Introduction
Melanoma is a rare, fatal type of skin tumor, which
comprises four main types: Lentigo maligna melanoma, superficial
spreading melanoma (SSM), nodular melanoma and acral lentiginous
melanoma (ALM) (1). ALM, which
generally affects the palms and soles of patients, has a low
incidence in the Caucasian population and occurs mainly in patients
of an Asian and African descent; up to 75% of all patients with
melanoma have ALM (1). Patients with
ALM usually have a poor prognosis due to difficulties in diagnosis
and ALM tends to be identified at an advanced clinical stage or
with high Breslow thickness (1–4). Genomic
instability and poor response to biological agents in ALM also
contribute to the poor outcome. Unlike SSM, in which BRAF mutation
is the most observed aberration, KIT proto-oncogene receptor
tyrosine kinase is the most mutated gene in ALM; however, this has
only been identified in 15% of patients (5). Therefore, identification of more
specific biomarkers for ALM is necessary.
Long non-coding RNAs (lncRNAs) have been
demonstrated to serve crucial roles in tumorigenesis by diverse
mechanisms and at various levels; for example, lncRNAs can act as
mediators to regulate gene expression, combine with proteins to
form a ribonucleoprotein complex and modify histones, recruit
enzymes to regulate proximal or distant genes or serve as a decoy
for transcription factors (6,7).
Although previous studies (8–21) have
reported that lncRNAs including HOTAIR, MALAT1, BANCR, ANRIL,
SPRY4-IT1, Llme23, UCA1, SLNCR1 and SAMMSON served oncogenic
functions in the progression and metastasis of melanoma, no studies
are currently available on lncRNAs specifically related to ALM, and
the mechanisms of lncRNA activity in ALM are still unclear.
Therefore, identification of lncRNAs in ALM may provide value for
early diagnosis and improved prognosis.
The present study aimed to investigate the role of
lncRNAs in the pathogenesis of ALM by performing microarray
analysis of the expression patterns of lncRNAs. This study may help
to clarify the function of lncRNAs in ALM and provide evidence of
their therapeutic and prognostic value.
Materials and methods
Tissue collection
A total of 12 samples, including six tumor and six
adjacent non-tumor tissues, were collected in pairs from six
patients with ALM (patient 1, male, 71 years; patient 2, male, 72
years; patient 3, female, 44 years; patient 4, female, 66 years;
female 5, female, 74 years and patient 6, male, 55 years) between
January 2017 and May 2018 at the Institute of Dermatology, Chinese
Academy of Medical Sciences and Peking Union Medical College
(Nanjing, China). The samples were immediately stored at −80°C. The
study was approved by the Ethics Committee of the Institute of
Dermatology, Chinese Academy of Medical Sciences and Peking Union
Medical College (approval no. 2013-LC/KY-033). All participating
patients gave informed consent.
RNA extraction and quality
control
According to the manufacturer's protocol, total RNA
was extracted using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.). RNA quantity and quality were measured by
NanoDrop ND-1000. Standard denaturing agarose gel electrophoresis
(1%) or Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.) was
used to assess integrity of RNA.
Microarray analysis
A total of 6 pairs of ALM and adjacent non-tumor
tissues were used for the microarray assay to determine
differentially expressed lncRNAs and mRNAs. Sample labeling and
array hybridization were performed according to the Agilent
One-Color Microarray-Based Gene Expression Analysis protocol
(Agilent Technology, Inc.). Arraystar Human LncRNA Microarray V4.0,
designed for the global profiling of human lncRNAs and
protein-coding transcripts, was used. The hybridized arrays were
washed and then scanned using Agilent Scanner G2505C (Agilent
Technology, Inc.). The acquired array images were analyzed using
Agilent Feature Extraction software (version 11.0.1.1). Quantile
normalization and subsequent data processing were performed using
the GeneSpring GX v12.1 software package (Agilent Technologies,
Inc.). Following quantile normalization of the raw data, lncRNAs
and mRNAs that had flags in Present or Marginal (‘All Targets
Value’) in ≥5 samples were selected for further analysis.
Differentially expressed lncRNAs and mRNAs between cancerous and
adjacent tissues were identified using the thresholds P<0.05 and
fold-change >2.
Gene ontology (GO) enrichment and
kyoto encyclopedia of genes and genomes (KEGG) analysis
GO analysis (http://www.geneontology.org) was performed to explore
the ‘biological processes’, ‘cellular components’ and ‘molecular
functions’ of the identified differentially expressed mRNAs, and
this was performed via top GO package in R environment. Using
hyper-geometric distribution, which was made using R language, the
significance between differentially expressed genes and KEGG could
be observed. Pathway analysis was performed using KEGG, Biocarta
and Reactome (http://www.genome.jp/kegg/). Fisher's exact test was
used to find out if the overlaps between the DE gene list and the
GO annotation list were greater than what was expected by chance.
The significance of each GO term and pathway association are
reflected by the P-value, and P<0.05 was considered to indicate
a statistically significant result. The -log10(P) was
used to determine the enrichment of each GO term in the
differentially expressed genes and the significance of the pathway
associations. A lower P-value was considered to indicate a more
significant correlation. The top 10 terms of GO analysis and KEGG
analysis were all characterized by P<0.05 and false discovery
rate (FDR)<0.05.
Reverse transcription-quantitative
(RT-q)PCR validation
A total of five randomly selected lncRNAs
(fold-change >2, P<0.05) were validated by RT-qPCR. These
five lncRNAs belonged to the top 40 according to FC and its
P<0.05. All these lncRNA_levels were gold level. Gold level
meant that these selected lncRNAs had been validated by specific
experiments and had relevant annotation, such as transcription
units, function mechanisms and subcellular localization. Following
RNA extraction, SuperScript™ III Reverse Transcriptase (Invitrogen;
Thermo Fisher Scientific, Inc.,) was used to synthesize cDNA
according to the manufacturer's protocol. Briefly, 2 µg RNA was
mixed with 1 µl lncRNA specific primer Mix, 1.6 µl dNTP Mix, and
RNase free water was added to a total volume of 13.5 µl. The sample
was placed in a water bath for 5 min at 65°C and on ice for 2 min.
Subsequently, 4 µl 5X First-Strand Buffer, 1 µl of 0.1 M DTT, 0.5
µl RNase Inhibitor and 1 µl SuperScript III RT were added. The
reaction was performed for 1 min at 37°C, for 60 min at 50°C and
for 15 min at 70°C. cDNA was eventually stored at −20°C. PCR was
performed using ViiA 7 Real-time PCR System (Applied Biosystems;
Thermo Fisher Scientific, Inc.) in a total volume of 10 µl,
including 0.5 µl forward primer (10 µM), 0.5 µl reverse primer (10
µM), 2 µl cDNA, 5 µl 2X Master Mix and 2 µl double-distilled water.
β-actin was used as an internal control. Primer sequences were as
follows: β-actin forward, 5′-GTGGCCGAGGACTTTGATTG-3′ and reverse,
5;-CCTGTAACAACGCATCTCATATT-3′; NR_004845 forward,
5′-TTGGCATACAGGTCTTTGTAGAT-3′ and reverse,
5′-TTGGCATACAGGTCTTTGTAGAT-3′; NR_026983 forward,
5′-ATCCCTGGTATTGAAGAGGTG-3′ and reverse,
5′-AGATTGTTTGGGCAGTGTTAG-3′; NR_034040 forward,
5′-TGACATCCGAATGCCATCCT-3′ and reverse,
5′-GCTGCTGACAAACAACCTGCT-3′; NR_036580 forward,
5′-AGCCCAGATTCTCCTACCAGC-3′ and reverse,
5′-CCTTCAGGTGGCTGTTTTGTAGT-3′; NR_037877 forward,
5′-ATGTTGACCATGCAGCCAATT-3′ and reverse,
5′-GTGTTTATCAGAGGTCATTTCCG-3′. The thermocycling conditions were as
follows: 95°C for 10 min, followed by 40 cycles of 95°C for 10 sec
and 60°C for 60 sec. The gene expression levels in tumor tissue
relative to adjacent tissue were calculated as fold-change using
the standard curve method (22). An
unpaired t-test was used and P<0.05 was considered to indicate a
statistically significant difference.
LncRNA and mRNA co-expression (CNC)
network
Using the validated lncRNAs and their target mRNAs,
the CNC network was constructed by Cytoscape software (version
2.8.3; The Cytoscape Consortium) with the criterion that the
Pearson correlation coefficient (PCC) of the lncRNA and mRNA
correlation analysis was ≥0.95. Additionally, PCC ≥0.95 was
considered to indicate meaningfully related pair. While performing
CNC analysis and calculating PCC, the P-value and FDR were also
obtained (P<0.05 and FDR<0.05), which further confirmed the
reliability of PCC.
Competing endogenous network
analysis
Based on the hypothesis that RNA transcripts can
crosstalk by competing for common microRNAs (miRNAs) and that miRNA
response elements (MREs), which were the foundation of this
interaction, the competing endogenous network was predicted. The
prediction of such lncRNA-miRNA-mRNA interaction was based on the
selected lncRNAs and related mRNAs. LncRNA/miRNA interactions were
predicted by miRcode (http://www.mircode.org) and miRNA/mRNA interactions
were predicted by miRanda (http://www.miranda.org) and TargetScan (http://www.targetscan.org).
Results
Differentially expressed lncRNA and
mRNA profiles detected by microarray
According to the microarray expression profiling
data and the filtering analysis including fold-change ≥2 and
P<0.05, a total of 4,490 lncRNAs and 3,915 mRNAs were identified
to be differentially expressed between the ALM and adjacent
non-tumor samples. Among them, 2,211 and 2,277 lncRNAs were
upregulated and downregulated in the ALM samples compared with
adjacent sections, respectively. In addition, 1,191 and 2,722 mRNAs
were upregulated and downregulated, respectively. The top 5
upregulated lncRNAs were T380070, ENST00000554431, T097678,
GSE61474_TCONS_00183926 and NR_015399, and the top 5 downregulated
lncRNAs were TCONS_00013495, T367528, ENST00000598924, T050010 and
TCONS_00010140. The top 5 upregulated mRNAs were ENST00000370287,
NM_005367, NM_001129826, NM_001922 and NM_000273, and the top 5
downregulated mRNAs were ENST00000433840, NM_206998, NM_014867,
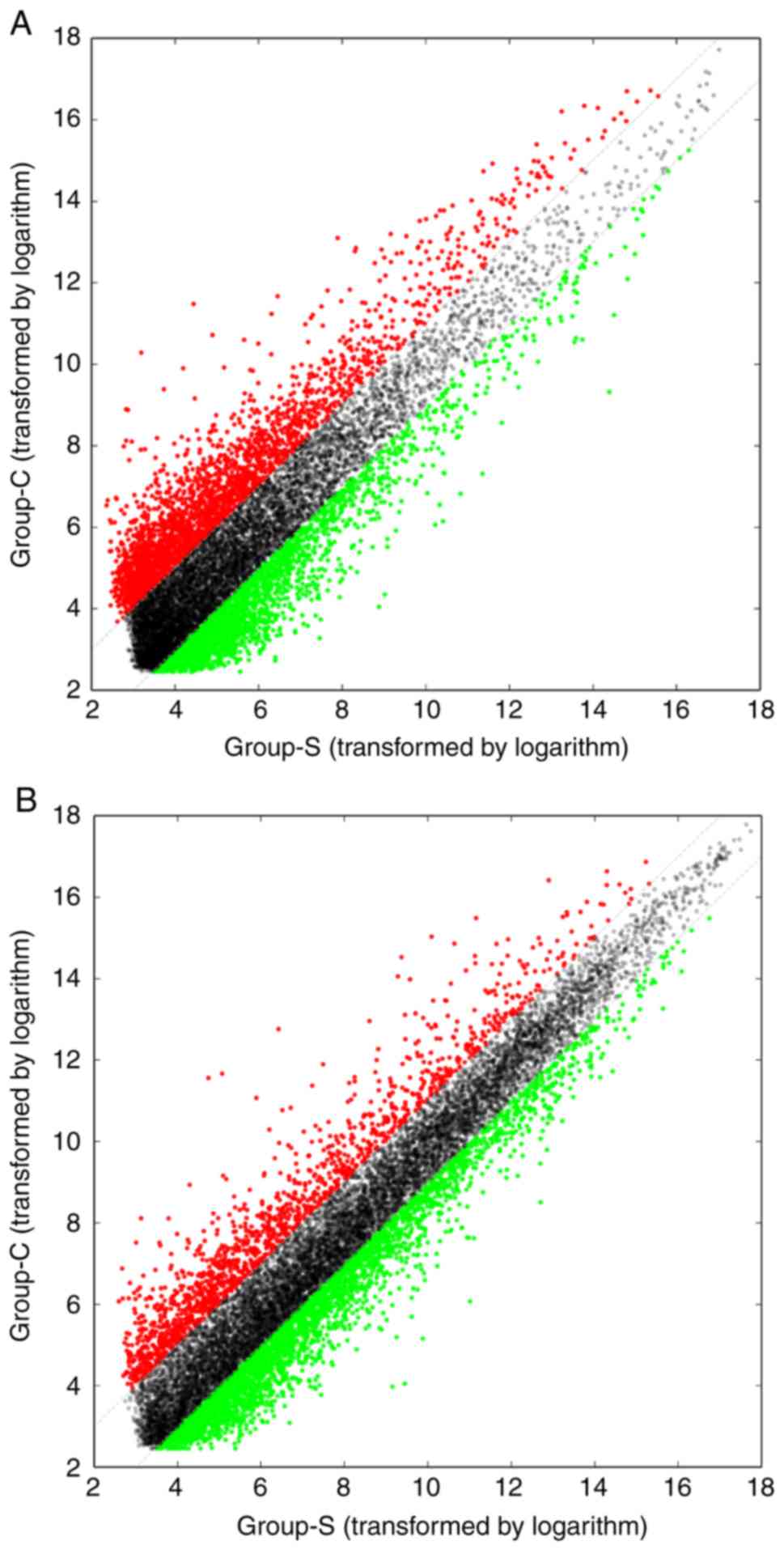
NM_173595 and NM_004202. The variations in the lncRNA (Fig. 1A) and mRNA (Fig. 1B) expression profiles between ALM and
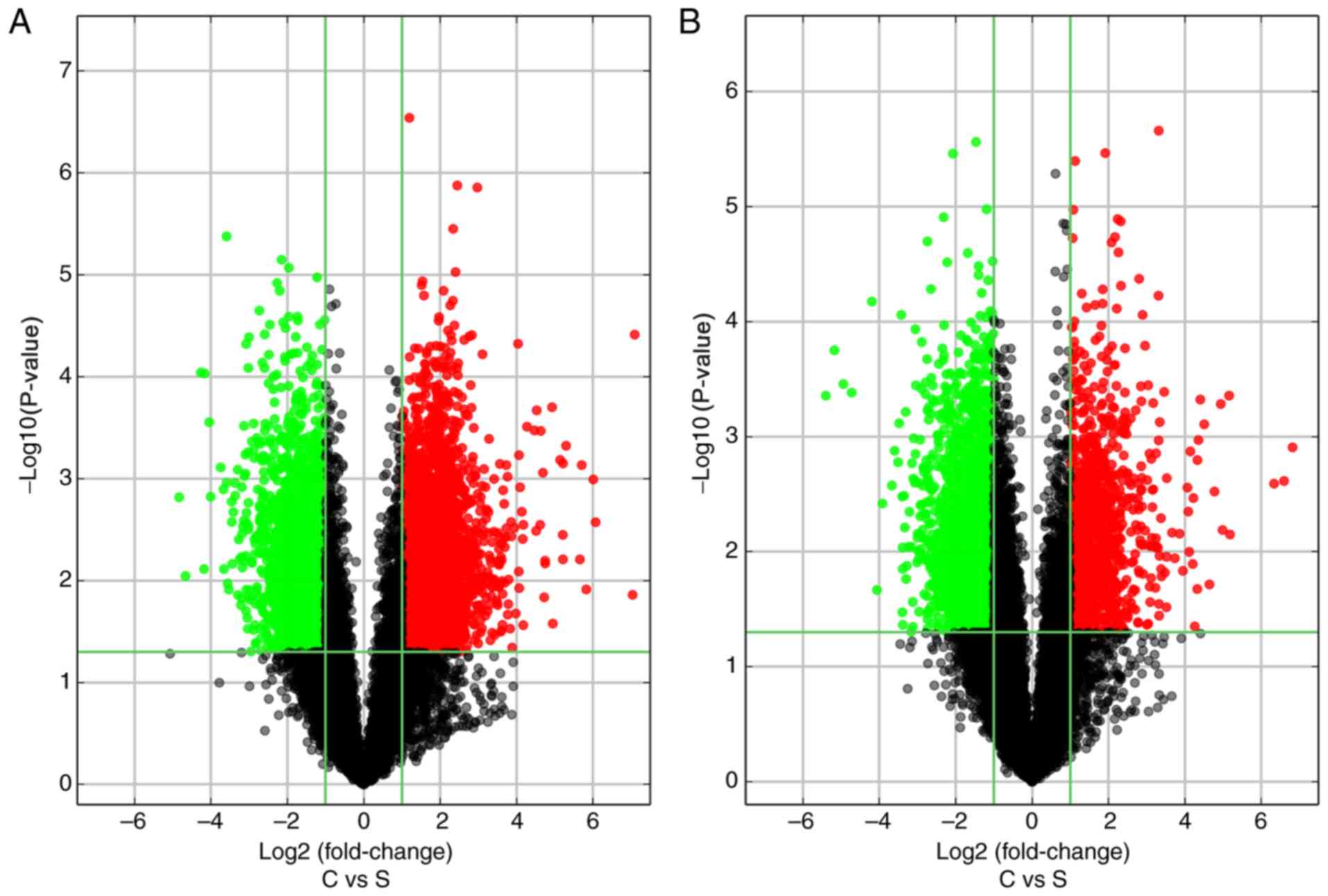
adjacent tissue samples were assessed by scatterplot analysis, and
volcano plots were constructed to demonstrate the association
between the fold-changes and the statistical significance of the
differentially expressed lncRNAs (Fig.
2A) and mRNAs (Fig. 2B). The
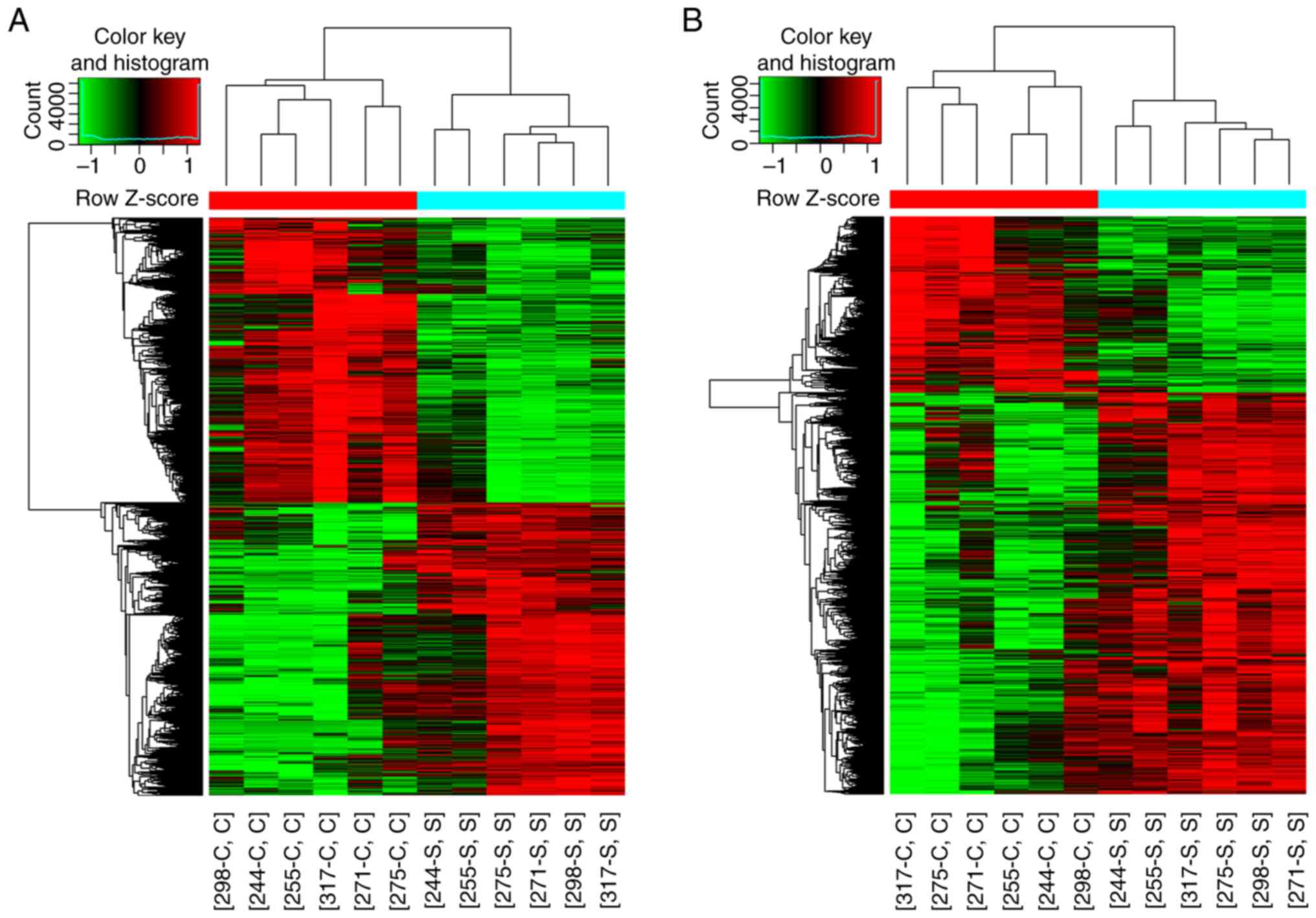
expression patterns of lncRNAs and mRNAs were also demonstrated in
hierarchical clustering (Fig.
3).
GO enrichment and KEGG pathway
analysis
As lncRNAs affected coding gene expression by
influencing mRNAs, GO enrichment analysis of significantly
differentially expressed mRNAs was performed to determine the
effects of these lncRNAs. GO categories of ‘biological process’
(BP), ‘cellular component’ (CC) and ‘molecular function’ (MF) were
analyzed to determine the gene and gene product enrichment. The
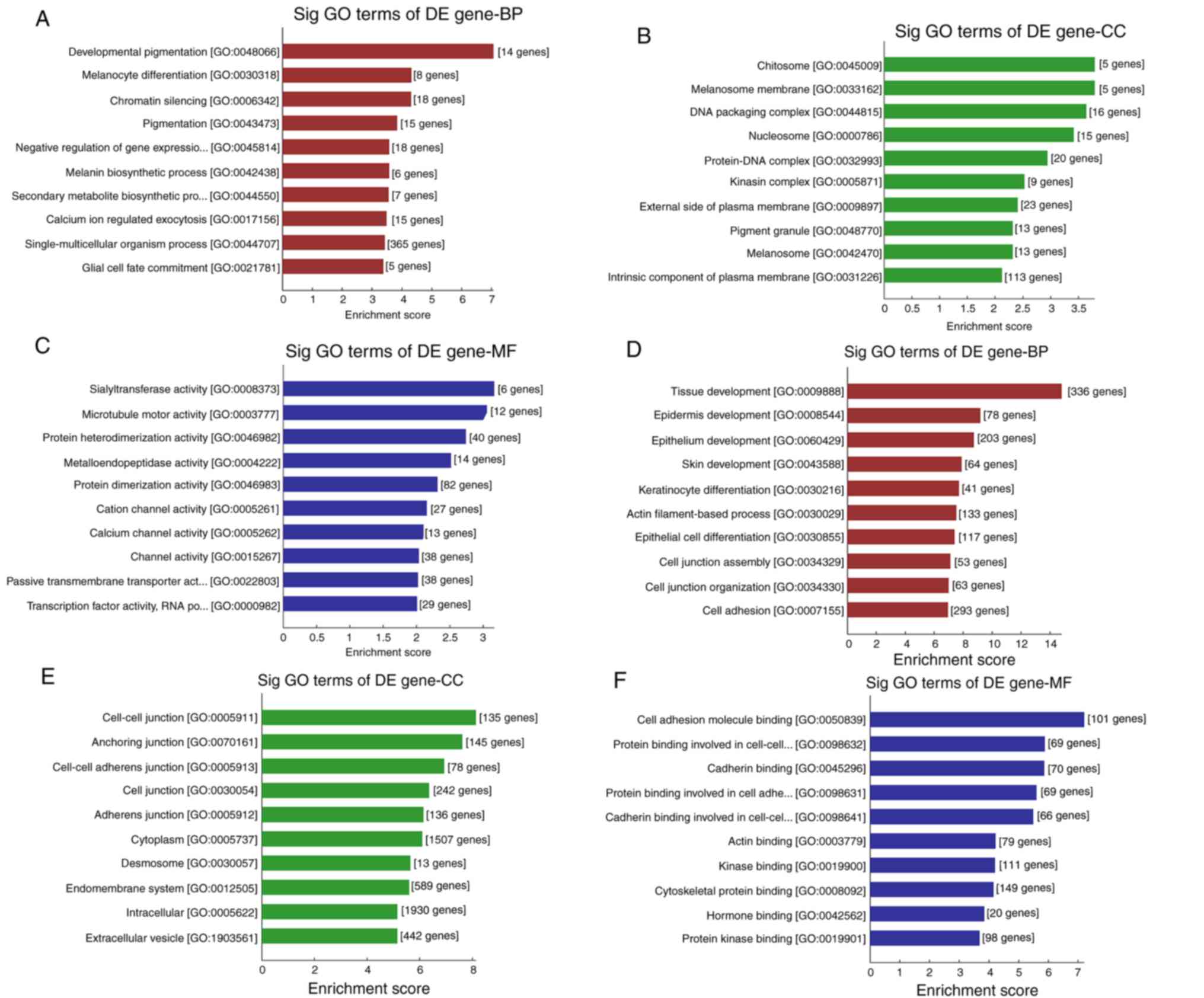
results of GO enrichment analysis are presented in Fig. 4. The analysis revealed that the
majority of the BPs associated with upregulated mRNAs were involved
in the events of pigmentation and melanocytes, such as
‘developmental pigmentation’, ‘melanocyte differentiation’,
‘pigmentation’ and ‘melanin metabolite biosynthetic process’
(Fig. 4A). The majority of the CCs
of the upregulated mRNAs were associated with ‘melanosome
membrane’, ‘pigment granule’ and ‘melanosome’ (Fig. 4B). Terms associated with channel
activity were enriched in the MF category, including ‘channel
activity’, ‘cation channel activity’ and ‘calcium channel activity’
(Fig. 4C). For the downregulated
mRNAs, the majority of the enriched terms were associated with the
cell junction and tissue development (Fig. 4D-F). For example, the top 3 terms in
BP were ‘tissue development’, ‘epidermis development’ and
‘epithelium development’. ‘Cell-cell junction’, ‘anchoring
junction’ and ‘cell-cell adhesion junction’ were the top three
terms in CC, whereas ‘cell adhesion molecule binding’, ‘protein
binding involved in cell-cell adhesion’, ‘cadherin binding’ and
‘kinase binding or actin binding’ were the most three enriched
terms in MF.
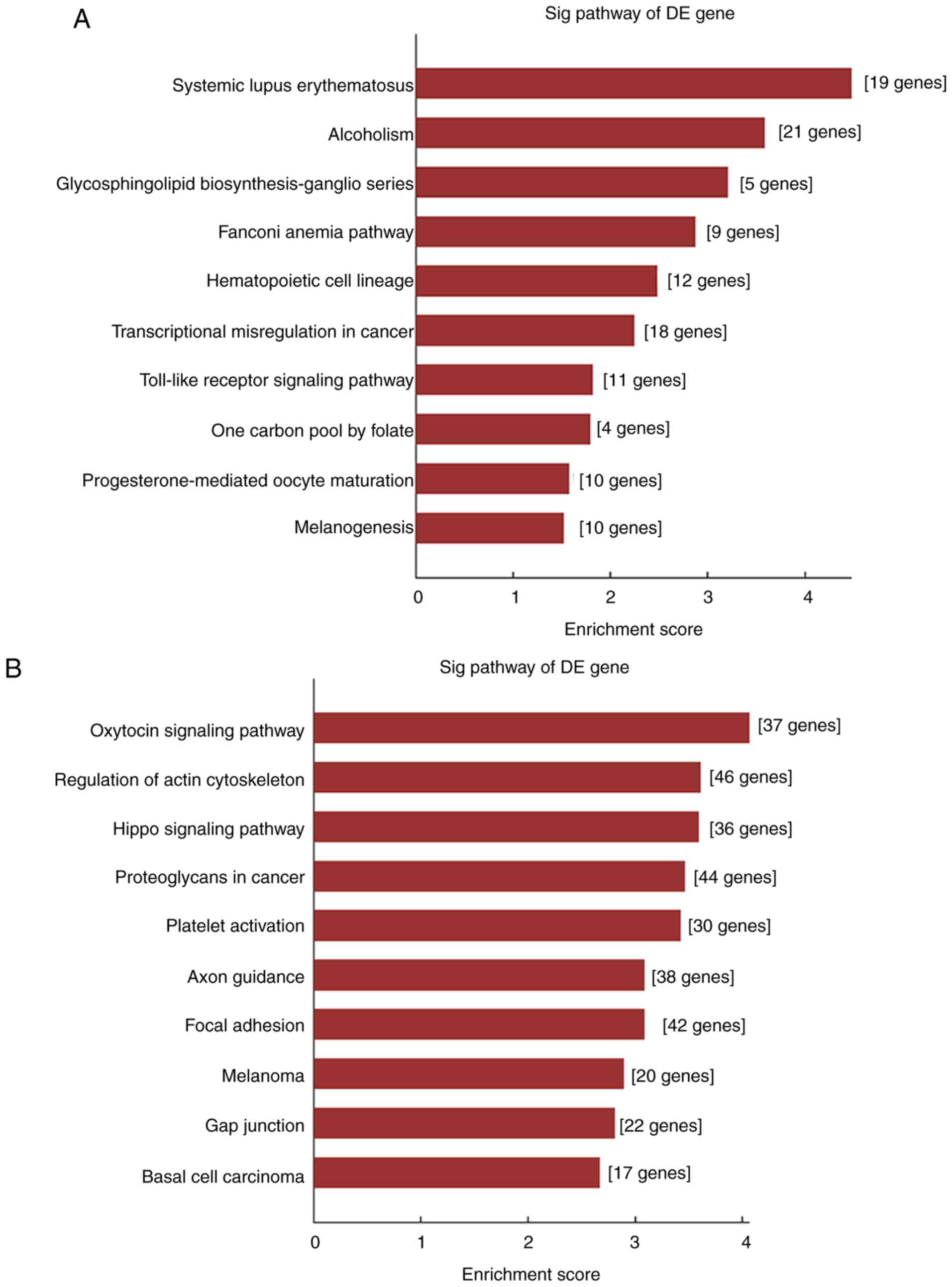
KEGG pathway enrichment analysis results are
presented in Fig. 5, including 10
pathways associated with upregulated mRNAs (Fig. 5A) and 10 pathways associated with
downregulated mRNAs (Fig. 5B). These
results demonstrated that the upregulated mRNAs may be involved in
‘glycosphingolipid biosynthesis-ganglio series’, ‘transcriptional
misregulation in cancer’, ‘toll-like receptor signaling pathway’
and ‘melanogenesis’. In addition, ‘oxytocin signaling pathway’,
‘regulation of actin cytoskeleton and hippo signaling pathway’,
‘focal adhesion’ and ‘gap junction’ pathways were associated with
the downregulated mRNAs.
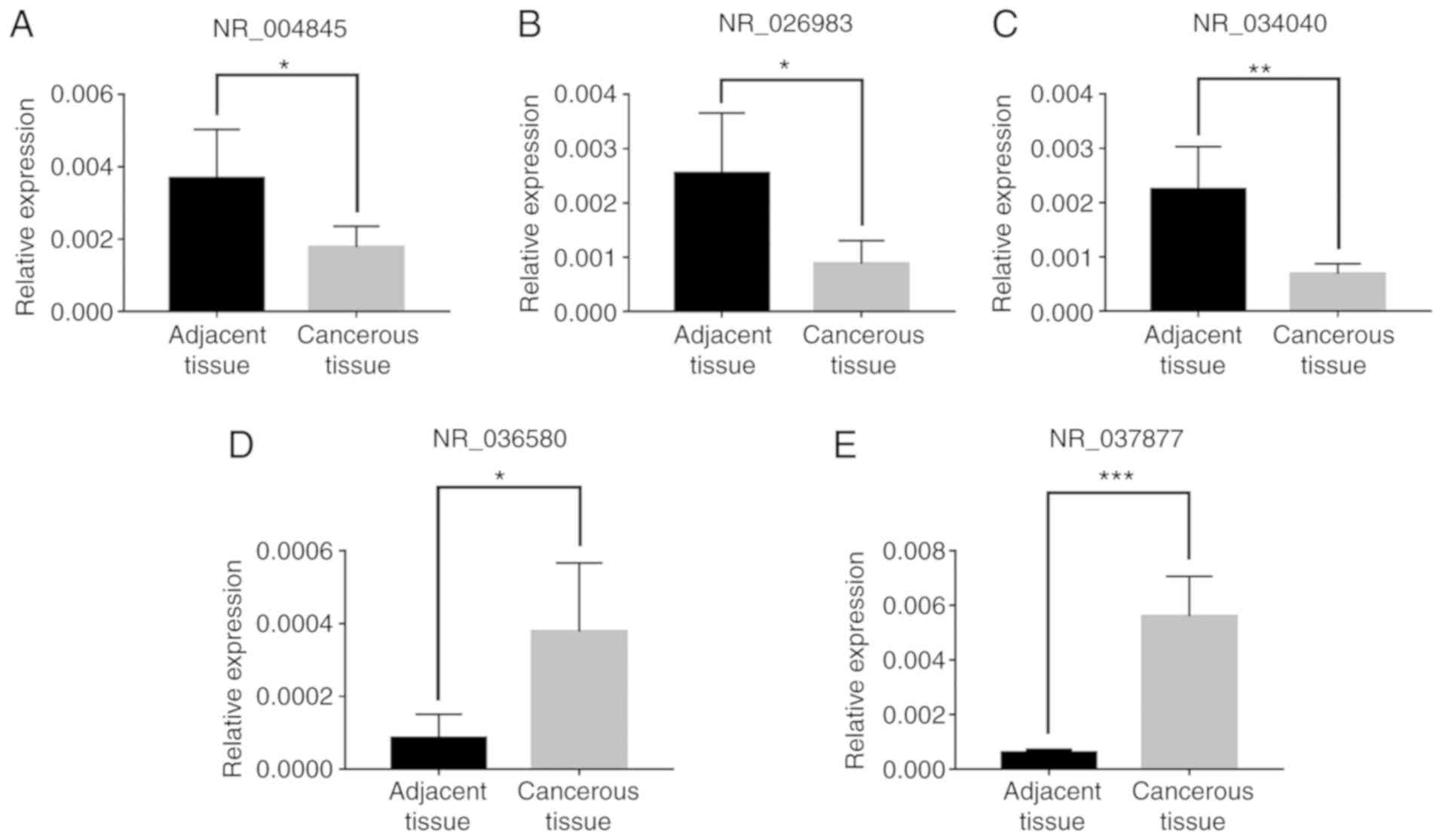
RT-qPCR validation
To confirm the previous results and detect the
function of lncRNAs in ALM, five randomly selected lncRNAs
(Table I) were validated by RT-qPCR,
including three downregulated lncRNAs NR_004845, NR_026983 and
NR_034040 (Fig. 6A-C) and two
upregulated lncRNAs NR_036580 and NR_037877 (Fig. 6D and E). The results were similar to
those of the microarray analysis (Fig.
6). Microarray analysis and RT-qPCR demonstrated downregulated
expression of NR_004845, NR_026983 and NR_034040 and upregulated
expression of NR_036580 and NR_037877. This provided reliable
confirmation of lncRNA changes determined by microarray
analysis.
 | Table I.Five randomly selected long noncoding
RNAs for reverse transcription-quantitative PCR confirmation,
construction for lncRNA and mRNA co-expression network as well as
competitive endogenous RNA network construction. |
Table I.
Five randomly selected long noncoding
RNAs for reverse transcription-quantitative PCR confirmation,
construction for lncRNA and mRNA co-expression network as well as
competitive endogenous RNA network construction.
| Seqname | GeneSymbol | P-value | Fold-change | Regulation | Chrom | Strand | Relationship |
|---|
| NR_004845 | LOC644936 | 0.000893591 | 8.5660466 | Down | chr5 | − | Intergenic |
| NR_026983 | BTF3P11 | 0.002967842 | 8.0696294 | Down | chr13 | + | Intergenic |
| NR_034040 | LGALS8-AS1 | 0.001504141 | 16.0657987 | Down | chr1 | − | Intronic
antisense |
| NR_036580 | DPP10-AS1 | 0.000199269 | 30.4236389 | Up | chr2 | − | Intronic
antisense |
| NR_037877 | LOC100505912 | 0.003565498 | 36.9781352 | Up | chr4 | − | Intergenic |
LncRNA and mRNA CNC network, GO
enrichment and KEGG analysis
Based on the five validated lncRNAs (Table I) and their target mRNAs, the
co-expression network consisting of 1,064 nodes and 1,312
connections was constructed (Fig.
S1). The downregulated lncRNAs NR_004845, NR_026983 and NR
_034040 correlated with 391, 141 and 150 mRNAs, respectively. The
upregulated lncRNAs NR_036580 and NR_037877 correlated with 249 and
373 mRNAs, respectively.
GO and KEGG analysis of the CNC network were
performed. The top 10 enriched GO terms in BP, CC and MF are
presented in Fig. 7A-C. The results
revealed that, in BP, target mRNAs were enriched in ‘adherens
junction organization’, ‘pigmentation’ and ‘adherens junction
assembly’. In CC, ‘intrinsic component of membrane’ and ‘membrane
part’ were the most significantly enriched terms. In addition,
terms associated with channel activity and DNA binding were
enriched in MF. These results were in accordance with the outcome
of the GO enrichment analysis of microarray results.
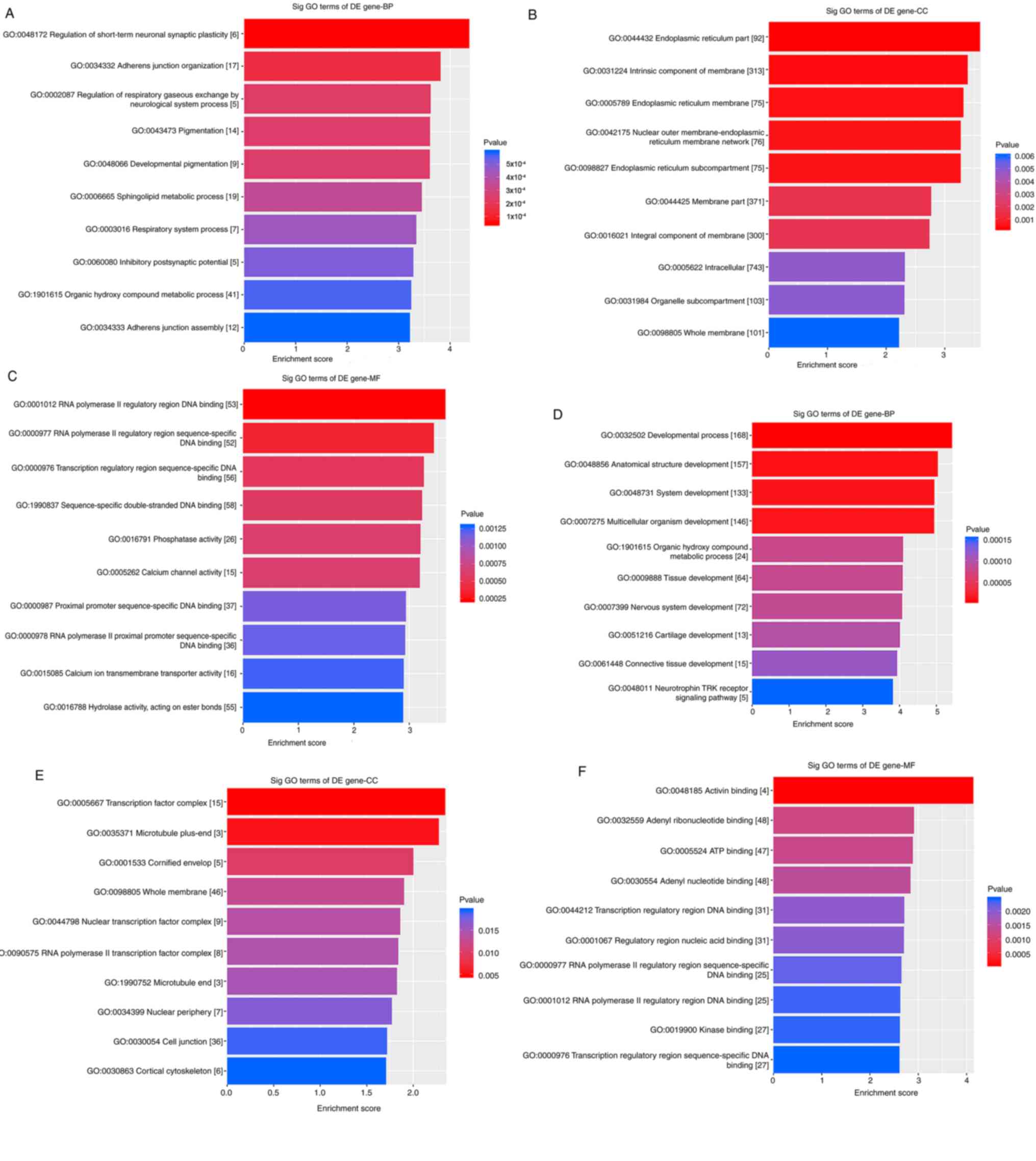 | Figure 7.GO enrichment analysis in the lncRNA
and mRNA co-expression network and the lncRNA/miRNA/mRNA ceRNA
network. (A-C) The top 10 significantly enriched GO terms in (A)
BP, (B) CC and (C) MF in the lncRNA and mRNA co-expression network.
(D-F) The top 10 significantly enriched GO terms in (D) BP, (E) CC
and (F) MF in the lncRNA/miRNA/mRNA ceRNA network. GO, gene
ontology; lncRNA, long non-coding RNA; miRNA, microRNA; ceRNA,
competing endogenous RNA; BP, biological process; CC, cellular
component; MF, molecular function; DE, differentially
expressed. |
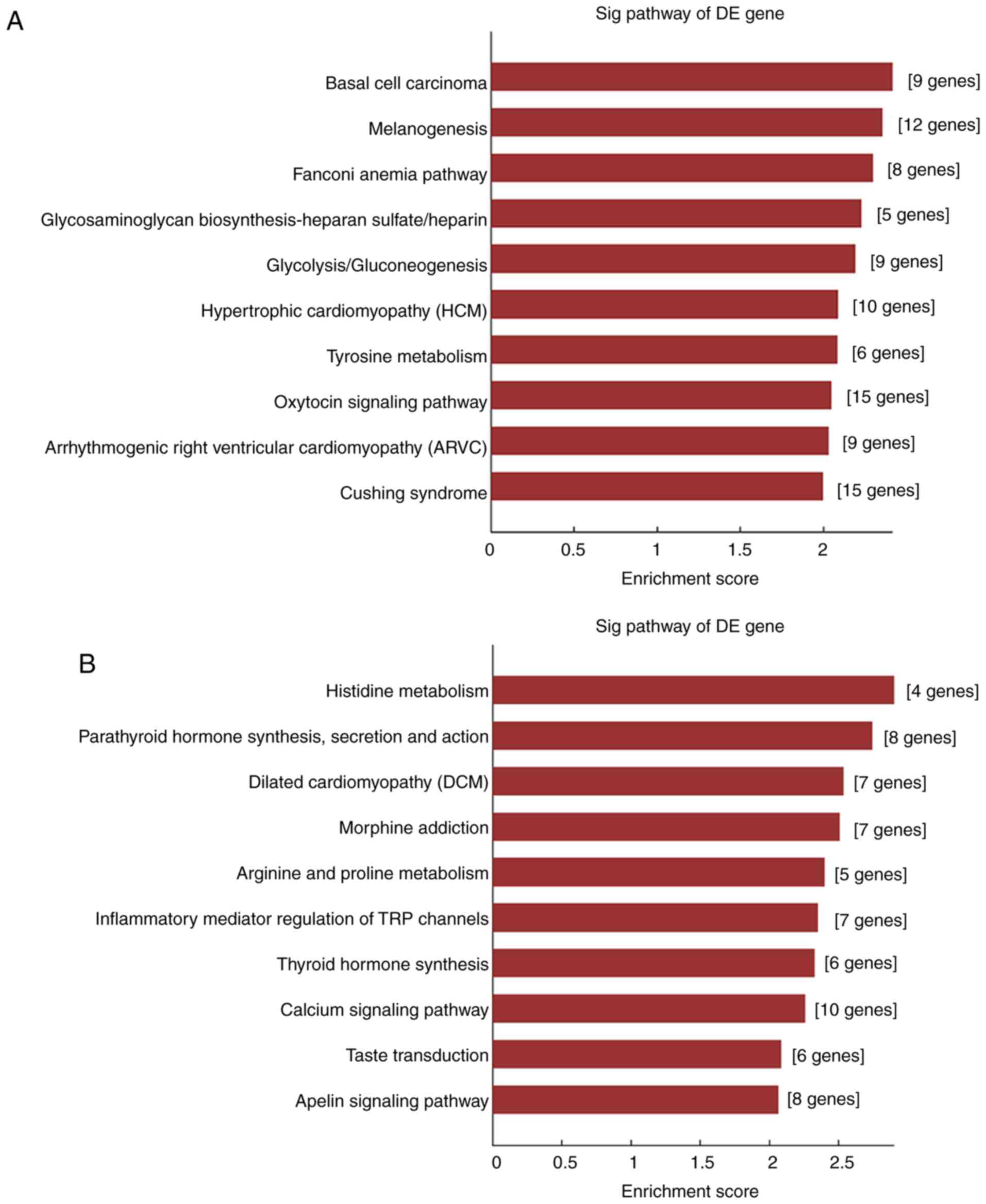
The KEGG analysis (Fig.
8A) demonstrated that the selected lncRNAs were associated with
pathways such as ‘melanogenesis’, ‘glycosaminoglycan
biosynthesis-heparan sulfate/heparin’, ‘glycolysis/gluconeogenesis’
and ‘oxytoxin signaling pathway’. These results also revealed that
the selected lncRNAs were representative.
Competing endogenous network analysis,
GO and KEGG analysis
The theory of competing endogenous RNA (ceRNA) has
revealed that a number of ceRNAs may serve a regulatory function
between coding and non-coding genes via the competition of MREs.
Therefore, based on the results of the microarray analysis, a ceRNA
network was constructed to determine whether lncRNAs may act as
ceRNAs and contribute to the occurrence of ALM. The aforementioned
five randomly selected lncRNAs were used to construct this network,
which is presented in Fig. S2. The
light green nodes represent lncRNAs, the red nodes represent miRNAs
and the light blue nodes represent mRNAs.
GO and KEGG analyses were performed to determine the
potential biological action of the ceRNA network. The results of
the GO enrichment analysis demonstrated that several terms
associated with tissue development, such as ‘developmental
process’, ‘anatomical structure development’ and ‘system
development’ were enriched in BP (Fig.
7D). In CC, terms such as ‘cell junction’ were enriched
(Fig. 7E). In addition, the terms
enriched in MF (Fig. 7F) were all
associated with binding. These results were consistent between the
microarray analysis and the ceRNA network. In KEGG analysis,
pathways including ‘inflammatory mediator regulation of TRP
channels’, ‘calcium signaling pathway’ and ‘apelin signaling
pathway’ were enriched (Fig.
8B).
Discussion
No sensitive or specific biomarker for the early
diagnosis and treatment of ALM is currently available. Considering
the high incidence and poor outcome of ALM in Asia, especially in
China, further investigations into the molecular mechanism of ALM
are crucial to improve the survival rate. However, existing studies
of gene mutations, epigenetics, immune abnormalities and tumor
microenvironment do not fully explain the malignant biological
behaviors of ALM. The high risk of recurrence and metastasis of ALM
in China has been previously attributed to patients' negligence of
the disease and repeated irritation of the lesion, which is not
convincing. Previously, lncRNA has emerged as a critical molecule
in human cancers, such as breast and colorectal cancer, prostate,
hepatocellular and basal cell carcinoma (23–26).
However, a limited number of studies on lncRNAs associated with the
pathogenesis, progression and metastasis of melanoma have been
published and studies on ALM are lacking.
In the present study, using the microarray analysis
technology, a preliminary molecular analysis of lncRNAs and mRNAs
in ALM was performed to facilitate further studies on the
pathogenesis of ALM and to explore whether the biological behavior
of ALM may be induced by unidentified lncRNAs. In addition, GO and
KEGG pathway enrichment analyses were concluded to identify the
potential functions of differentially expressed mRNAs.
The comparison of the expression profiles of lncRNAs
and mRNAs in ALM and adjacent non-tumor tissues demonstrated that
4,490 lncRNAs and 3,915 mRNAs were differentially expressed in
these samples. These results were inconsistent with previous
studies (8–21) of lncRNAs associated with melanoma.
This may be attributed to the unique properties of ALM. The most
upregulated or downregulated lncRNAs and mRNAs may help identify
the molecular markers for early diagnosis of ALM. A total of five
lncRNAs were randomly selected RT-qPCR; the results were consistent
with those of the microarray analysis, which suggested that the
results of the microarray analysis were reliable.
GO enrichment analysis was used to identify the
functions of the lncRNAs through the mRNA expression patterns.
Among the upregulated mRNAs, most of the BP terms were associated
with pigmentation and melanocytes. Previous reports have
demonstrated that in animal models, the inhibition of Wnt/β-catenin
signaling may lead to a decrease in melanocytes; however,
inhibitors of the Wnt/β-catenin signaling pathway do not prevent
the process of pigmentation in melanoma cells (27,28). One
inhibitor, ICG-001, exhibited a positive effect on pigmentation
(28). Another study has
demonstrated that increased pigmentation is a feature of primary
melanoma with a BRAF mutation; in addition, pigmentation within the
sentinel node (SN) may be associated with increased SN tumor burden
and prognosis (29). The presence of
pigmentation may be associated with a worse clinical outcome.
The enriched CC terms were associated with
organelles that promoted the formation of melanin, such as pigment
granule and melanosome. These results agree with previous studies
(30,31). Compared with non-melanoma cells,
melanoma cells are often characterized by different production of
melanosomes. According to their morphology, melanosomes can be
divided into four stages; stage IV suggests that the melanosome may
be damaged (30,31). In addition to melanin synthesis, the
melanosome also serves a role in clearing toxic by-products and
waste during the process of melanin synthesis; this mechanism may
promote the occurrence of drug resistance (30). The mediators of drug resistance may
be associated with protein products such as
microphthalmia-associated transcription factor, G-protein coupled
receptor 143 and premelanosome protein gp100 (31). A previous study has demonstrated that
silencing the expression of genes which regulate the development of
the melanosome improves the sensitivity of melanoma cells to
certain drugs (30). Therefore,
considering the high probability of drug resistance in ALM, further
studies focusing on whether the number of melanosomes in ALM is
different from other types of melanoma may be useful.
In MF, channel activity-related terms were
associated with upregulated mRNAs. Previous studies have
demonstrated that BKCa channels regulate cell morphology and
progression, as well as the migration of tumor cells (32). In addition, the expression of
Na+ channels in tumor cells increase
Na+-Ca2+ exchange, which further increases
the intracellular concentration of Ca2+, enhancing the
metastatic ability of tumor cells. Additionally, it also
demonstrated that Na+-Ca2+ exchange is partly
regulated by the mammalian target of rapamycin signaling pathway,
which affects the proliferation and metastasis of melanoma cells
(33). This is consistent with the
results of the GO enrichment analysis in the present study.
For downregulated mRNAs, the BP, CC and MF terms
were associated with tissue development, cell-cell junction and
cell adhesion molecule binding. These terms may be associated with
the progression and metastasis of ALM. Previous studies have
reported that epithelial (E)-cadherin, placental (P)-cadherin and
heart (H)-cadherin affect the physiological conditions of
melanocytes and keratinocytes (34,35).
E-cadherin and H-cadherin are often located in the basal layer of
the epidermis, whereas P-cadherin is located in hair follicles.
Loss of E-cadherin and expression of neural (N)-cadherin in
melanoma cells are the early events in melanoma formation and
metastasis (34). The expression of
N-cadherin may enable melanoma cells to interact with dermal
fibroblasts, which results in their migration into the dermis
(35). In addition, the term ‘cell
adhesion’ suggested that there may be numerous cell adhesion
molecules associated with melanoma that may serve as targets for
inhibiting growth or invasion and improving prognosis in ALM. A
previous study has demonstrated that low molecular weight heparin
(LMWH) inhibited the adhesion of melanoma cells through the protein
kinase C α (PKCα)/JNK signaling pathway (36). Additionally, the integrin very late
antigen-4 (VLA-4), which is a crucial molecule for the invasion of
melanoma cells, is inhibited by heparin. Cyr61 also serves a role
in tumor formation by activating integrin-like VLA-4. A binding
site for heparin has been identified in Cyr61, thus a VLA-4/Cyr61
axis may be speculated (37). This
axis may be a promising target of heparin treatment in melanoma.
Nitric oxide-releasing nonsteroidal anti-inflammatory drugs
inhibited the function of VLA-4 and its ligand vascular cell
adhesion molecule-1, thus serving as an anti-metastasis drug for
melanoma (38). Hyaluronan, which is
a component of the extracellular matrix, may regulate the
metastasis of melanoma through cell adhesion. Overexpression of
hyaluronan synthase 3 increased the amount of hyaluronan on the
cell surface and induced cell cycle arrest at G1/G0, resulting in
the blockage of cell adhesion and further metastasis (39). Other molecules such as activated
leukocyte cell adhesion molecule, carcinoembryonic antigen-related
cell adhesion molecule 1, PRL-3/PTP4A3 phosphatase and vascular
endothelial growth factor may regulate cell adhesion in melanoma
and its long-term prognosis (40–43).
Cell adhesion detection has also been applied in the clinical
diagnosis of melanoma. The expression products of genes such as β-3
integrin, cellular tumor antigen p53, laminin B1 chain and
tissue-type plasminogen activator may serve a role in cell adhesion
and sentinel lymph node metastasis (44). Combined detection of several of the
aforementioned genes may be more effective at predicting the
likelihood of nodal metastasis.
KEGG pathway enrichment analysis revealed that
pathways such as ‘glycosphingolipid biosynthesis-ganglio series’,
‘toll-like receptor (TLR) signaling pathway’ and ‘melanogenesis’
were associated with the upregulated mRNAs; these pathways have
previously been demonstrated to be involved in the pathogenesis of
melanoma. ‘Sialyltransferase activity’ is also associated with
melanoma. A previous study has demonstrated that GM3 α2,
8-sialyltransferase (GD3 synthase) served a role in the
biosynthesis of gangliosides, especially GD3 (45). In melanoma, ganglioside GD3 had been
identified as a tumor-specific antigen (46). The expression of the GD3 synthase
gene is activated by nuclear factor κB (45). In addition, previous studies have
reported that TLR2, 3, 4, 7, 8 and 9 are expressed on melanoma
cells and may interact with the development of melanoma (47,48);
TLR4 agonist lipopolysaccharide increases the proliferation of
TLR4-positive melanoma cells. In addition, knockdown of TLR4
inhibited the migratory ability of melanoma cells (49). These results suggested that TLR4
signaling may contribute to melanoma progression.
Pathways such as ‘oxytocin signaling pathway’,
‘regulation of actin cytoskeleton’, ‘hippo signaling pathway’,
‘focal adhesion’ and ‘gap junction’ were associated with the
downregulated mRNAs. Certain enhanced pathways, such as ‘regulation
of actin cytoskeleton’, are also associated with LMWH (36). The inhibition of adhesion in melanoma
cells through the PKCα/JNK signaling pathway often involves changes
in the actin cytoskeleton. Another enhanced pathway was ‘hippo
signaling pathway’ (50–54); most of the components of this pathway
are tumor-suppressor molecules. Once the pathway is activated,
phosphorylated Yes-associated protein (YAP) and paralog protein TAZ
accumulate in the cell plasma and induce cell cycle arrest; when
these molecules are located in the nucleus, they promote cell
proliferation (50). YAP and TAZ
have been identified in melanoma, and the activated hippo signaling
pathway may have an inhibitory effect on the development of
melanoma. The expression of TAZ in invasive melanoma is higher
compared with YAP, but studies speculated that patients with
melanoma with high expression of YAP tend to exhibit poor prognosis
(51,55,56). In
addition, single nucleotide polymorphisms such as TEA domain
transcription factor (TEAD) 1 and TEAD4 may also influence the
survival of patients with melanoma. Therefore, these two molecules
may serve as therapeutic targets for melanoma (54). A previous study has speculated that
the pathogenesis of melanoma was associated with the crosstalk
between hippo and mitogen associated protein kinase signaling
pathways via the interaction of Raf-1 proto-oncogene
serine/threonine kinase and serine/threonine kinase 3 (52).
LncRNAs affect the pathogenesis of various diseases
through epigenetic regulation. To determine the exact mechanism of
lncRNAs involved in ALM, a ceRNA network between lncRNAs, miRNAs
and mRNAs was constructed as lncRNAs may disturb the activity of
certain miRNAs, which would subsequently affect their target mRNAs.
In this ceRNA network, five lncRNAs interacted with 417 mRNAs
through 252 miRNAs. The results of GO and KEGG analysis were
similar to those of the genes identified by microarray
analysis.
Limitations existed in our study: What has been done
in the present study was just a microarray analysis and its related
CNC network, ceRNA network, GO analysis and KGEG analysis based on
the predicted targeted genes. The further research of lncRNA
function would be performed in the authors' future investigation
and perhaps at that time, more evidence would be found.
In conclusion, to the best of our knowledge, the
present study is the first to reveal lncRNA expression patterns in
ALM using microarray analysis. The results of the present study
suggested genes implicated in tissue development, pigmentation,
cell adhesion activity, organelles related to melanin formation and
channel activity may be involved in the pathogenesis and metastasis
of ALM. In addition, the CNC and ceRNA network analysis results
suggested that dysregulated lncRNAs and mRNAs may serve a role in
tumor formation and development, and lncRNAs may also act as ceRNAs
to disturb the pathogenesis of ALM. These molecules may be
promising therapeutic targets for patients with ALM and further
studies are needed to explore the precise mechanisms of ALM.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
CAMS Innovation Fund for Medical Sciences (CIFMS-2017-12M-1-017)
and the PUMC Youth Fund (grant no. 3332017168).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HS, JX, HC, YW and JS conceived this study. HS, JX,
CX and WB performed the experiments and collected the data. Data
were analyzed and interpreted by HC, YW and JS. HS wrote the paper.
The paper was reviewed by HC, YW and JS. All authors have read and
approved the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Institute of Dermatology, Chinese Academy of Medical
Sciences and Peking Union Medical College (2013-LC/KY-033). All
patients provided informed consents.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lino-Silva LS, Zepeda-Najar C,
Salcedo-Hernández RA and Martinez-Said H: Acral lentiginous
melanoma: Survival analysis of 715 cases. J Cutan Med Surg.
23:38–43. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Häfliger EM, Ramelyte E, Mangana J, Kunz
M, Kazakov DV, Dummer R and Cheng PF: Metastatic acral lentiginous
melanoma in a tertiary referral center in Switzerland: A systematic
analysis. Melanoma Res. 28:442–450. 2018.PubMed/NCBI
|
|
3
|
Kim HJ, Seo JW, Roh MS, Lee JH and Song
KH: Clinical features and prognosis of Asian patients with acral
lentiginous melanoma who have nodal nevi in their sentinel lymph
node biopsy specimen. J Am Acad Dermatol. 79:706–713. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakamura Y and Fujisawa Y: Diagnosis and
management of acral lentiginous melanoma. Curr Treat Options Oncol.
19:422018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wada M, Ito T, Tsuji G, Nakahara T,
Hagihara A, Furue M and Uchi H: Acral lentiginous melanoma versus
other melanoma: A single-center analysis in Japan. J Dermatol.
44:932–938. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hombach S and Kretz M: Non-coding RNAs:
Classification, biology and functioning. Adv Exp Med Biol.
937:3–17. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kondo Y, Shinjo K and Katsushima K: Long
non-coding RNAs as an epigenetic regulator in human cancers. Cancer
Sci. 108:1927–1933. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu X, Zheng H, Tse G, Chan MT and Wu WK:
Long non-coding RNAs in melanoma. Cell Prolif. 51:e124572018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang L, Zhang W, Su B and Yu B: Long
noncoding RNA HOTAIR is associated with motility, invasion, and
metastatic potential of metastatic melanoma. Biomed Res Int.
2013:2510982013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun L, Sun P, Zhou QY, Gao X and Han Q:
Long noncoding RNA MALAT1 promotes uveal melanoma cell growth and
invasion by silencing of miR-140. Am J Transl Res. 8:3939–3946.
2016.PubMed/NCBI
|
|
11
|
Flockhart RJ, Webster DE, Qu K,
Mascarenhas N, Kovalski J, Kretz M and Khavari PA: BRAFV600E
remodels the melanocyte transcriptome and induces BANCR to regulate
melanoma cell migration. Genome Res. 22:1006–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li R, Zhang L, Jia L, Duan Y, Li Y, Bao L
and Sha N: Long non-coding RNA BANCR promotes proliferation in
malignant melanoma by regulating MAPK pathway activation. PLoS One.
9:e1008932014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu S, Wang H, Pan H, Shi Y, Li T, Ge S,
Jia R, Zhang H and Fan X: ANRIL lncRNA triggers efficient
therapeutic efficacy by reprogramming the aberrant INK4-hub in
melanoma. Cancer Lett. 381:41–48. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pasmant E, Laurendeau I, Héron D, Vidaud
M, Vidaud D and Bièche I: Characterization of a germ-line deletion,
including the entire INK4/ARF locus, in a melanoma-neural system
tumor family: Identification of ANRIL, an antisense noncoding RNA
whose expression coclusters with ARF. Cancer Res. 67:3963–3969.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mazar J, Zhao W, Khalil AM, Lee B, Shelley
J, Govindarajan SS, Yamamoto F, Ratnam M, Aftab MN, Collins S, et
al: The functional characterization of long noncoding RNA SPRY4-IT1
in human melanoma cells. Oncotarget. 5:8959–8969. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khaitan D, Dinger ME, Mazar J, Crawford J,
Smith MA, Mattick JS and Perera RJ: The melanoma-upregulated long
noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer
Res. 71:3852–3862. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu CF, Tan GH, Ma CC and Li L: The
non-coding RNA llme23 drives the malignant property of human
melanoma cells. J Genet Genomics. 40:179–188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wei Y, Sun Q, Zhao L, Wu J, Chen X, Wang
Y, Zang W and Zhao G: LncRNA UCA1-miR-507-FOXM1 axis is involved in
cell proliferation, invasion and G0/G1 cell cycle arrest in
melanoma. Med Oncol. 33:882016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tian Y, Zhang X, Hao Y, Fang Z and He Y:
Potential roles of abnormally expressed long noncoding RNA UCA1 and
Malat-1 in metastasis of melanoma. Melanoma Res. 24:335–341. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schmidt K, Joyce CE, Buquicchio F, Brown
A, Ritz J, Distel RJ, Yoon CH and Novina CD: The lncRNA SLNCR1
mediates melanoma invasion through a conserved SRA1-like region.
Cell Rep. 15:2025–2037. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leucci E, Vendramin R, Spinazzi M,
Laurette P, Fiers M, Wouters J, Radaelli E, Eyckerman S, Leonelli
C, Vanderheyden K, et al: Melanoma addiction to the long non-coding
RNA SAMMSON. Nature. 531:518–522. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Larionov A, Krause A and Miller W: A
standard curve based method for relative real time PCR data
processing. BMC Bioinformatics. 6:622005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Forrest ME and Khalil AM: Review:
Regulation of the cancer epigenome by long non-coding RNAs. Cancer
Lett. 407:106–112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu N, Wang F, Lv M and Cheng L: Microarray
expression profile analysis of long non-coding RNAs in human breast
cancer: A study of Chinese women. Biomed Pharmacother. 69:221–227.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sand M, Bechara FG, Sand D, Gambichler T,
Hahn SA, Bromba M, Stockfleth E and Hessam S: Long-noncoding RNAs
in basal cell carcinoma. Tumour Biol. 37:10595–10608. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xue Y, Ma G, Gu D, Zhu L, Hua Q, Du M, Chu
H, Tong N, Chen J, Zhang Z and Wang M: Genome-wide analysis of long
noncoding RNA signature in human colorectal cancer. Gene.
556:227–234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dorsky RI, Moon RT and Raible DW: Control
of neural crest cell fate by the Wnt signalling pathway. Nature.
396:370–373. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim KI, Jeong DS, Jung EC, Lee JH, Kim CD
and Yoon TJ: Wnt/β-catenin signaling inhibitor ICG-001 enhances
pigmentation of cultured melanoma cells. J Dermatol Sci.
84:160–168. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
van Lanschot CG, Koljenovic S, Grunhagen
DJ, Verhoef C and van Akkooi AC: Pigmentation in the sentinel node
correlates with increased sentinel node tumor burden in melanoma
patients. Melanoma Res. 24:261–266. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen KG, Leapman RD, Zhang G, Lai B,
Valencia JC, Cardarelli CO, Vieira WD, Hearing VJ and Gottesman MM:
Influence of melanosome dynamics on melanoma drug sensitivity. J
Natl Cancer Inst. 101:1259–1271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hertzman Johansson C, Azimi A, Frostvik
Stolt M, Shojaee S, Wiberg H, Grafström E, Hansson J and Egyházi
Brage S: Association of MITF and other melanosome-related proteins
with chemoresistance in melanoma tumors and cell lines. Melanoma
Res. 23:360–365. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tajima N, Itokazu Y, Korpi ER, Somerharju
P and Käkelä R: Activity of BK(Ca) channel is modulated by membrane
cholesterol content and association with Na+/K+-ATPase in human
melanoma IGR39 cells. J Biol Chem. 286:5624–5638. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Y, Luo Z, Hao Y, Ba W, Wang R, Wang
W, Ding X and Li C: mTOR-mediated Na+/Ca2+
exchange affects cell proliferation and metastasis of melanoma
cells. Biomed Pharmacother. 92:744–749. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kuphal S and Bosserhoff AK: E-cadherin
cell-cell communication in melanogenesis and during development of
malignant melanoma. Arch Biochem Biophys. 524:43–47. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rossier-Pansier L, Baruthio F, Rüegg C and
Mariotti A: Compartmentalization in membrane rafts defines a pool
of N-cadherin associated with catenins and not engaged in cell-cell
junctions in melanoma cells. J Cell Biochem. 103:957–971. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chalkiadaki G, Nikitovic D, Katonis P,
Berdiaki A, Tsatsakis A, Kotsikogianni I, Karamanos NK and
Tzanakakis GN: Low molecular weight heparin inhibits melanoma cell
adhesion and migration through a PKCa/JNK signaling pathway
inducing actin cytoskeleton changes. Cancer Lett. 312:235–244.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schmitz P, Gerber U, Schütze N, Jüngel E,
Blaheta R, Naggi A, Torri G and Bendas G: Cyr61 is a target for
heparin in reducing MV3 melanoma cell adhesion and migration via
the integrin VLA-4. Thromb Haemost. 110:1046–1054. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cheng H, Mollica MY, Lee SH, Wang L,
Velazquez-Martinez CA and Wu S: Effects of nitric oxide-releasing
nonsteroidal anti-inflammatory drugs (NONO-NSAIDs) on melanoma cell
adhesion. Toxicol Appl Pharmacol. 264:161–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Takabe P, Bart G, Ropponen A, Rilla K,
Tammi M, Tammi R and Pasonen-Seppänen S: Hyaluronan synthase 3
(HAS3) overexpression downregulates MV3 melanoma cell
proliferation, migration and adhesion. Exp Cell Res. 337:1–15.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Valcárcel M, Mendoza L, Hernández JJ,
Carrascal T, Salado C, Crende O and Vidal-Vanaclocha F: Vascular
endothelial growth factor regulates melanoma cell adhesion and
growth in the bone marrow microenvironment via tumor
cyclooxygenase-2. J Transl Med. 9:1422011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Foy M, Anézo O, Saule S and Planque N:
PRL-3/PTP4A3 phosphatase regulates integrin β1 in adhesion
structures during migration of human ocular melanoma cells. Exp
Cell Res. 353:88–99. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
van Kilsdonk JW, Takahashi N, Weidle U,
Burtscher H, Jarry J, Daha MR, Swart GW and van Kempen LC:
Modulation of activated leukocyte cell adhesion molecule-mediated
invasion triggers an innate immune gene response in melanoma. J
Invest Dermatol. 132:1462–1470. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ullrich N, Löffek S, Horn S, Ennen M,
Sánchez-Del-Campo L, Zhao F, Breitenbuecher F, Davidson I, Singer
BB, Schadendorf D, et al: MITF is a critical regulator of the
carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1)
in malignant melanoma. Pigment Cell Melanoma Res. 28:736–740. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Meves A, Nikolova E, Heim JB, Squirewell
EJ, Cappel MA, Pittelkow MR, Otley CC, Behrendt N, Saunte DM,
Lock-Andersen J, et al: Tumor cell adhesion as a risk factor for
sentinel lymph node metastasis in primary cutaneous melanoma. J
Clin Oncol. 33:2509–2515. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kang NY, Kim CH, Kim KS, Ko JH, Lee JH,
Jeong YK and Lee YC: Expression of the human CMP-NeuAc:GM3
alpha2,8-sialyltransferase (GD3 synthase) gene through the
NF-kappaB activation in human melanoma SK-MEL-2 cells. Biochim
Biophys Acta. 1769:622–630. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Miyata M, Ichihara M, Tajima O, Sobue S,
Kambe M, Sugiura K and Furukawa K and Furukawa K: UVB-irradiated
keratinocytes induce melanoma-associated ganglioside GD3 synthase
gene in melanocytes via secretion of tumor necrosis factor α and
interleukin 6. Biochem Biophys Res Commun. 445:504–510. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Goto Y, Arigami T, Kitago M, Nguyen SL,
Narita N, Ferrone S, Morton DL, Irie RF and Hoon DS: Activation of
Toll-like receptors 2, 3, and 4 on human melanoma cells induces
inflammatory factors. Mol Cancer Ther. 7:3642–3653. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Saint-Jean M, Knol AC, Nguyen JM, Khammari
A and Dréno B: TLR expression in human melanoma cells. Eur J
Dermatol. 21:899–905. 2011.PubMed/NCBI
|
|
49
|
Feng R, Gong J, Wu L, Wang L, Zhang B,
Liang G, Zheng H and Xiao H: MAPK and Hippo signaling pathways
crosstalk via the RAF-1/MST-2 interaction in malignant melanoma.
Oncol Rep. 38:1199–1205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kim JE, Finlay GJ and Baguley BC: The role
of the hippo pathway in melanocytes and melanoma. Front Oncol.
3:1232013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Menzel M, Meckbach D, Weide B, Toussaint
NC, Schilbach K, Noor S, Eigentler T, Ikenberg K, Busch C,
Quintanilla-Martinez L, et al: In melanoma, Hippo signaling is
affected by copy number alterations and YAP1 overexpression impairs
patient survival. Pigment Cell Melanoma Res. 27:671–673. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nallet-Staub F, Marsaud V, Li L, Gilbert
C, Dodier S, Bataille V, Sudol M, Herlyn M and Mauviel A:
Pro-invasive activity of the Hippo pathway effectors YAP and TAZ in
cutaneous melanoma. J Invest Dermatol. 134:123–132. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Takazawa Y, Kiniwa Y, Ogawa E, Uchiyama A,
Ashida A, Uhara H, Goto Y and Okuyama R: Toll-like receptor 4
signaling promotes the migration of human melanoma cells. Tohoku J
Exp Med. 234:57–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yuan H, Liu H, Liu Z, Zhu D, Amos CI, Fang
S, Lee JE and Wei Q: Genetic variants in Hippo pathway genes YAP1,
TEAD1 and TEAD4 are associated with melanoma-specific survival. Int
J Cancer. 137:638–645. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hall CA, Wang R, Miao J, Oliva E, Shen X,
Wheeler T, Hilsenbeck SG, Orsulic S and Goode S: Hippo pathway
effector Yap is an ovarian cancer oncogene. Cancer Res.
70:8517–8525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kang W, Tong JH, Chan AW, Lee TL, Lung RW,
Leung PP, So KK, Wu K, Fan D, Yu J, et al: Yes-associated protein 1
exhibits oncogenic property in gastric cancer and its nuclear
accumulation associates with poor prognosis. Clin Cancer Res.
17:2130–2139. 2011. View Article : Google Scholar : PubMed/NCBI
|