Introduction
Lung cancer is a common malignant tumor, and is the
leading cause of cancer-related death in the United States and
worldwide (1). The cancer occurs
below the bronchi, and there are no typical clinical symptoms in
the early stage. According to the characteristics of pathological
tissues, lung cancer can be classified into two major categories:
Small cell lung cancer (SCLC) and non-small cell lung cancer
(NSCLC). NSCLC is the most common subtype, accounting for >80%
of all lung cancer cases (2). The
lung squamous cell cancer (LUSC) subtype accounts for 25–30% of all
lung cancers, and is mainly presented as central lung cancer
(3). Studies have shown that
squamous cell carcinoma is associated with smoking, and is common
in men (1,2). However, not all smokers develop lung
cancer. Only 10–20% of smokers develop lung cancer in their
lifetime, which may be due to genetic differences in susceptibility
(4). In recent years, great progress
has been made in molecular targeted therapy for LUSC; however,
further research is required. Unfortunately, some patients do not
benefit from conventional chemotherapy and radiotherapy due to drug
resistance and toxic effects. Studies have shown that changes in
neoplasms at the molecular level occur earlier than various
clinical features (5,6). Therefore, research focusing on the
molecular level may be more conducive to the early diagnosis and
treatment of cancer to improve the prognosis. New diagnostic
markers and therapeutic targets are urgently needed to further
improve the diagnosis and treatment of LUSC, and to reduce the
fatality rate.
Although progress continues to be made in the
treatment of non-squamous NSCLC, the needs of patients with
squamous NSCLC remain unmet. With regard to targeted chemotherapy
drugs, there has been a focus on non-squamous cell disease in terms
of most regulatory approvals for advanced NSCLC, and updates to
clinical practice guidelines. Although the progress that has been
made with the therapeutic strategies of squamous NSCLC is limited,
the identification of more effective treatments for this patient
group is gaining momentum. Therefore, further research on LUSC will
provide new directions for treatment.
With the use of the current advanced DNA methylation
and RNA sequence research methods, great progress has also been
made in research on the relationship between DNA methylation and
gene expression during the onset and infiltration of neoplasms. In
a study of the integrative analysis of DNA methylation and mRNA
expression, Shi et al (7)
revealed the function of epigenetic changes on LUSC. However, the
mechanism contributing to oncogenesis is unclear, and in view of
this, Gevaert (8) developed
MethylMix, a new computational algorithm implemented in R software,
to identify abnormal methylated genes and predict changes in
transcription. The Cancer Genome Atlas (TCGA) (9), a well-known database on the cancer
genome, provides a large amount of genetic information and clinical
data, which can assist in understanding the clinical
characteristics of molecular information.
In the present study, LUSC-related genes with
abnormal methylation were identified in the TCGA database, and
associated differential genes of abnormal methylation in LUSC were
determined. Gene expression and abnormal methylation gene data of
LUSC samples from the TCGA database were analyzed. Four candidate
genes [DQX1 (DEAQ-box RNA dependent ATPase 1), GPR75
(probable G-protein coupled receptor 75), STX12 (syntaxin
12), and TRIM61 (putative tripartite motif-containing
protein 61)] were identified from 52 methylation-driven genes
(P<0.05), which may be independent prognostic biomarkers.
Furthermore, the genes ALG1L (ALG1
chitobiosyldiphosphodolichol beta-mannosyltransferase like),
DQX1, and ZNF418 (zinc finger protein 418) were
confirmed to meaningfully predict prognosis by integrative survival
analysis. In addition, a significant association between site
methylation and survival was found.
Materials and methods
Data acquisition and
preprocessing
Methylation and mRNA expression data of LUSC
patients were downloaded from the TCGA database (https://portal.gdc.cancer.gov/) (accessed March,
2019) (10). The methylation data
were obtained from 573 samples, including 69 normal samples and 504
cancer samples, and the mRNA expression data were obtained from 551
samples, including 49 normal samples and 502 LUSC samples. Clinical
survival data were also included. The LIMMA 3.40.6 package
(http://www.bioconductor.org/packages/release/bioc/html/limma.html)
(11) in R language was employed to
identify aberrant methylated genes and differentially expressed
genes between lung cancer and normal tissues.
Identification of methylation-driven
gene
The MethylMix (http://www.bioconductor.org/packages/release/bioc/html/MethyIMix.html),
an algorithm implemented by R 3.5.2 software (http://www.rproject.org/) was used to analyze the
correlation between gene methylation and gene expression and to
screen methylation-driven genes. The following three data files are
required: DNA methylation data for normal group; DNA methylation
data from cancer group; and matched gene expression data for cancer
group. Based on the MethylMix algorithm, the correlation between
the level of methylation and gene expression was calculated. Next,
genes significantly associated were identified and the
hypomethylation and hypermethylation genes were also determined by
the β-mixed model. Finally, methylation-driven genes were
identified (8,12). P<0.05 and Cor<0.3 were the
selected criteria for screening.
Gene Ontology (GO) enrichment
analysis
ConsensusPathDB (http://cpdb.molgen.mpg.de/CPDB, version 34) is an
online software integrating interaction networks in Homo
sapiens, including binary and complex signaling, gene
regulatory and drug-target interactions, as well as biochemical
pathways (13,14). The methylation-driven genes list was
submitted following the instructions on the website, the submit
list was clicked and the data were downloaded. Finally, the GOplot
R package was used to plot the enrichment results. P<0.05 was
set as the cut-off criterion for GO enrichment analysis.
Pathway analysis
ConsensusPathDB was also used to perform pathway
enrichment analysis of methylation-driven genes (14,15). The
pathway enrichment analysis of methylation-driven genes was
performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG)
database (http://www.genome.ad.jp/kegg/). The pathways were
selected with P<0.05 as the cut-off criterion.
Survival analysis of driver genes and
methylated sites
The methylation levels of driver genes were
extracted, followed by Kaplan-Meier analysis using the survival
analysis package in R software to compare the effects of different
methylation levels of driver genes on survival (16). The P-value was obtained using the
long-rank test. The level of gene methylation was then combined
with gene expression data to analyze the combined effect on
survival, which was also performed using the survival R package. In
addition, the methylation information on the related sites of the
methylation-driven genes was extracted, based on the downloaded
methylation and clinical data of squamous cell carcinoma from the
TCGA to further determine the value of the methylation-driven genes
for prognostic evaluation. The survival curve was drawn using the
survival R package. P<0.05 was considered to indicate a
statistically significant value.
Results
Screening of methylation-driven
genes
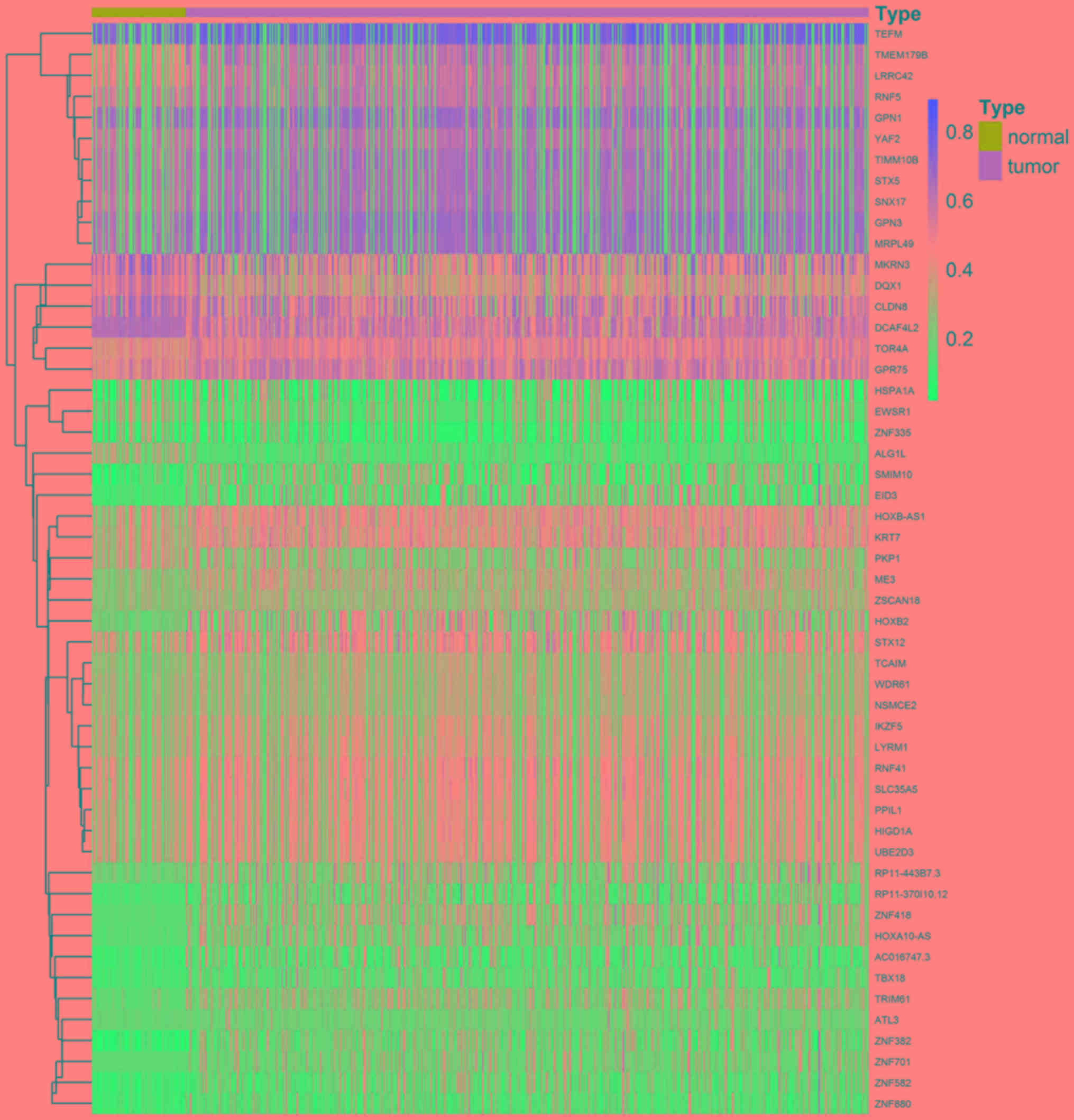
A total of 52 methylation-driven genes were
identified by comparing the levels of methylation in tumor and
normal tissues, and the genes were visualized using a heatmap
(Fig. 1). The methylation level of
44 genes in the cancer group was higher compared with that in the
normal group, and the methylation level of 8 genes was lower
compared with that of the normal group. Five methylation-driven
genes with the smallest P-values were selected to plot a
distribution map of the degree of methylation (Fig. 2A-E). The distribution of the
remainder of the genes is shown in Fig.
S1.
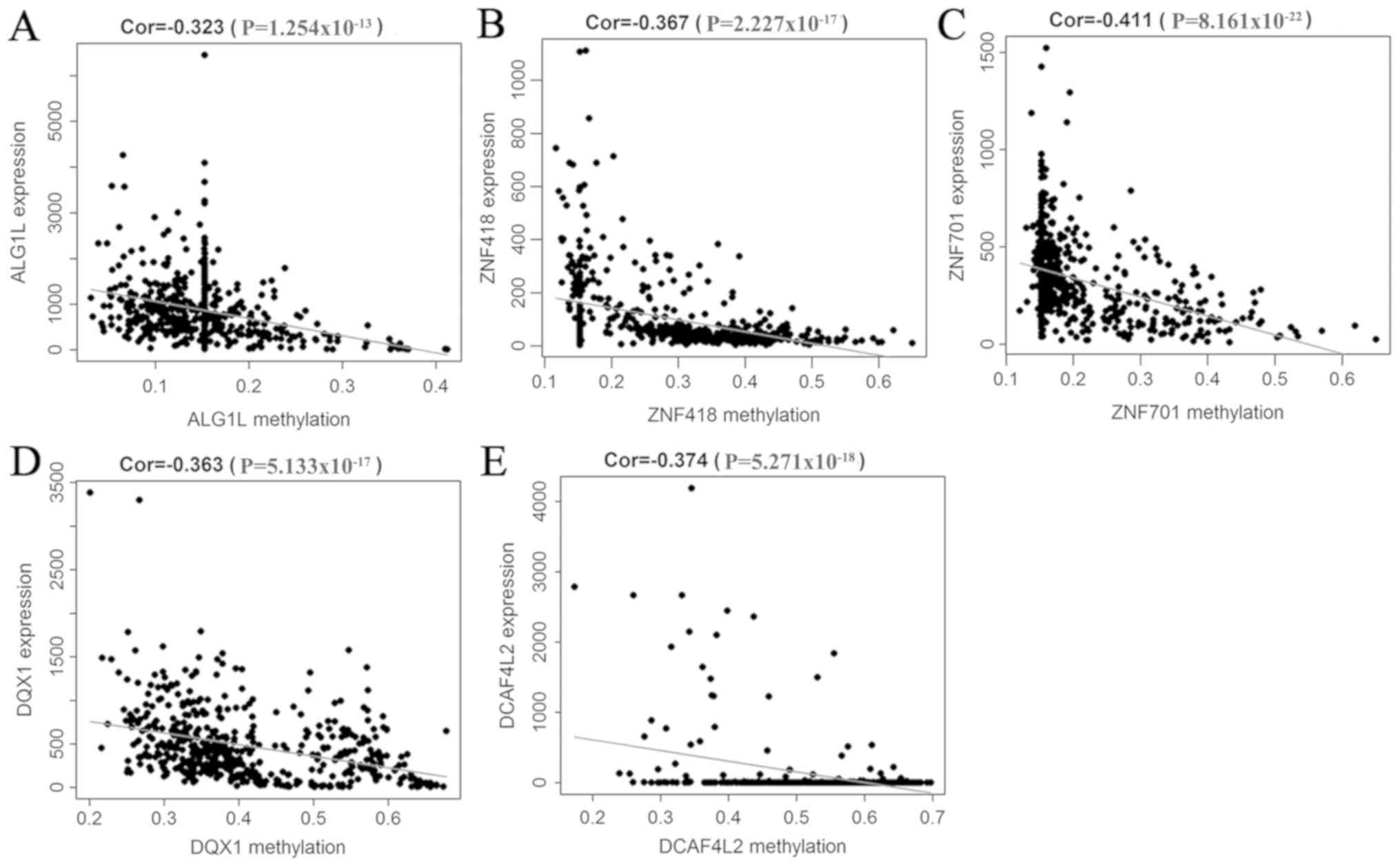
Correlation analysis between gene
methylation and expression
A correlation analysis between the methylation level
of 52 methylation-driven genes and their expression was performed,
which indicated that the level of methylation was negatively
correlated with the respective expression of these genes. Five
genes were selected to generate their scatterplots, and to estimate
the correlation coefficient. These genes were the first 5 with the
smallest P-values in the correlation test (Fig. 3A-E).
GO enrichment analysis
GO enrichment analysis was conducted using the
ConsensusPathDB online software. Methylation-driven genes were
enriched in ‘intracellular membrane-bounded organelle’, ‘Smc5-Smc6
complex’, and ‘SUMO ligase complex’ (Fig. 4).
Pathway analysis
The pathway enrichment analysis is shown in Fig. 5, and was conducted in ConsensusPathDB
online. In total, 13 pathways were enriched. These genes were
significantly linked to ‘BARD1 signaling events’, ‘Nicotine Pathway
(Dopaminergic Neuron)’ and ‘Pharmacodynamics’ (Fig. 5).
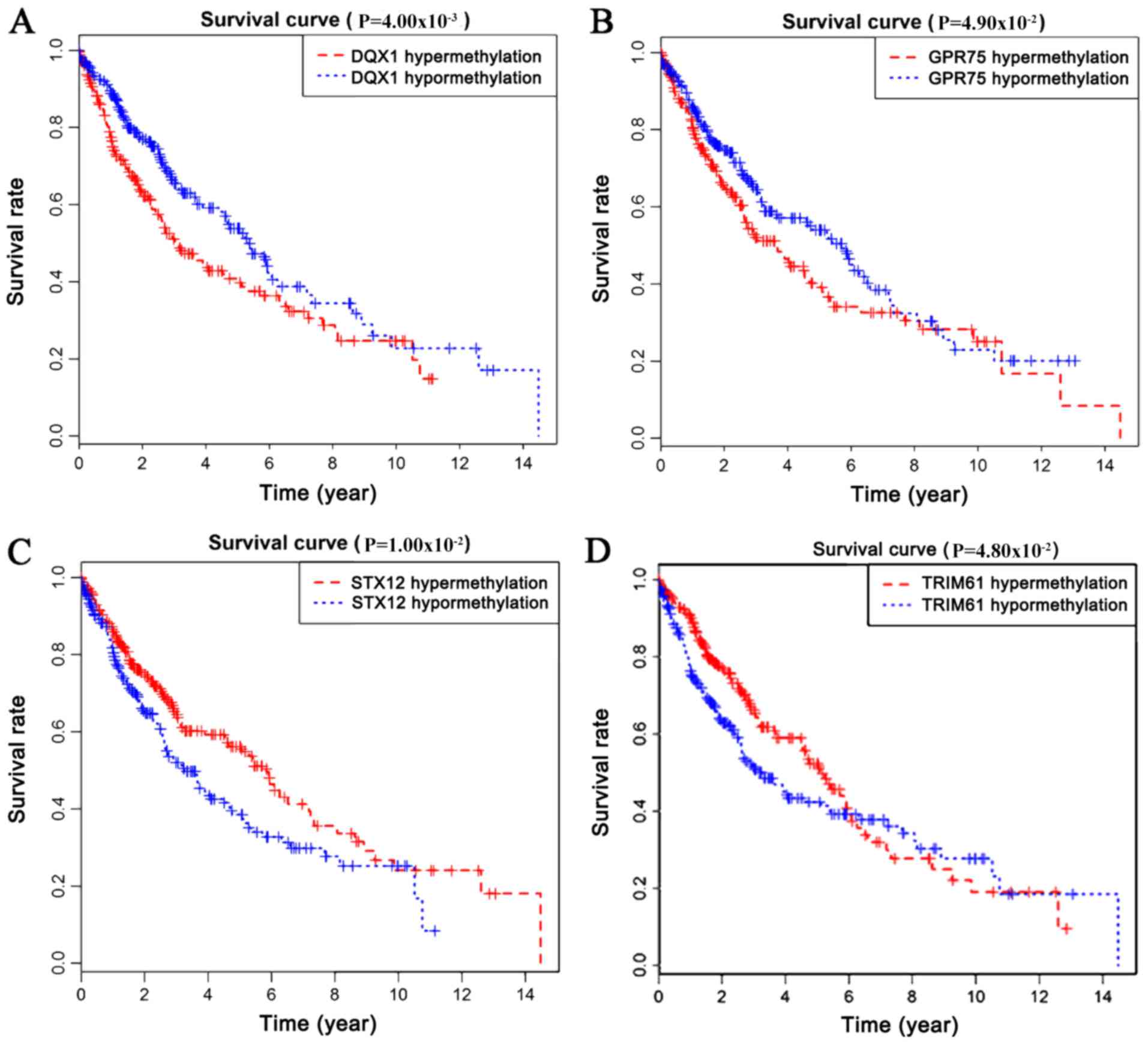
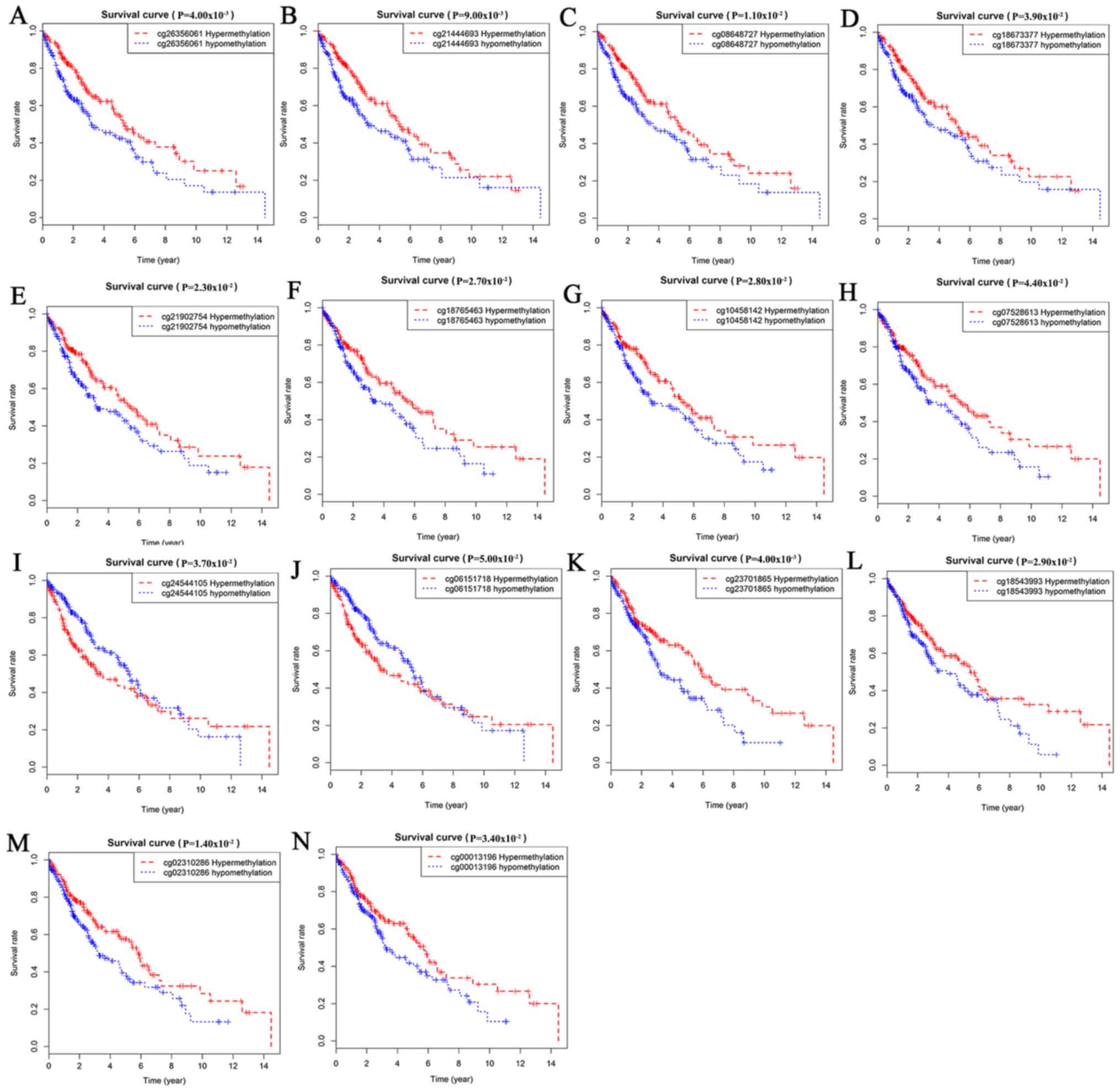
Survival analysis of
methylation-driven genes in LUSC
Survival analysis was statistically significant when
the P-value was <0.05. By analyzing the degree of gene
methylation, it was found that the genes DQX1, GPR75, STX12,
and TRIM61 significantly affected the survival and prognosis
of lung cancer patients (Fig. 6).
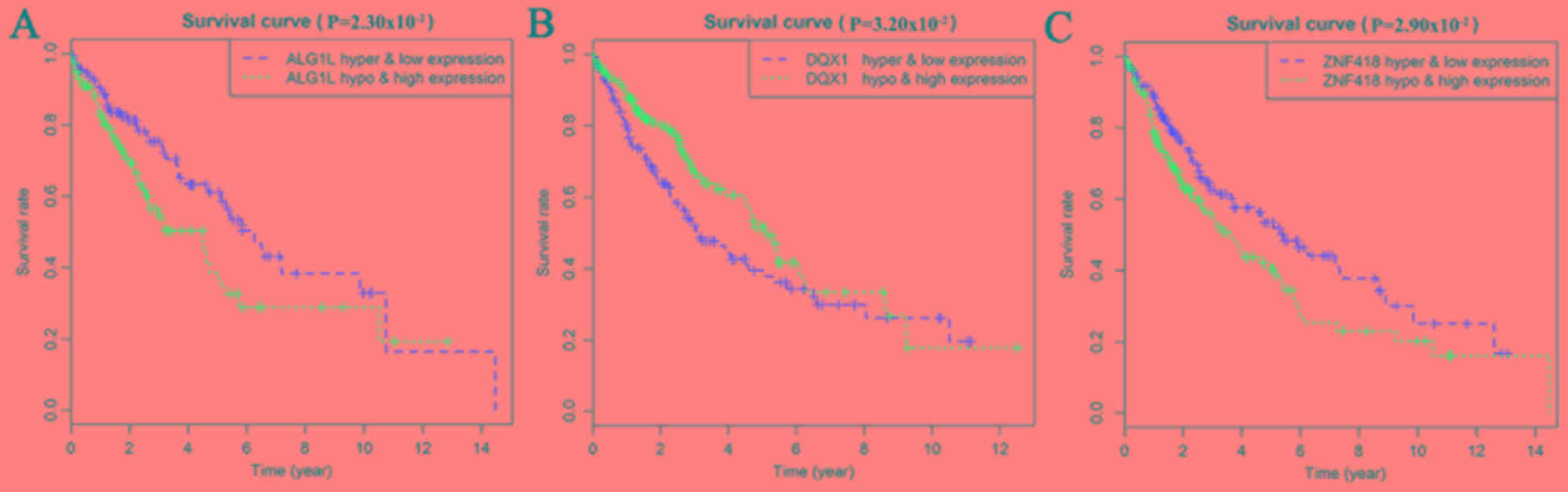
Taking genes methylation and expression as comprehensive factors,
it was confirmed that the genes ALG1L, DQX1, and
ZNF418 were closely related to prognosis (Fig. 7). The methylation levels of multiple
sites of ZNF418, ZNF701, DQX1 and DCAF4L2 were found
to be correlated with patient survival (Fig. 8).
Discussion
LUSC, which usually has a poor prognosis, is one of
the major subtypes of lung cancer, and is difficult to treat as
patients tend to be older and have a higher incidence of
comorbidities (17,18). The occurrence of lung cancer is an
intricate biological process involving multiple steps and factors,
and is closely related to changes in genetic information.
Epigenetic changes are involved in important aspects of lung cancer
development, and its regulatory mechanisms mainly involve DNA
methylation, and non-coding RNA expression regulation and histone
modification (19).
In recent years, significant advances have been made
in the molecular biological mechanisms of LUSC, early diagnostic
evaluation markers, as well as specific genetic alterations. DNA
methylation is an epigenetic mechanism that leads to tumorigenesis,
and has attracted a lot of attention from researchers. DNA
methylation abnormalities have been found at the genome level in
the majority of tumors, including lung cancer (20,21), and
the frequency of CpG island hypermethylation in tumor cells is much
higher compared with that of gene mutation (22). Abnormalities in DNA methylation often
occur in the early stages of cancer, and persist throughout the
development of the cancer. Methylation or demethylation may lead to
tumor gene activation or inactivation of cancer suppressor genes.
DNA methylation is considered to be a vitally important mechanism
that causes cells to change from a normal to malignant state
(23), and is the possible cause of
tumor treatment tolerance (24).
However, some studies have found that differentially methylated
genes serve as potential cancer driver genes (25,26).
Previous research findings have verified that
enhanced expression of genes caused by hypomethylation, and
decreased expression caused by hypermethylation, serve an important
role in the regulation and development of malignant carcinoma
(27,28). Methylation is a major epigenetic
modification of genomic DNA, and an important means of regulating
genomic function. Although epigenetic modifications are reversible,
they have great potential as effective therapeutic targets
(29). Therefore, the detection and
treatment of DNA methylation as a target will result in new
strategies for the diagnosis and treatment of lung cancer. For
example, Sugimoto et al (30)
found that patients benefit from aberrant methylation of
GRWD1 (glutamate rich WD repeat containing 1) in tumor
development, due to activity of the GRWD1 gene being
inhibited by its own methylation in tumor cells, whereas expression
of the GRWD1 gene can benefit tumor cell growth. In contrast, Chen
et al (31) found that
hypermethylation of the AGTR1 promoter is more common in
patients with LUSC. Ni et al (32) showed that the methylation of gene
SHOX2 (short stature homeobox 1) is more pronounced in lung
cancer, especially in LUSC, and is a potential non-invasive
biomarker of lung cancer. A study by Guo et al (33) found that the hypermethylated state of
WIF-1 gene, commonly found in NSCLC, is not only more likely to
occur in squamous cell carcinoma, but its expression was also
correlated with poor clinical prognosis. In a study on tumor and
corresponding non-malignant lung tissue specimens, Kim et al
(34) demonstrated that high
methylation of the Wrap53α promoter predicts a worse
prognosis in patients with borderline significance. Zhang et
al (35) reported that
PAX6 gene hypermethylation is an independent prognostic
indicator, and is significantly correlated with an overall low
survival rate of NSCLC, which may therefore be a potentially
attractive biomarker for prognostic assessment in NSCLC patients.
Thus, comprehensive functional and survival analysis of
methylation-driven genes is able to provide a deeper understanding
of their underlying mechanisms, and to identify novel strategies
for lung cancer therapy.
In the present study, our aim was to investigate
methylation-driven genes in patients with LUSC by analyzing data on
gene methylation downloaded from TCGA, and to assess their
relationship with prognosis. Fifty-two methylation-driven genes
were identified via the MethylMix package of R software. Enrichment
analysis was performed to investigate cellular functions and
pathways significantly associated with these genes, and to further
reveal the biological mechanism of these methylation-driven genes.
Functional enrichment analysis revealed that these genes are mainly
associated with ‘intracellular membrane-bounded organelle’,
‘Smc5-Smc6 complex’, and a variety of other functions, such as
‘SUMO ligase complex’. These genes were found to be involved in
‘BARD1 signaling events’, the ‘Nicotine Pathway (Dopaminergic
Neuron)’, and ‘Pharmacodynamics’ by enrichment pathway analysis.
These analyses revealed the function of these genes, and their
relationships with each other.
Survival analysis confirmed that methylation of the
genes DXQ1, GPR75, STX12 and TRIM61 is closely
related to prognosis. Survival analysis for combined gene
expression and methylation indicated that ALG1L, DQX1, and
ZNF418 could serve as markers to predict prognosis, and may
even be targets in future targeted therapy of LUSC. The methylation
levels of multiple sites were shown to be correlated with patient
survival.
A previous study found that ZNF418 is
expressed in a variety of tissues, such as lung, pancreas, muscle,
and heart (36). ZNF418 is
significantly down-regulated in gastric carcinoma tissues (37). In the present study, integrated
survival and gene expression and methylation analysis revealed that
the gene ZNF418 tends to predict a poor prognosis in
patients with LUSC. This suggests that ZNF418 may be
involved in the mechanism of progression in LUSC. DCAF4L2 is
a member of the WD-repeat domain-containing protein family, which
is an intermediary of protein-protein interaction. The levels of
DCAF4L2 have been previously reported to be elevated in lung
cancer and in human colorectal cancer, leading to worse clinical
staging, involving lymphatic and distant metastases. These findings
confirm its oncogenic role (38).
GPR75, a G protein-coupled receptor, is significantly
hypermethylated in colorectal neoplasia (39). Consistently, the present study also
showed that it is hypermethylated in LUSC, and this
hypermethylation is associated with infaust outcome of LUSC. STX12
is a member of the syntaxin family of soluble
N-ethylmaleimide-sensitive factor attachment protein
receptors (SNAREs). A previous study reported that STX12 is able to
mediate tumor cell invasion (40).
The present study has revealed that the expression of STX12
is negatively correlated with methylation, and hypermethylation of
STX12 is associated with favorable clinical results in LUSC.
These results are theoretically consistent with LUSC; however,
further validation of our findings is needed in future studies.
The study of methylation-driven genes has important
clinical significance for the early diagnosis and prognosis of lung
cancer. Methylation-driven genes are expected to be identified as
novel tumor markers for clinical application in the future. It is
plausible that our study provides credible potential targets for
the biological mechanism and clinical management of LUSC. However,
further research is required to confirm our findings.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Scientific
Research Projects of Chongqing Municipal Health Bureau
(2013-2-174), China.
Availability of data and materials
The datasets generated and/or analyzed in the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
PH and QL drafted the manuscript, QL collected and
organized the data, PH contributed to bioinformatics analysis by R
software, and JX contributed to the research design. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lemjabbar-Alaoui H, Hassan OU, Yang YW and
Buchanan P: Lung cancer: Biology and treatment options. Biochim
Biophys Acta. 1856:189–210. 2015.PubMed/NCBI
|
|
4
|
Lorenzo-González M, Fernández-Villar A and
Ruano-Ravina A: Disentangling tobacco-related lung
cancer-genome-wide interaction study of smoking behavior and
non-small cell lung cancer risk. J Thorac Dis. 11:10–13. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jordan EJ, Kim HR, Arcila ME, Barron D,
Chakravarty D, Gao J, Chang MT, Ni A, Kundra R, Jonsson P, et al:
Prospective comprehensive molecular characterization of lung
adenocarcinomas for efficient patient matching to approved and
emerging therapies. Cancer Discov. 7:596–609. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aoki MN, Amarante MK, de Oliveira CEC and
Watanabe MAE: Biomarkers in non-small cell lung cancer:
Perspectives of individualized targeted therapy. Anticancer Agents
Med Chem. 18:2070–2077. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi YX, Wang Y, Li X, Zhang W, Zhou HH,
Yin JY and Liu ZQ: Genome-wide DNA methylation profiling reveals
novel epigenetic signatures in squamous cell lung cancer. BMC
Genomics. 18:9012017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gevaert O: MethylMix: An R package for
identifying DNA methylation-driven genes. Bioinformatics.
31:1839–1841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tomczak K, Czerwinska P and Wiznerowicz M:
The cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
10
|
Grossman RL, Heath AP, Ferretti V, Varmus
HE, Lowy DR, Kibbe WA and Staudt LM: Toward a shared vision for
cancer genomic data. N Engl J Med. 375:1109–1112. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Law CW, Alhamdoosh M, Su S, Dong X, Tian
L, Smyth GK and Ritchie ME: RNA-seq analysis is easy as 1-2-3 with
limma, Glimma and edgeR. Version 3. F1000Res. 5:2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cedoz PL, Prunello M, Brennan K and
Gevaert O: MethylMix 2.0: An R package for identifying DNA
methylation genes. Bioinformatics. 34:3044–3046. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kamburov A, Pentchev K, Galicka H,
Wierling C, Lehrach H and Herwig R: ConsensusPathDB: Toward a more
complete picture of cell biology. Nucleic Acids Res. 39((Database
issue)): D712–D717. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kamburov A, Stelzl U, Lehrach H and Herwig
R: The ConsensusPathDB interaction database: 2013 update. Nucleic
Acids Res. 41((Database issue)): D793–D800. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herwig R, Hardt C, Lienhard M and Kamburov
A: Analyzing and interpreting genome data at the network level with
ConsensusPathDB. Nat Protoc. 11:1889–1907. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dudley WN, Wickham R and Coombs N: An
introduction to survival statistics: Kaplan-meier analysis. J Adv
Pract Oncol. 7:91–100. 2016.PubMed/NCBI
|
|
17
|
Langer CJ, Obasaju C, Bunn P, Bonomi P,
Gandara D, Hirsch FR, Kim ES, Natale RB, Novello S, Paz-Ares L, et
al: Incremental innovation and progress in advanced squamous cell
lung cancer: Current status and future impact of treatment. J
Thorac Oncol. 11:2066–2081. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Paes de Araújo R, Bertoni N, Seneda AL,
Felix TF, Carvalho M, Lewis KE, Hasimoto ÉN, Beckmann M, Drigo SA,
Reis PP and Mur LAJ: Defining metabolic rewiring in lung squamous
cell carcinoma. Metabolites. 9:E472019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim D and Kim DH: Epigenome-based
precision medicine in lung cancer. Methods Mol Biol. 1856:57–85.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qazi TJ, Quan Z, Mir A and Qing H:
Epigenetics in Alzheimer's disease: Perspective of DNA methylation.
Mol Neurobiol. 55:1026–1044. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pan Y, Liu G, Zhou F, Su B and Li Y: DNA
methylation profiles in cancer diagnosis and therapeutics. Clin Exp
Med. 18:1–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schmiemann V, Böcking A, Kazimirek M,
Onofre AS, Gabbert HE, Kappes R, Gerharz CD and Grote HJ:
Methylation assay for the diagnosis of lung cancer on bronchial
aspirates: A cohort study. Clin Cancer Res. 11:7728–7734. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baylin SB and Ohm JE: Epigenetic gene
silencing in cancer - a mechanism for early oncogenic pathway
addiction? Nat Rev Cancer. 6:107–116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Da Costa EM, McInnes G, Beaudry A and
Raynal NJ: DNA methylation-targeted drugs. Cancer J. 23:270–276.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Manolakos A, Ochoa I, Venkat K, Goldsmith
AJ and Gevaert O: CaMoDi: A new method for cancer module discovery.
BMC Genomics. 15 (Suppl 10):S82014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gevaert O, Villalobos V, Sikic BI and
Plevritis SK: Identification of ovarian cancer driver genes by
using module network integration of multi-omics data. Interface
Focus. 3:201300132013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bernstein BE, Meissner A and Lander ES:
The mammalian epigenome. Cell. 128:669–681. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Duruisseaux M and Esteller M: Lung cancer
epigenetics: From knowledge to applications. Semin Cancer Biol.
51:116–128. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jones PA, Issa JP and Baylin S: Targeting
the cancer epigenome for therapy. Nat Rev Genet. 17:630–641. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sugimoto N, Maehara K, Yoshida K,
Yasukouchi S, Osano S, Watanabe S, Aizawa M, Yugawa T, Kiyono T,
Kurumizaka H, et al: Cdt1-binding protein GRWD1 is a novel
histone-binding protein that facilitates MCM loading through its
influence on chromatin architecture. Nucleic Acids Res.
43:5898–5911. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen R, Hong Q, Jiang J, Chen X, Jiang Z,
Wang J, Liu S, Duan S and Shi S: AGTR1 promoter hypermethylation in
lung squamous cell carcinoma but not in lung adenocarcinoma. Oncol
Lett. 14:4989–4994. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ni S, Ye M and Huang T: Short stature
homeobox 2 methylation as a potential noninvasive biomarker in
bronchial aspirates for lung cancer diagnosis. Oncotarget.
8:61253–61263. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo H, Zhou S, Tan L, Wu X, Wu Z and Ran
R: Clinicopathological significance of WIF1 hypermethylation in
NSCLC, a meta-analysis and literature review. Oncotarget.
8:2550–2557. 2017.PubMed/NCBI
|
|
34
|
Kim DS, Lee WK and Park JY: Promoter
methylation of Wrap53α, an antisense transcript of p53, is
associated with the poor prognosis of patients with non-small cell
lung cancer. Oncol Lett. 16:5823–5828. 2018.PubMed/NCBI
|
|
35
|
Zhang X, Yang X, Wang J, Liang T, Gu Y and
Yang D: Down-regulation of PAX6 by promoter methylation is
associated with poor prognosis in non small cell lung cancer. Int J
Clin Exp Pathol. 8:11452–11457. 2015.PubMed/NCBI
|
|
36
|
Dang DT, Pevsner J and Yang VW: The
biology of the mammalian Krüppel-like family of transcription
factors. Int J Biochem Cell Biol. 32:1103–1121. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hui HX, Hu ZW, Jiang C, Wu J, Gao Y and
Wang XW: ZNF418 overexpression protects against gastric carcinoma
and prompts a good prognosis. Onco Targets Ther. 11:2763–2770.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang H, Chen Y, Han J, Meng Q, Xi Q, Wu G
and Zhang B: DCAF4L2 promotes colorectal cancer invasion and
metastasis via mediating degradation of NFκb negative regulator
PPM1B. Am J Transl Res. 8:405–418. 2016.PubMed/NCBI
|
|
39
|
Ashktorab H, Daremipouran M, Goel A, Varma
S, Leavitt R, Sun X and Brim H: DNA methylome profiling identifies
novel methylated genes in African American patients with colorectal
neoplasia. Epigenetics. 9:503–512. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Williams KC and Coppolino MG:
SNARE-dependent interaction of Src, EGFR and β1 integrin regulates
invadopodia formation and tumor cell invasion. J Cell Sci.
127:1712–1725. 2014. View Article : Google Scholar : PubMed/NCBI
|