YY1 suppresses proliferation and
migration of pancreatic ductal adenocarcinoma by regulating the
CDKN3/MdM2/P53/P21 signaling pathway. Int J Cancer. 142:1392–1404.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu C, Cao H, He X, Sun P, Feng Y, Chen L
and Gong H: Cyclin-dependent kinase inhibitor 3 (CDKN3) plays a
critical role in prostate cancer via regulating cell cycle and DNA
replication signaling. Biomed Pharmacother. 96:1109–1118. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nalepa G, Barnholtz-Sloan J, Enzor R, Dey
D, He Y, Gehlhausen JR, Lehmann AS, Park SJ, Yang Y, Yang X, et al:
The tumor suppressor CDKN3 controls mitosis. J Cell Biol.
201:997–1012. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song H, Hanlon N, Brown NR, Noble ME,
Johnson LN and Barford D: Phosphoprotein-protein interactions
revealed by the crystal structure of kinase-associated phosphatase
in complex with phosphoCDK2. Mol Cell. 7:615–626. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yeh CT, Lu SC, Chao CH and Chao ML:
Abolishment of the interaction between cyclin-dependent kinase 2
and Cdk-associated protein phosphatase by a truncated KAP mutant.
Biochem Biophys Res Commun. 305:311–314. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang L, Sun L, Huang J and Jiang M:
Cyclin-dependent kinase inhibitor 3 (CDKN3) novel cell cycle
computational network between human non-malignancy associated
hepatitis/cirrhosis and hepatocellular carcinoma (HCC)
transformation. Cell Prolif. 44:291–299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barrón EV, Roman-Bassaure E,
Sánchez-Sandoval AL, Espinosa AM, Guardado-Estrada M, Medina I,
Juárez E, Alfaro A, Bermúdez M, Zamora R, et al: CDKN3 mRNA as a
biomarker for survival and therapeutic target in cervical cancer.
PLoS One. 10:e01373972015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zang X, Chen M, Zhou Y, Xiao G, Xie Y and
Wang X: Identifying CDKN3 gene expression as a prognostic biomarker
in lung adenocarcinoma via meta-analysis. Cancer Inform. 14 (Suppl
2):S183–S191. 2015.
|
|
17
|
Wang H, Chen H, Zhou H, Yu W and Lu Z:
Cyclin-dependent kinase inhibitor 3 promotes cancer cell
proliferation and tumorigenesis in nasopharyngeal carcinoma by
targeting p27. Oncol Res. 25:1431–1440. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deng M, Wang J, Chen Y, Zhang L, Xie G,
Liu Q, Zhang T, Yuan P and Liu D: Silencing cyclin-dependent kinase
inhibitor 3 inhibits the migration of breast cancer cell lines. Mol
Med Rep. 14:1523–1530. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu CQ, Zhu ST, Wang M, Guo SL, Sun XJ,
Cheng R, Xing J, Wang WH, Shao LL and Zhang ST: Pathway analysis of
differentially expressed genes in human esophageal squamous cell
carcinoma. Eur Rev Med Pharmacol Sci. 19:1652–1661. 2015.PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu N, Clifford RJ, Yang HH, Wang C,
Goldstein AM, Ding T, Taylor PR and Lee MP: Genome wide analysis of
DNA copy number neutral loss of heterozygosity (CNNLOH) and its
relation to gene expression in esophageal squamous cell carcinoma.
BMC Genomics. 11:5762010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen YK, Tung CW, Lee JY, Hung YC, Lee CH,
Chou SH, Lin HS, Wu MT and Wu IC: Plasma matrix metalloproteinase 1
improves the detection and survival prediction of esophageal
squamous cell carcinoma. Sci Rep. 6:300572016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lai MW, Chen TC, Pang ST and Yeh CT:
Overexpression of cyclin-dependent kinase-associated protein
phosphatase enhances cell proliferation in renal cancer cells. Urol
Oncol. 30:871–878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang SL, Chen TJ, Lee YE, Lee SW, Lin LC
and He HL: CDKN3 expression is an independent prognostic factor and
associated with advanced tumor stage in nasopharyngeal carcinoma.
Int J Med Sci. 15:992–998. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Y, Ji S, Fu LY, Jiang T, Wu D and Meng
FD: Knockdown of Cyclin-dependent kinase inhibitor 3 inhibits
proliferation and invasion in human gastric cancer cells. Oncol
Res. 25:721–731. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dai W, Miao H, Fang S, Fang T, Chen N and
Li M: CDKN3 expression is negatively associated with pathological
tumor stage and CDKN3 inhibition promotes cell survival in
hepatocellular carcinoma. Mol Med Rep. 14:1509–1514. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang J, Che W, Wang W, Su G, Zhen T and
Jiang Z: CDKN3 promotes tumor progression and confers cisplatin
resistance via RAD51 in esophageal cancer. Cancer Manag Res.
11:3253–3264. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu J, Min L, Zhu S, Guo Q, Li H, Zhang Z,
Zhao Y, Xu C and Zhang S: Cyclin-dependent kinase inhibitor 3
promoted cell proliferation by driving cell cycle from G1 to S
phase in esophageal squamous cell carcinoma. J Cancer.
10:1915–1922. 2019. View Article : Google Scholar : PubMed/NCBI
|