Introduction
Colorectal cancer (CRC) is the fourth most
frequently diagnosed cancer and the second leading cause of
cancer-associated mortality worldwide according to the global
cancer statistics from 2018 (1). In
the United States, approximately 135,430 new cases are diagnosed
each year and approximately 50,260 patients die of CRC, which
accounts for approximately 9% of all cancer-associated mortalities
(2). In 2015, CRC was considered as
the fifth most common type of cancer in terms of occurrence and
mortality in China (3). Cancer stage
is a major factor contributing to the overall survival of patients
with CRC. The 3-year survival rate is 80–90% for patients with CRC
who are diagnosed with stages I or II, whereas the 3-year survival
rate is reduced to 20% for patients with CRC diagnosed with stage
IV (4).
Over the past decade, large-scale sequencing studies
have investigated the genetic basis of CRC and uncovered key
pathways involved in the pathogenesis of CRC, including WNT,
RAS-MAPK, PI3K, TGF-β, P53 and DNA mismatch repair pathways
(5–8). The Cancer Genome Atlas program
conducted a genome-scale analysis of 276 CRC samples using
multi-omics sequencing results and discovered that 16% of
hypermutated CRC samples are characterized by high microsatellite
instability, hypermethylation, mutL homolog 1 (MLH1)
silencing or somatic mutations in mismatch repair genes and
polymerases. In addition to the known mutations in the genes APC
regulator of WNT signaling pathway (APC), tumor protein 53
(TP53), SMAD family member 4 (SMAD4),
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha and KRAS proto-oncogene GTPase, 19 new recurrently mutated
genes have been detected, including AT-rich interaction domain 1A
(ARID1A), SRY-box transcription factor 9 (SOX9) and
APC membrane recruitment protein 1, and amplifications of the genes
erb-b2 receptor tyrosine kinase 2 and insulin like growth factor 2
have been identified (9).
Although our understanding of genomic alterations of
oncology has rapidly improved (5–9), the
uncommonly mutated genes in CRC, especially in the Asian
population, remain unknown. In the present study, targeted region
sequencing (508 genes) on 22 CRC and 10 paired non-cancerous
samples from patients with CRC was performed to study genetic
changes. The conclusion of this study may provide further insight
into genomic changes in CRC and determine potential therapeutic
targets in CRC.
Materials and methods
Patients and samples
A total of 22 primary CRC and 10 paired normal
colorectal tissues were surgically resected at Sanming Hospital
between May 2014 and August 2015 and immediately frozen. Patients
with secondary CRC were excluded from the present study. The mean
age of patients at diagnosis was 66.7±10.3 years and ranged between
35 and 80 years old. Written informed consent was obtained from all
patients and the study was approved by the Ethics Review Board of
the Sanming First Hospital. The clinical characteristics of
patients are presented in Table
I.
 | Table I.Clinical and demographic
characteristics of the 22 patients with CRC. |
Table I.
Clinical and demographic
characteristics of the 22 patients with CRC.
| Variable | CRC (n=22) |
|---|
| Sex |
|
|
Male | 9 |
|
Female | 13 |
| Tumor/adenoma/Polyp
location |
|
|
Rectum | 7 |
|
Sigmoideum | 1 |
|
Ascending colon | 7 |
|
Transverse colon | 1 |
|
Rectosigmoid | 1 |
|
Descending colon | 5 |
| Pathologic
diagnosis |
|
|
Protruding adenocarcinoma | 14 |
|
Protruding and mucinous
adenocarcinoma | 7 |
|
Protruding moderately
differentiated adenocarcinoma | 1 |
| Pathologic
stage |
|
| I | 6 |
| II | 11 |
|
III | 3 |
| IV | 2 |
| Tumor stage |
|
| T1 | 6 |
| T2 | 12 |
| T3 | 2 |
| T4 | 2 |
| Nodal stage |
|
| N0 | 18 |
| N1 | 1 |
| N2 | 3 |
| Metastasis
stage |
|
| M0 | 20 |
| M1 | 2 |
DNA extraction, library preparation
and sequencing
Genomic DNA was extracted from the 22 CRC and 10
paired normal colorectal tissues using the QIAamp DNA Blood Midi
Kit (Qiagen GmbH) according to the manufacturer's protocol. A
NanoDrop 2000 (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.) was used to assess DNA quality and concentration. Total DNA
samples (400 ng) were used to construct sequencing libraries
following the improved protocols for Illumina sequencing (10). In brief, genomic DNA samples were
fragmented and ligated with Illumina standard adapters to both
fragments ends. Adapter-ligated fragments are then PCR-amplified
and gel-purified. Libraries were pooled (11) and hybridized using the Oseq-T panel
(508 genes; BGI Genomics Co. Ltd.) for capturing. The process of
hybridization was performed as previously described (12). The hybridized product was sequenced
using the Illumina MiSeq platform (Illumina, Inc.) using a 100
paired-end (PE) sequencing strategy.
Variant calling and filtering
High-quality reads were aligned to the human
reference genome 19 (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/chromosomes/)
using MEM algorithms of Burrows-Wheeler-Aligner v.0.7.17
(http://bio-bwa.sourceforge.net) with
default parameters, except that the number of threads was set to 2
to accelerate this mapping process (-t 2) (13). Read deduplication was performed
utilizing Picard v.1.98 software (http://picard.sourceforge.net/), using the main
parameters as follows: ASSUME_SORTED=true and
VALIDATION_STRINGENCY=LENIENT. Local realignment around
insertion-deletions were performed using GATK v3.3.0
RealignerTargetCreator and IndelRealigner model (https://software.broadinstitute.org/gatk/) (14). Base quality score recalibration was
also performed by GATK v3.3.0 BaseRecalibrator and PrintReads model
(https://software.broadinstitute.org/gatk/) (13). Single nucleotide variant and
insertion-deletion (indel) mutations were detected by 3.3.0
HaplotypeCaller of GATK (https://software.broadinstitute.org/gatk/). New
variants and variants with maximum allele frequency <1% reported
in the 1000 genome project (15) and
dbsnp v141 database (16) were
considered as high confidence variants. High confidence variants
that were detected specifically in CRC samples but not in normal
colon tissues were regarded as somatic mutations. The SnpEff tool
(http://snpeff.sourceforge.net/SnpEff_manual.html)
was applied to annotate somatic mutations (17). The final somatic variants and
annotation results were used in the downstream analysis.
Driver gene prediction and functional
annotation
Driver genes were predicted using three distinct
computational tools, including MutSigCV (v1.0) (https://cloud.genepattern.org/gp/pages/index.jsf)
(18), oncodriveFM (v0.0.1)
(https://www.intogen.org) (19) and Integrated CAncer GEnome Score
(iCAGES) (http://icages.wglab.org) (20). All parameters were set to default
values. Driver genes were determined according to the following
criteria: i) Genes with q-value <0.1 in the MutSigCV analysis
(21); ii) genes with q-value less
<0.05 in the oncodriveFM analysis (19); and iii) genes predicted to be drivers
by iCAGES, with icagesGeneScores of predicted drivers >0.5
(22). To functionally annotate
these driver genes, the Database for Annotation, Visualization and
Integrated Discovery (https://david.ncifcrf.gov) (23) was applied to analyse the significant
Gene Ontology (GO; http://david.ncifcrf.gov) terms and Kyoto Encyclopedia
of Genes and Genomes (KEGG; http://david.ncifcrf.gov) (24) pathways in which driver genes were
enriched. Bonferroni adjusted P-value <0.05 indicated a
statistical significance.
Copy number variation analysis
Focal copy number variations (CNVs) were detected
using CONTRA v2.0.8 software (25)
between CRC and paired normal samples using default parameters,
(eg: contra.py -t *.bed -s *tumor.bam -c *normal.bam -f hg19.fasta
-o output-removeDups), except for ‘-removeDups’. For CRC samples
without paired normal tissues, deduplicated bam files of 10 normal
samples were merged to form one bam file. Focal CNVs were detected
between deduplicated bam files of CRC samples and the merged bam
file of 10 normal samples. All parameters were set to default
values. Focal CNVs with P-values <0.05 were considered
statistically significant. Fraction of CNV (FCNV) was computed as
follows: FCNV=(size of significant CNVs)/size of exons.
Statistical analyses
In the MutSigCV analysis, P-value was calculated for
the gene by convoluting the background distributions of all the
mutation types, and determining the probability of meeting or
exceeding that score by background mutation alone.
Benjamini-Hochberg false discovery rate procedure was used to
correct for P-values of multiple tests. In the oncodrive FM
analysis, the method randomly sampled one milliongroups of the same
number of observed mutations. P-value referred to the fraction of
functional impact scores equal to or greater than the observed
average functional impact score of the gene. Bonferroni correction
was applied to calculate the q-value for each gene. The Bonferroni
correction was used to adjust the P-values computed by Fisher's
exact test in the GO and KEGG pathway enrichment analyses.
Difference of quantitative values was compared between two groups
using Wilcoxon rank-sum test in R 3.2.0 (www.r-project.org).
Results
Clinical and demographic
characteristics of the 22 patients with CRC
The 22 patients with CRC comprised 13 female and 9
male patients. The primary sites of CRC included rectum (7 cases),
sigmoideum (1 case), ascending colon (7 cases), transverse colon (1
case), rectosigmoid (1 case) and descending colon (5 cases). The
pathological types of the 22 patients comprised 14 cases of
protruding adenocarcinoma, 7 cases of protruding and mucinous
adenocarcinoma and 1 case of protruding moderately differentiated
adenocarcinoma. In addition, 6, 11, 3 and 2 patients with CRC were
diagnosed with stages I, II, III and IV, respectively. Furthermore,
2 and 4 patients presented positive metastasis in lymph nodes and
distant organs, respectively (Table
I).
Summary statistics of alignment to
target regions
A 3.65 Mb target region comprising 508 genes was
captured from genomic DNA and used to perform variant calling. On
average, 98.87% reads were mapped successfully. The duplicated
reads were removed, resulting in an average of 4,312,287 (425.73
Mb) effective reads. Of the total effective bases, 69.95% was
mapped to target regions (capture specificity), and the mean
sequencing depth in the target regions was 81.57-fold. On average,
99.37% of targeted bases had at least 1× coverage, and 97.15% of
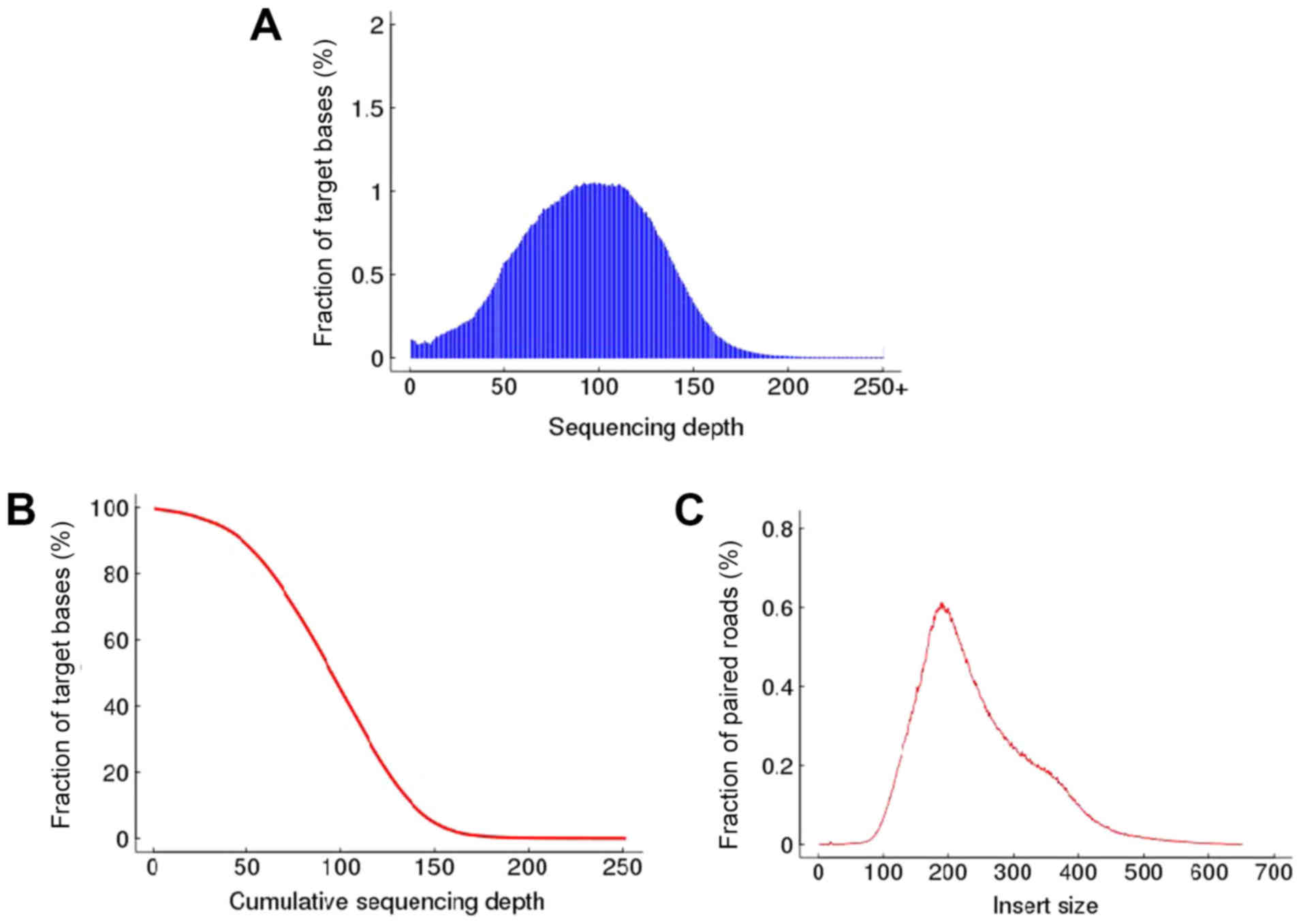
the targeted bases had at least 10× coverage (Table SI). In addition, the distributions
of per-base sequencing depth, cumulative sequencing depth and
insert size are presented in Fig. 1.
The density of sequencing depth on the target region followed
normal distribution, with a mean of 100X. As expected, the peak of
the insertion size distribution of the sequencing library was ~200
bp.
Somatic mutations in colorectal
cancer
To characterize the mutational spectrum, a targeted
region sequencing (3.65 Mb) on the 22 CRC and 10 paired normal
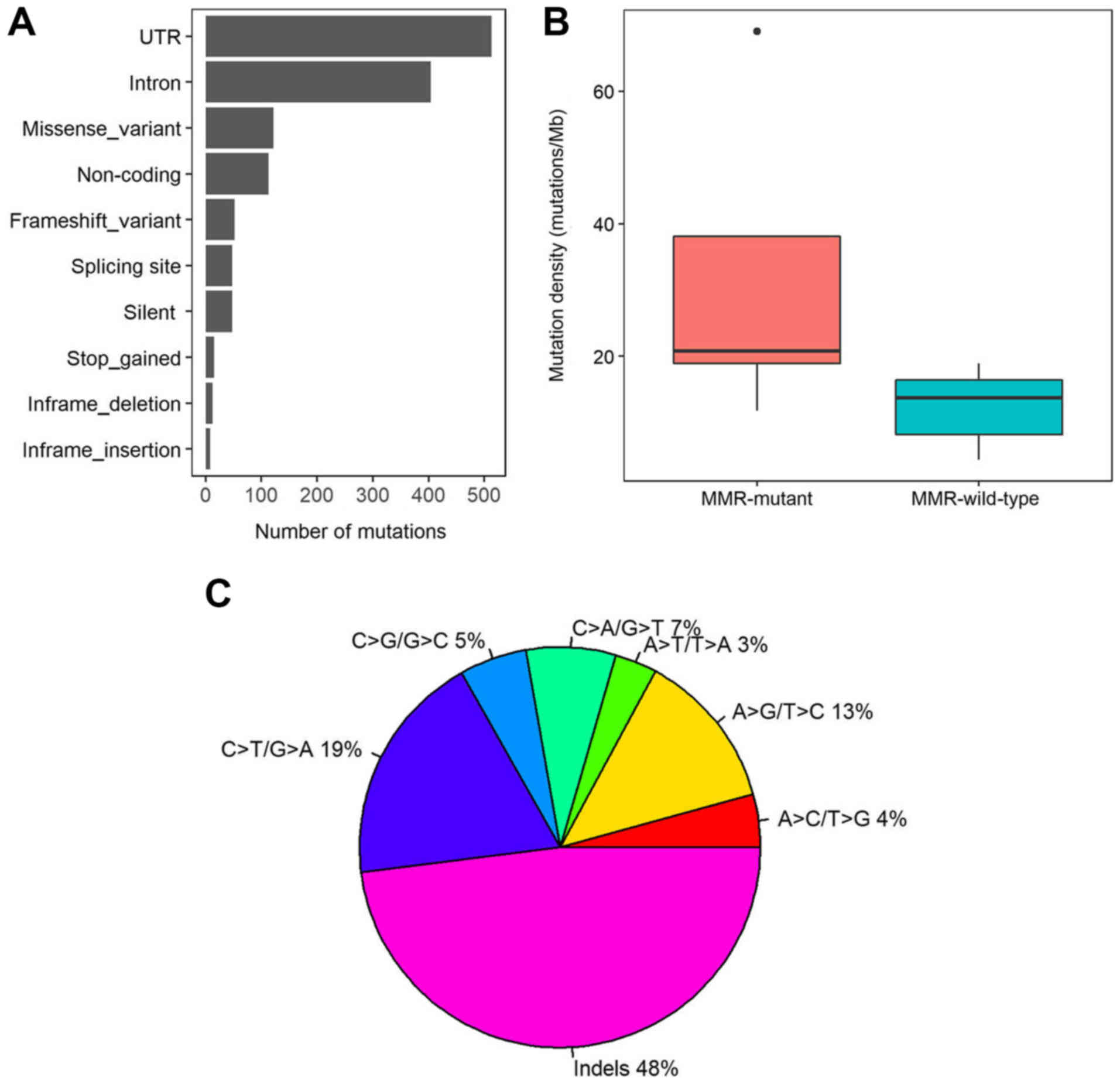
tissues was performed. A total of 1,335 somatic mutations were
identified, including 122 missense mutations, 48 silent variants,
48 splicing site variants, 13 inframe deletions and 8 inframe
insertions. Furthermore, 513, 404 and 113 variants were located in
the untranslated region, intron and noncoding regions,
respectively. In total, 52 variants caused a frame shift and 15
variants led to stop gain (Fig. 2A).
The somatic mutation rates varied considerably among the samples.
The average mutation density was 16.63 mutations/Mb in the 508-gene
panel (range, 4.38–69.04 mutations/Mb). To analyse the cause for
the different mutation densities, the mutation statuses in DNA
mismatch repair (MMR) pathway genes was assessed, including
MLH1, MLH3, mutS homolog 2 (MSH2), MSH3, MSH6
and PMS1 homolog 2. Five patients with CRC had mutations in any of
the MMR genes, and the average mutation density in MMR mutant
patients was significantly higher compared with MMR wild-type
patients (31.73 vs. 12.18 mutations/Mb; P=0.01; Wilcoxon rank sum
test; Fig. 2B). In addition, indels,
C>T/G>A and A>G/T>C were the three most prevalent
mutation types with mutation rates of 48, 18. 8, and 12.8%,
respectively (Fig. 2C).
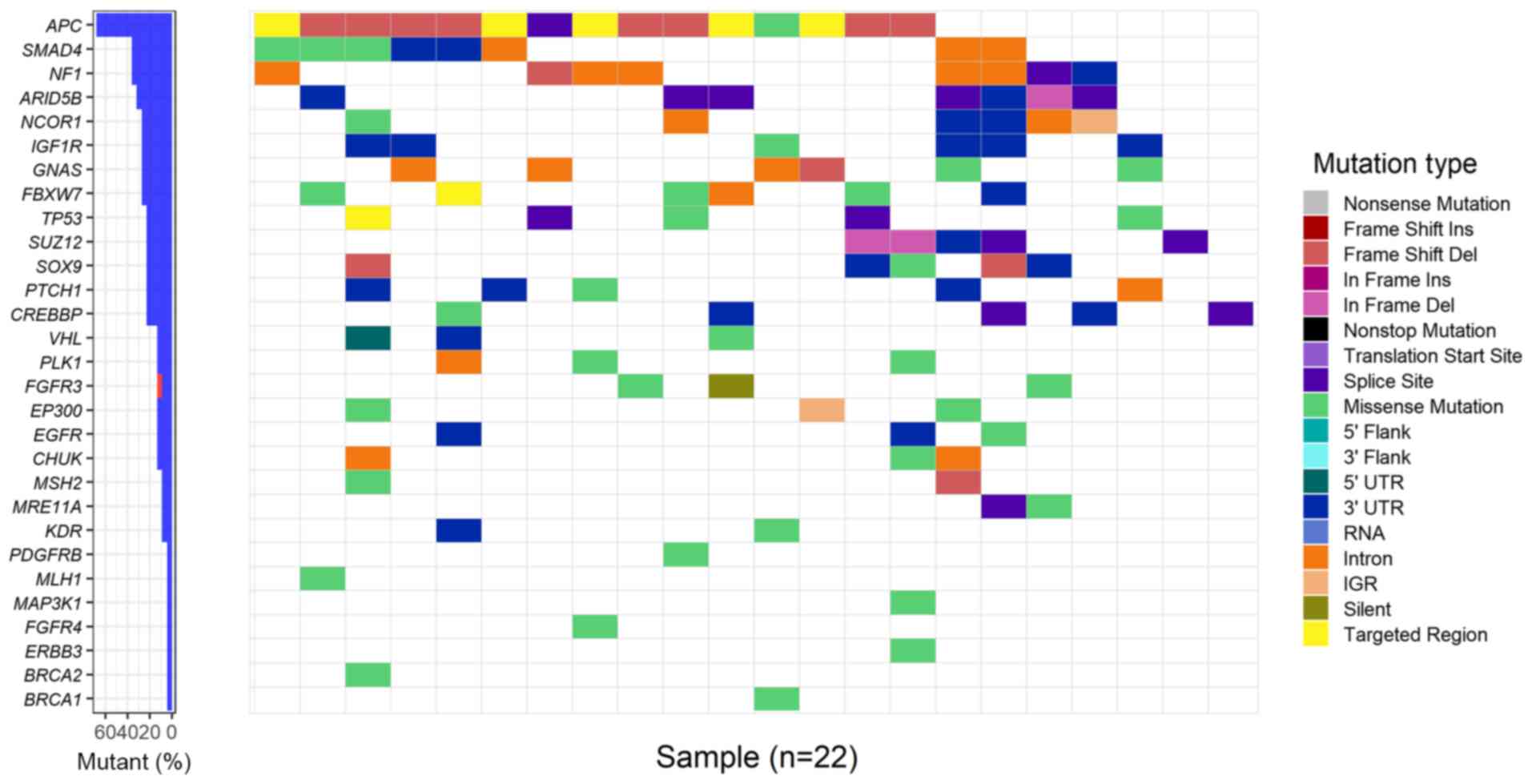
Driver genes in colorectal cancer
MutSigCV analysis identified four recurrently
mutated genes, named APC, TP53, SUZ12 polycomb repressive
complex 2 subunit (SUZ12) and AT-rich interaction domain 5B
(ARID5B), with significant statistical evidence (q-value
<0.2). Furthermore, OncodriveFM and iCAGES detected five and 24
driver genes, respectively. In total, 29 unique driver genes were
detected using these three computational tools. TP53 was the
overlapping gene among the three sets of driver genes. APC,
SMAD4, neurofibromin 1 (NF1), ARID5B and nuclear
receptor corepressor 1 (NCOR1) were the top five most
frequently mutated genes in patients with CRC with mutation rates
of 68, 36, 36, 32 and 27%, respectively (Fig. 3). In addition, the tumour-suppressor
genes APC and SMAD4 had an excessively high number of
frameshift or nonsense mutations. The average mutation rate was 18%
in the 29 driver genes (Table SII).
Certain driver genes, including mitogen-activated protein kinase
kinase kinase 1 (MAP3K1), FGFR4 and PDGFR-β,
were mutated at low frequencies (5% for all three genes), and
firstly reported as driver genes in CRC samples.
To functionally annotate the 29 driver genes, GO
term and KEGG pathway enrichment analyses were performed. The
results from GO enrichment analysis demonstrated that the 29 driver
genes were significantly enriched in 152 GO terms, including
‘positive regulation of cell proliferation’,
‘phosphatidylinositol-mediated signalling’, ‘cellular response to
DNA damage stimulus’ and ‘MAPK cascade’ (Bonferroni-adjusted
P<0.05; data not shown). Furthermore, the driver genes were
significantly enriched in 9 KEGG pathways, including ‘Wnt
signalling pathways’, ‘cell cycle’, ‘colorectal cancer’, ‘melanoma,
glioma’, ‘pancreatic and endometrial cancer’ (Bonferroni-adjusted
P<0.05; Table SIII).
Copy number variation analysis in CRC
samples
To further analyse the copy number variation in
driver genes, CONTRA was used to detect focal CNVs at the exon
level. The results demonstrated that 12 driver genes had FCNV
>20% in all CRC sample, including GNAS complex locus
(GNAS), BRCA2 DNA repair associated (BRCA2),
SOX9, fibroblast growth factor receptor 4 (FGFR4),
SMAD4, SUZ12, FGFR3, epidermal growth factor receptor
(EGFR), E1A binding protein p300, NCOR1, TP53 and
insulin like growth factor 1 receptor. In particular, GNAS
presented a significant focal gain >20% in 45% (10/22) of CRC
samples compared with normal samples. In addition, tumour
suppressor genes, including TP53, presented a significant
focal loss >20% in 22.7 (5/22) and 4.5% (1/22) of CRC samples
compared with normal samples. The EGFR oncogene exhibited a
significant focal gain >20% in 9.1% (2/22) of CRC samples
compared with normal samples (Table
II).
| Table II.Fraction of significant CNVs of
driver genes in colorectal cancer samples. |
Table II.
Fraction of significant CNVs of
driver genes in colorectal cancer samples.
| Genes | Sample | Fraction of
CNV | CNV type |
|---|
| GNAS | 13CB | 0.54 | Gain |
| GNAS | 15CB | 0.55 | Gain |
| GNAS | 16CB | 0.86 | Gain |
| GNAS | 18CB | 0.56 | Gain |
| GNAS | 19CB | 0.56 | Gain |
| GNAS | 20CB | 0.23 | Gain |
| GNAS | 21CB | 0.65 | Gain |
| GNAS | 31CB | 0.33 | Gain |
| GNAS | 36CB | 0.56 | Gain |
| GNAS | 41CB | 0.93 | Gain |
| BRCA2 | 17CB | 0.58 | Gain |
| BRCA2 | 28CB | 0.88 | Gain |
| BRCA2 | 30CB | 0.56 | Gain |
| BRCA2 | 32CB | 0.49 | Loss |
| BRCA2 | 35CB | 0.48 | Loss |
| SMAD4 | 19CB | 0.31 | Loss |
| SMAD4 | 30CB | 0.27 | Loss |
| SMAD4 | 31CB | 0.22 | Loss |
| SMAD4 | 40CD | 0.22 | Loss |
| SOX9 | 17CB | 0.29 | Loss |
| SOX9 | 18CB | 0.83 | Gain |
| SUZ12 | 19CB | 0.23 | Loss |
| SUZ12 | 30CB | 0.22 | Gain |
| FGFR3 | 11CB | 0.20 | Gain |
| FGFR3 | 18CB | 0.22 | Gain |
| EGFR | 16CB | 0.73 | Gain |
| EGFR | 31CB | 0.33 | Gain |
| EP300 | 16CB | 0.42 | Gain |
| NCOR1 | 19CB | 0.23 | Loss |
| TP53 | 39CB | 0.27 | Loss |
| IGF1R | 42CB | 0.25 | Loss |
| FGFR4 | 19CB | 0.20 | Gain |
Discussion
The activation of driver mutations might serve a
crucial role in cancer development (26). Genes that carry these driver
mutations are considered as driver genes and are essential to
tumorigenesis (27–31). The most common approach to predict
driver genes in numerous cancer samples is the identification of
genes that present significantly higher mutation frequencies
compared with the background mutation rate (18,32), via
the MutSigCV model for example. However, many driver genes may
occur at a low frequency. (<1%) in tumours (8). Novel computational tools have therefore
been developed to detect driver genes with middle or low mutation
frequencies. OncodriveFM first evaluates the functional impact of a
somatic mutation using the three different tools SIFT (33), PolyPhen2 (34) and MutationAssessor (35), and applies the transFIC method
(http://bg.upf.edu/transfic) to transform
the three functional scores into a uniform score (36). To identify driver genes, oncodriveFM
(19) compares the actual functional
impact with a null distribution model generated by 1,000,000
permutations and computes the bias towards the accumulation of
variants with high functional impact.
The iCAGES is a novel statistical framework that can
predict driver variants by integrating contributions from coding,
noncoding and structural variants. iCAGES can therefore identify
driver genes by combining genomic information and biological
knowledge to generate prioritized drug treatments (20). iCAGES consists of three consecutive
layers. The first layer prioritizes personalized cancer driver
coding, noncoding and structural variations. The second layer
associates these mutations to genes using a statistical model with
prior biological knowledge of cancer driver genes for specific
subtypes of cancer. The third layer generates a list of drugs
targeting the repertoire of these potential driver genes. Three
computational tools, including MutSigCV, oncodriveFM and iCAGES,
were applied to detect driver genes based on complementary
principles independently of somatic mutation recurrence. The
combination of the three tools enables the detection of recurrently
and rarely mutated driver genes in a more comprehensive manner than
with MutSigCV alone (9).
Over the last decade, large-scale sequencing
projects have been completed to characterize the mutation profiling
of CRC (9,37,38).
Recurrent gene mutations, including APC, KRAS
proto-oncogene, GTPase and titin genes, and dysregulated signalling
pathways, including the Wnt, tumour growth factor-β,
phosphoinositide 3-kinase and P53 signalling pathways, have been
reported to contribute to CRC carcinogenesis (9,37,38).
Giannakis et al (37)
identified recurrently mutated genes in CRC, including BCL9 like,
RNA binding motif protein 10, CCCTC-binding factor and Kruppel like
factor 5, which were not previously reported in CRC. R-spondin
(RSPO) gene fusions are present in 10% of colon tumours and are
mutually exclusive with APC mutations, which suggests that
they might be involved in the activation of Wnt signalling pathway
(38). In the present study, the use
of three computational algorithms, including MutSigCV, oncodriveFM
and iCAGES allowed the detection of 29 driver genes on 22 CRC
samples using somatic mutations. The results demonstrated that
APC, SMAD4, NF1, ARID5B and TP53 genes were frequently
mutated in patients with CRC, which was similar to results from a
previous study (9). Among the 29
driver genes, EGFR, kinase insert domain receptor and
platelet derived growth factor receptor beta (PDGFR-β) are
oncogenes, whereas von Hippel-Lindau tumor suppressor, NF1,
SUZ12, BRCA1 and BRCA2 are tumour suppressor genes,
according to the curated oncogene (39) and tumour suppressor gene (40) databases. In particular, certain
driver genes, including mitogen-activated protein kinase kinase
kinase 1 (MAP3K1), FGFR4 and PDGFR-β, had low
mutation frequencies. To the best of our knowledge, these three
genes were reported as driver genes in CRC samples for the first
time. MAP3K1 is a Ser/Thr protein kinase that belongs to the
mitogen-activated protein kinase kinase kinase 1
(MEKK)/serine/threonine protein kinase subgroup of the MEKK family.
Silencing MAP3K1 expression significantly enhances
paclitaxel-induced cell proliferation inhibition in breast cancer
cells, (41) pancreatic cancer cells
(42) and medulloblastoma cells
(43) and inhibits the human
pancreatic cancer cell invasive and migratory abilities (43). In accordance to previous studies
(41–44), this study suggests that MAP3K1
may have oncogenic role in cancers. It has been demonstrated that
FGFR4 serves the role of oncogene and regulator of drug or
radiation resistance in CRC (45).
Furthermore, FGFR4 silencing can inhibit colon cancer cell
line proliferation and induces caspase-dependent apoptosis
(45). In addition, FGFR4 can
regulate FLICE-like inhibitory protein expression via Signal
transducer and activator of transcription 3, conferring
resistance to 5-fluorouracil (5-FU) and oxaliplatin chemotherapy in
colon cancer cell lines (45).
Furthermore, it has been reported that FGFR induces
resistance to radiation therapy in CRC by downregulating RAD51
recombinase level and inhibiting cancer cell proliferation
(46). PDGFR-β is a cell
surface tyrosine kinase receptor for members of the PDGF family
(47). It has been demonstrated that
PDGFR-β high expression is positively correlated with
lymphatic metastasis and advanced UICC stages, and that
PDGFR-β high expression might be considered as a negative
factor for the prognosis of patients with colon cancer (48–50).
Decreased expression of PDGFR-β can inhibit the invasion and
proliferation of CRC cell lines (HCT116 and DLD-1) in a
dose-dependent manner (48). PDGFR-β
expression may therefore be considered as a risk factor for
recurrence in CRC, suggesting that PDGFR inhibitor could represent
a useful therapeutic agent for CRC (48). These newly identified driver genes
may be considered as novel candidates that could be used for
functional validation in future investigation.
In conclusion, the present study screened over 500
genes in order to analyse the genetic alterations present in 22 CRC
samples. A total of 29 driver genes were identified. Driver genes
with significant copy number variations, such as GNAS and
TP53, may be crucial in CRC oncogenesis. These findings
discoveries may serve as basis for further investigation on CRC
diagnosis and oncogenesis.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Mr. Licheng Cai and
Mr. Yanyang Si from BGI-Shenzhen for their contribution on sample
collection and project management.
Funding
The study was supported by the Natural Science
Foundation of Fujian Province (grant nos. 2015J01568 and
2016J01656.
Availability of data and materials
The raw sequencing data that support the findings of
this study have been deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb (accession
number, CNP0000601: published on 2019/09/30).
Author's contributions
YH designed the entire study. HX, YT and SZ
conducted library preparation and targeted region sequencing. JLuo,
SZ, JLi and MT were responsible for the bioinformatics analyses,
manuscript preparation and manuscript revision. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Review Board
of the Sanming First Hospital [SFHRB(2014) No 3] and all patients
provided informed consent prior to the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CNV
|
copy number variation
|
|
CRC
|
colorectal cancer
|
|
GO
|
Gene ontology
|
|
iCAGES
|
Integrated CAncer GEnome Score
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
MMR
|
mismatch repair
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–32. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Benitez Majano S, Di Girolamo C, Rachet B,
Maringe C, Guren MG, Glimelius B, Iversen LH, Schnell EA, Lundqvist
K, Christensen J, et al: Surgical treatment and survival from
colorectal cancer in Denmark, England, Norway, and Sweden: A
population-based study. Lancet Oncol. 20:74–87. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fearon ER: Molecular genetics of
colorectal cancer. Annu Rev Pathol. 6:479–507. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bass AJ, Lawrence MS, Brace LE, Ramos AH,
Drier Y, Cibulskis K, Sougnez C, Voet D, Saksena G, Sivachenko A,
et al: Genomic sequencing of colorectal adenocarcinomas identifies
a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 43:964–968. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sjöblom T, Jones S, Wood LD, Parsons DW,
Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al:
The consensus coding sequences of human breast and colorectal
cancers. Science. 314:268–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wood LD, Parsons DW, Jones S, Lin J,
Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bronner IF, Quail MA, Turner DJ and
Swerdlow H: Improved protocols for Illumina sequencing. Curr Protoc
Hum Genet. 80:18.2.1–42. 2014. View Article : Google Scholar
|
|
11
|
Wei X, Sun Y, Xie J, Shi Q, Qu N, Yang G,
Cai J, Yang Y, Liang Y, Wang W and Yi X: Next-generation sequencing
identifies a novel compound heterozygous mutation in MYO7A in a
Chinese patient with Usher Syndrome 1B. Clin Chim Acta.
413:1866–1871. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shao D, Lin Y, Liu J, Wan L, Liu Z, Cheng
S, Fei L, Deng R, Wang J, Chen X, et al: A targeted next-generation
sequencing method for identifying clinically relevant mutation
profiles in lung adenocarcinoma. Sci Rep. 6:223382016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gibbs RA, Boerwinkle E, Doddapaneni H, Zhu
H, Alkan C, Dal E, Kahveci F, Garrison EP, Kural D, Lee WP and
Dermitzakis ET: A global reference for human genetic variation.
Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cingolani P, Platts A, Wang le L, Coon M,
Nguyen T, Wang L, Land SJ, Lu X and Ruden DM: A program for
annotating and predicting the effects of single nucleotide
polymorphisms, SnpEff: SNPs in the genome of Drosophila
melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6:80–92.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lawrence MS, Stojanov P, Polak P, Kryukov
GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH,
Roberts SA, et al: Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 499:214–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gonzalez-Perez A and Lopez-Bigas N:
Functional impact bias reveals cancer drivers. Nucleic Acids Res.
40:e1692012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dong C, Guo Y, Yang H, He Z, Liu X and
Wang K: iCAGES: Integrated CAncer GEnome Score for comprehensively
prioritizing driver genes in personal cancer genomes. Genome Med.
8:1352016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tokheim CJ, Papadopoulos N, Kinzler KW,
Vogelstein B and Karchin R: Evaluating the evaluation of cancer
driver genes. Proc Natl Acad Sci USA. 113:14330–14335. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao X, Lei Y, Li G, Cheng Y, Yang H, Xie
L, Long H and Jiang R: Integrative analysis of cancer driver genes
in prostate adenocarcinoma. Mol Med Rep. 19:2707–2715.
2019.PubMed/NCBI
|
|
23
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drugs. Nucleic Acids Res. 45(D1): D353–D361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li J, Lupat R, Amarasinghe KC, Thompson
ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG and
Gorringe KL: CONTRA: Copy number analysis for targeted
resequencing. Bioinformatics. 28:1307–1313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Greenman C, Stephens P, Smith R, Dalgliesh
GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C,
et al: Patterns of somatic mutation in human cancer genomes.
Nature. 446:153–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barbieri CE, Baca SC, Lawrence MS,
Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van
Allen E, Stransky N, et al: Exome sequencing identifies recurrent
SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet.
44:685–689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grasso CS, Wu YM, Robinson DR, Cao X,
Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC,
et al: The mutational landscape of lethal castration-resistant
prostate cancer. Nature. 487:239–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Collisson EA, Campbell JD, Brooks AN,
Berger AH, Lee W, Chmielecki J, Beer DG, Cope L, Creighton CJ,
Danilova L, et al: Comprehensive molecular profiling of lung
adenocarcinoma. Nature. 511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sato Y, Yoshizato T, Shiraishi Y, Maekawa
S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki
H, et al: Integrated molecular analysis of clear-cell renal cell
carcinoma. Nat Genet. 45:860–867. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cancer Genome Atlas Research Network, .
Integrated genomic characterization of papillary thyroid carcinoma.
Cell. 159:676–690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dees ND, Zhang Q, Kandoth C, Wendl MC,
Schierding W, Koboldt DC, Mooney TB, Callaway MB, Dooling D, Mardis
ER, et al: MuSiC: Identifying mutational significance in cancer
genomes. Genome Res. 22:1589–1598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sim NL, Kumar P, Hu J, Henikoff S,
Schneider G and Ng PC: SIFT web server: Predicting effects of amino
acid substitutions on proteins. Nucleic Acids Res. 40((Web Server
Issue)): W452–W457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Reva B, Antipin Y and Sander C: Predicting
the functional impact of protein mutations: Application to cancer
genomics. Nucleic Acids Res. 39:e1182011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
González-Pérez A and López-Bigas N:
Improving the assessment of the outcome of nonsynonymous SNVs with
a consensus deleteriousness score, Condel. Am J Hum Genet.
88:440–449. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Giannakis M, Mu XJ, Shukla SA, Qian ZR,
Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et
al: Genomic correlates of immune-cell infiltrates in colorectal
carcinoma. Cell Rep. 15:857–865. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Seshagiri S, Stawiski EW, Durinck S,
Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman
V, Jaiswal BS, et al: Recurrent R-spondin fusions in colon cancer.
Nature. 488:660–664. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu Y, Sun J and Zhao M: ONGene: A
literature-based database for human oncogenes. J Genet Genomics.
44:119–121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao M, Sun J and Zhao Z: TSGene: A web
resource for tumor suppressor genes. Nucleic Acids Res.
41((Database Issue)): D970–D976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hu P, Huang Q, Li Z, Wu X, Ouyang Q, Chen
J and Cao Y: Silencing MAP3K1 expression through RNA interference
enhances paclitaxel-induced cell cycle arrest in human breast
cancer cells. Mol Biol Rep. 41:19–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hirano T, Shino Y, Saito T, Komoda F,
Okutomi Y, Takeda A, Ishihara T, Yamaguchi T, Saisho H and
Shirasawa H: Dominant negative MEKK1 inhibits survival of
pancreatic cancer cells. Oncogene. 21:5923–5928. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Antonucci L, Di Magno L, D'Amico D, Manni
S, Serrao SM, Di Pastena F, Bordone R, Yurtsever ZN, Caimano M,
Petroni M, et al: Mitogen-activated kinase kinase kinase 1 inhibits
hedgehog signaling and medulloblastoma growth through GLI1
phosphorylation. Int J Oncol. 54:505–514. 2019.PubMed/NCBI
|
|
44
|
Su F, Li H, Yan C, Jia B, Zhang Y and Chen
X: Depleting MEKK1 expression inhibits the ability of invasion and
migration of human pancreatic cancer cells. J Cancer Res Clin
Oncol. 135:1655–1663. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Turkington RC, Longley DB, Allen WL,
Stevenson L, McLaughlin K, Dunne PD, Blayney JK, Salto-Tellez M,
Van Schaeybroeck S and Johnston PG: Fibroblast growth factor
receptor 4 (FGFR4): A targetable regulator of drug resistance in
colorectal cancer. Cell Death Dis. 5:e10462014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ahmed MA, Selzer E, Dörr W, Jomrich G,
Harpain F, Silberhumer GR, Müllauer L, Holzmann K, Grasl-Kraupp B,
Grusch M, et al: Fibroblast growth factor receptor 4 induced
resistance to radiation therapy in colorectal cancer. Oncotarget.
7:69976–69990. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Heldin CH and Lennartsson J: Structural
and functional properties of platelet-derived growth factor and
stem cell factor receptors. Cold Spring Harb Perspect Biol.
5:a0091002013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fujino S, Miyoshi N, Ohue M, Takahashi Y,
Yasui M, Hata T, Matsuda C, Mizushima T, Doki Y and Mori M:
Platelet-derived growth factor receptor-β gene expression relates
to recurrence in colorectal cancer. Oncol Rep. 39:2178–2184.
2018.PubMed/NCBI
|
|
49
|
Mezheyeuski A, Bradic Lindh M, Guren TK,
Dragomir A, Pfeiffer P, Kure EH, Ikdahl T, Skovlund E, Corvigno S,
et al: Survival-associated heterogeneity of marker-defined
perivascular cells in colorectal cancer. Oncotarget. 7:41948–41958.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schimanski C, Wehler T, Galle P, Gockel I
and Moehler M: PDGFR-α/β expression correlates with the metastatic
behavior of human colorectal cancer-A rationale for a molecular
targeting strategy? J Clin Oncol. 26:220192008. View Article : Google Scholar
|