Introduction
Esophageal cancer (ESCA) ranks sixth in terms of
cancer-associated mortality worldwide (1). In 2012, 455,000 patients were diagnosed
with ESCA worldwide, translating to an incidence rate of 5.9 per
100,000 (2). The 5-year survival
rate of patients with ESCA is only 18%, because ESCA typically
exhibits no symptoms and is therefore diagnosed at an advanced
clinical stage (3). At present, the
top predictive characteristic of ESCA prognosis is TNM staging
(4). Although TNM stage is useful,
it varies among patients with the same cancer stage (5). Furthermore, clinicopathological
characteristics are molecular comprehensive reflections, including
proteins and genes. Patients with ESCA with homogeneous clinical
stage may be further classified according to various molecular
patterns, like a 6-microRNA signature reported by Lan et al
(6). It is therefore crucial to
determine novel biomarkers for identifying patients with high risk
of mortality.
Long non-coding RNA (lncRNA) is defined as an RNA
transcript that is not translated into proteins and of >200
nucleotides in length (7).
Conversely, with protein coding genes, the gene expression levels
of reported lncRNAs are much lower (8). Furthermore, the role of lncRNAs is
crucial in tumor growth, progression and treatment response
(9), and most mRNAs serve critical
roles in fundamental cellular processes. Numerous studies reported
that integrated analysis of mRNAs and lncRNAs can contribute to the
prognosis evaluation of breast and hepatocellular carcinoma
(10,11). However, only a few studies have
focused on the integrated assessment of mRNAs and lncRNAs in
ESCA.
The present study aimed to analyze the gene
expression profile of ESCA samples from The Cancer Genome Atlas
(TCGA) database, and a signature was constructed by integrating
mRNA and lncRNA expression for the prediction of ESCA
prognosis.
Materials and methods
Data source and preprocessing
The TCGA database (accessed January 2019; http://portal.gdc.cancer.gov/) provided the RNA
sequencing (RNA-seq) data and clinicopathological characteristics
of patients with ESCA collected between December 2011 and December
2013 (12). The inclusion criteria
were as follows: i) Patients with ESCA, RNA-seq data and
clinicopathological information; and ii) patients with ESCA with at
least 30 days follow-up. A total of 117 samples were selected in
the present study, including 106 tumor tissues and 11 normal
tissues (Table SI). Only RNAs with
transcript per million value >0.1 in ≥50% of ESCA samples were
enrolled for further investigation. A total of 16,368 mRNAs and
7,347 lncRNAs were annotated according to GENCODE datasets
(www.gencodegenes.org). The
differentially expressed (DE) mRNAs and lncRNAs in tumor tissues
compared with normal tissues were analyzed using the ‘limma’
package (version 3.36.5; http://bioconductor.org/packages/release/bioc/html/limma.html)
in R (version 3.5.2; http://www.r-project.org/). The results were
visualized by volcano plot. Due to the gap between the count of
mRNAs and lncRNAs, different thresholds were set for identifying DE
mRNAs and lncRNAs. DEmRNAs with P<0.05 and |log fold change
(FC)|>2.0, and DElncRNAs with P<0.05 and |log FC|>1.0 were
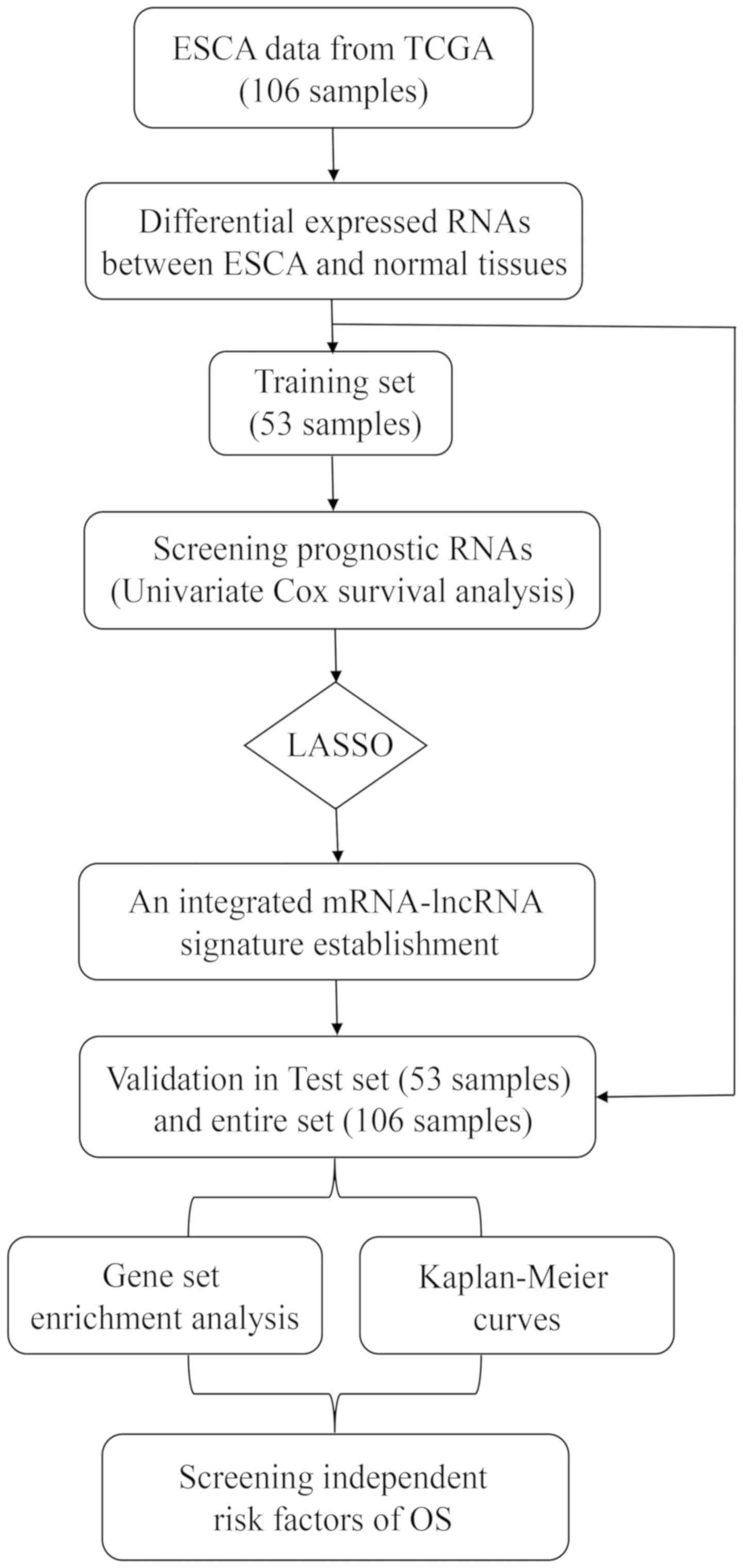
considered as significant. The research process scheme is presented
in Fig. 1. Since the TCGA database
information is publicly available, no ethical approval was
required.
Construction and validation of the
mRNA-lncRNA signature
The prognostic values of mRNAs and lncRNAs were
determined using univariate Cox proportional hazards regression
analysis. According to previous studies, P<0.1 was considered to
indicate a statistically significant difference (6,13,14).
Subsequently, a total of 106 tumor samples were randomly divided
into two sets: A training set (n=53) and a test set (n=53; Table I).
 | Table I.Characteristics of patients in the
training and test set. |
Table I.
Characteristics of patients in the
training and test set.
| Variables | Training set, n
(%) | Test set, n
(%) | P-value |
|---|
| Number | 53 | 53 |
|
| Age, years (mean ±
SD) | 65.00±12.41 | 64.06±12.34 | 0.695 |
| Sex |
|
|
|
|
Female | 11 (20.8) | 8 (15.1) | 0.613 |
|
Male | 42 (79.2) | 45 (84.9) |
|
| Height, cm |
|
|
|
|
<175 | 28 (57.1) | 29 (59.2) | >0.999 |
|
>175 | 21 (42.9) | 20 (40.8) |
|
| No
value | 4 | 4 |
|
| Weight, kg |
|
|
|
|
<80 | 32 (60.4) | 27 (51.9) | 0.434 |
|
>80 | 21 (39.6) | 25 (48.1) |
|
| No
value | 0 | 1 |
|
| Ethnicity |
|
|
|
|
Non-white | 3 (7.1) | 7 (15.2) | 0.320 |
|
White | 39 (92.9) | 39 (84.8) |
|
| No
value | 11 | 7 |
|
| Pathology |
|
|
|
|
Adenocarcinoma | 40 (75.5) | 35 (66.0) | 0.393 |
|
Squamous | 13 (24.5) | 18 (34.0) |
|
| Alcohol
history |
|
|
|
| No | 17 (32.7) | 15 (28.3) | 0.675 |
|
Yes | 35 (67.3) | 38 (71.7) |
|
| No
value | 1 | 0 |
|
| Barrett's
disease |
|
|
|
| No | 34 (69.4) | 40 (81.6) | 0.240 |
|
Yes | 15 (30.6) | 9 (18.4) |
|
| No
value | 4 | 4 |
|
| T |
|
|
|
|
1+2 | 23 (44.2) | 22 (42.3) | >0.999 |
|
3+4 | 29 (55.8) | 30 (57.7) |
|
| No
value | 1 | 1 |
|
| N |
|
|
|
| 0 | 16 (33.3) | 16 (33.3) | >0.999 |
|
1+3 | 32 (66.7) | 32 (66.7) |
|
| No
value | 5 | 5 |
|
| M |
|
|
|
| 0 | 42 (87.5) | 36 (83.7) | 0.766 |
| 1 | 6 (12.5) | 7 (16.3) |
|
| No
value | 5 | 10 |
|
| Stage |
|
|
|
|
I+II | 29 (54.7) | 24 (48.0) | 0.557 |
|
III+IV | 24 (45.3) | 26 (52.0) |
|
| No
value | 0 | 3 |
|
| Survival
status |
|
|
|
|
Alive | 27 (50.9) | 33 (62.3) | 0.327 |
|
Dead | 26 (49.1) | 20 (37.7) |
|
| Survival time,
years (mean ± SD) | 1.47±1.35 | 1.34±1.16 | 0.606 |
The least absolute shrinkage and selection operator
(LASSO) method, which is appropriate for high-dimensional data
analysis (15), can select an
optimal group of genes lacking collinearity through penalty
imposing and most regression coefficients shrinking to zero. LASSO
was therefore used in the training set to determine and confirm the
selected mRNAs and lncRNAs. The prognostic mRNA-lncRNA signature
risk score in patients was calculated using each prognostic RNA
expression level and its associated coefficient. The formula used
was as follow: Risk score=β1 × gene 1 + β2 × gene 2 +…+ βn × gene
n, where β indicates the coefficient of each gene and gene
indicates the expressed gene value.
Based on the median risk score cut-off (2.541),
patients were divided into a high-risk group and a low-risk group.
Kaplan-Meier (KM) and log-rank methods were used to test the
difference in the two groups by using the ‘survival’ R package
(version 2.43; http://cran.r-project.org/web/packages/survival/index.html).
To validate the signature sensitivity and precision, the ‘timeROC’
R package (version 0.3; http://cran.r-project.org/web/packages/timeROC/index.html)
was used to calculate the area under the receiver operating
characteristic (ROC) curves. This signature was further validated
in the test set and the entire set. Subsequently, stratified
analysis based on the clinicopathological characteristics of
patients with ESCA was carried out in the entire set.
Development of the nomogram
The prognostic significance of the signature was
evaluated with clinicopathologic characteristics, including age,
sex, height, weight, pathology, alcohol consumption history,
Barrett's disease and TNM stage, by univariate and multivariate Cox
proportional hazard regressions analysis. In order to provide
clinicians with a quantitative tool to predict the individual
probability of survival time, the R package ‘rms’ (version 5.1–3;
http://cran.r-project.org/web/packages/rms/) was used
to build the nomogram associated with the variables, including a
mRNA-lncRNA signature and TNM stage, derived from the previous
analysis. Furthermore, the nomogram predictive performance was
calculated using concordance index and bootstrapping validation
calibration (1,000 bootstrap resamples) to reduce the potential of
overfitting.
mRNA-lncRNA signature function
prediction
To study the potential biological mechanism between
low- and high-risk groups, gene set enrichment analysis (GSEA;
http://software.broadinstitute.org/gsea) was
performed. The BioCarta (c2.cp.biocarta.v6.2.symbols.gmt) and
Reactome (c2.cp.reactome.v6.2.symbols.gmt) datasets (http://software.broadinstitute.org/gsea/msigdb/collections.jsp)
were selected as the reference gene sets. A gene with P<0.05 and
enrichment score >0.5 was considered as significantly
enriched.
Results
DEmRNAs and DElncRNAs in patients with
ESCA
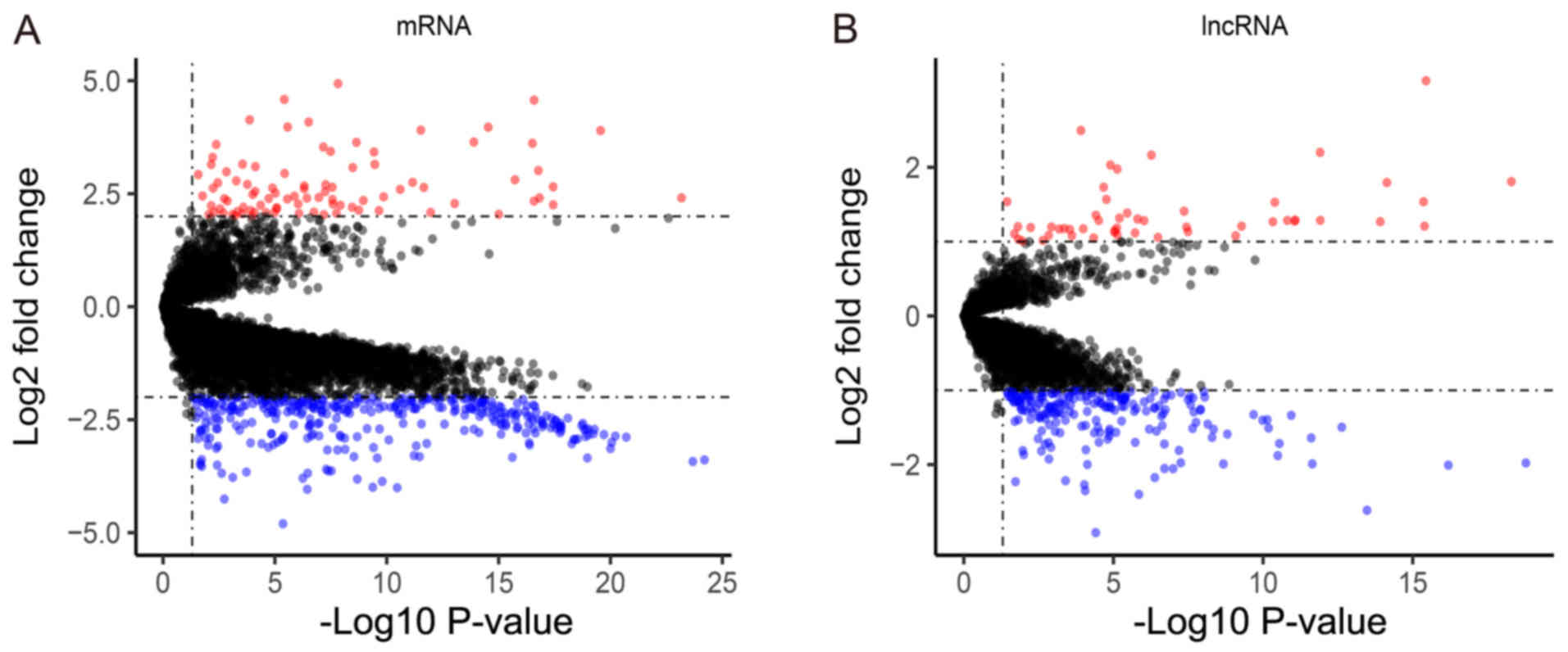
A total of 440 mRNAs were identified as
significantly different between tumor and normal tissues, of which
93 mRNAs were upregulated and 347 were downregulated (Fig. 2A). In addition, 263 DElncRNAs (51
upregulated; 212 downregulated) were identified and selected for
further analysis (Fig. 2B).
Signature development in the training
set
A total of 52 DEmRNAs and 38 DElncRNAs were
identified in the training set as being associated with prognosis
following univariate Cox proportional hazards regression analysis
(Table SII). Furthermore, using the
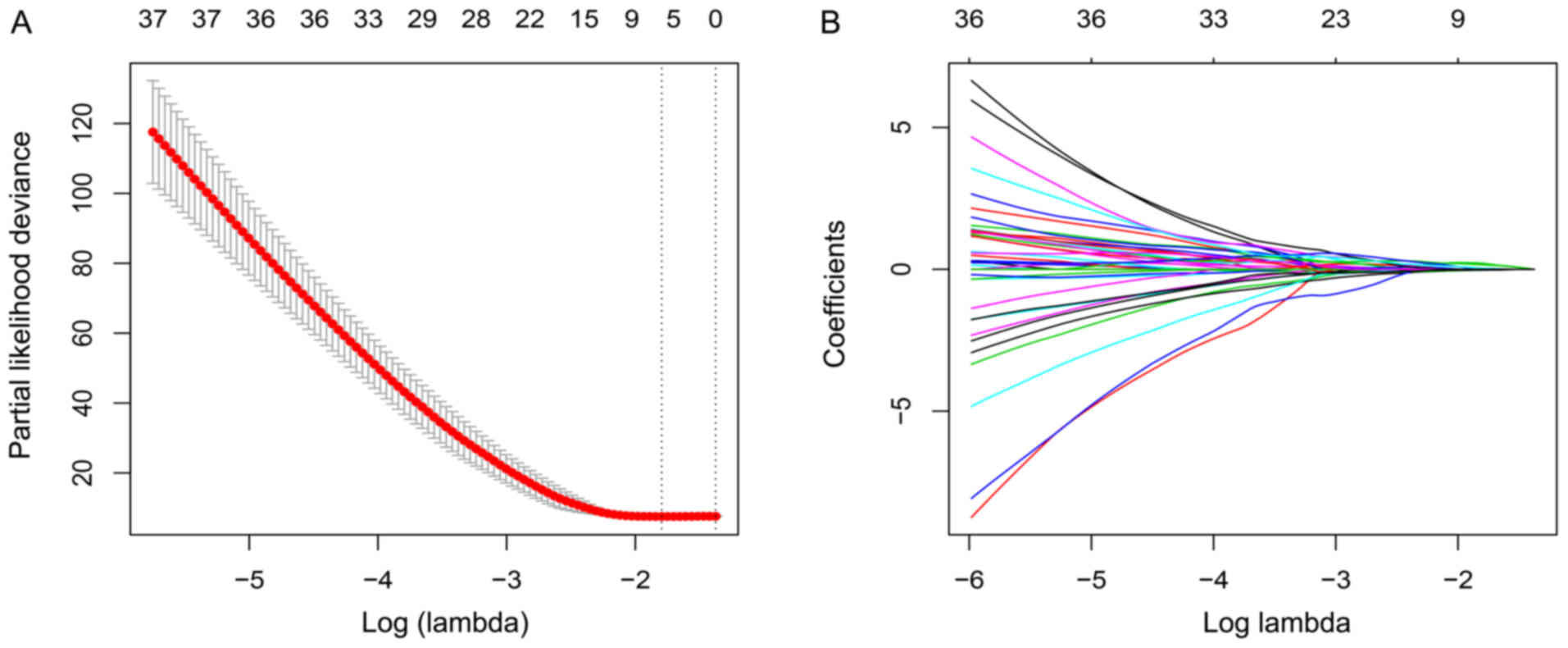
LASSO method, three mRNAs, PCNA, TNS4, SLC26A9, and two lncRNAs,
ZFAS1, AC104041.1, were identified (Fig.
3B). An appropriate value of lambda was set as 0.166 using
cross validation (Fig. 3A). The risk
score calculated from the five expressed genes weighted by their
coefficients was set as below: Risk score=(0.209095 × expression of
PCNA) + (−0.023333 × expression of TNS4) + (−0.025136 × expression
of SLC26A9) + (0.160318 × expression of ZFAS1) + (0.035372 ×
expression of AC104041.1).
According to the median risk score, patients were
divided into high- and low-risk groups. The risk scores, survival
time distributions and patients' status in the training set are
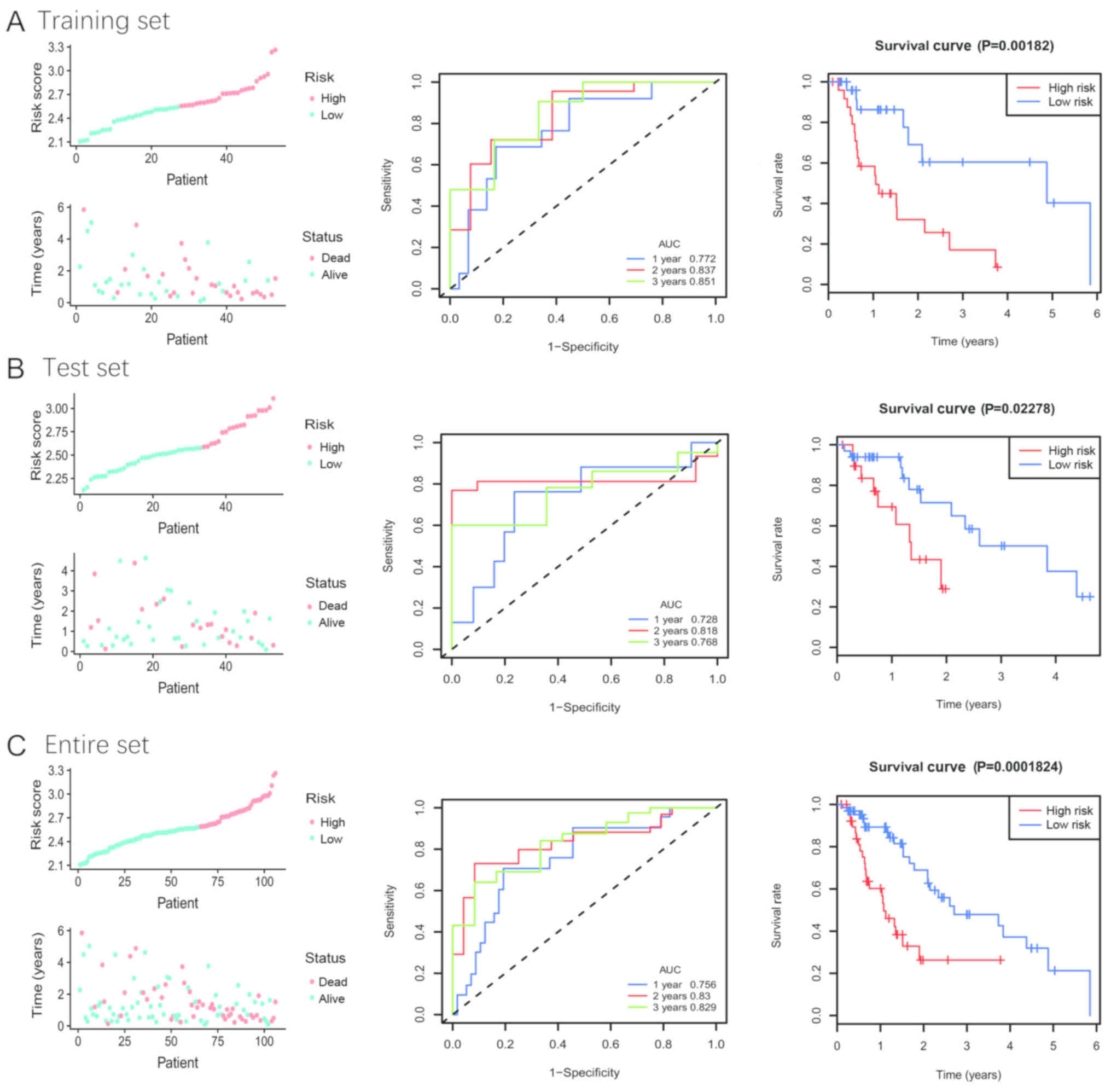
presented in Fig. 4A (left panel).
The results from KM survival analysis demonstrated that the two
groups had significantly different outcomes (Fig. 4A; right panel). In addition, 1-, 2-
and 3-year time-dependent ROC analyses were performed to evaluate
the mRNA-lncRNA signature prognostic sensitivity and specificity
(Fig. 4A; middle panel). The AUCs of
the mRNA-lncRNA signature were 0.772, 0.837 and 0.851 at 1-, 2- and
3-year survival times, respectively, suggesting that this signature
may have a high prognostic accuracy.
Validation of the signature in the
test and entire sets
By using the established cut-off point, patients
were assigned to a low- or high-risk group in the test and entire
sets. The patients' risk score distribution and survival status
were ranked by the risk scores in the test set (Fig. 4B, left panel) and the entire set
(Fig. 4C, left panel). The results
from KM analysis for overall survival (OS) indicated that patients
with low risk may exhibit improved clinical outcomes compared with
patients with high risk (Fig. 4B and
C, right panel). To confirm the accuracy of the signature, the
areas under ROC curves were calculated, and the results were 0.728,
0.818 and 0.768 at 1-, 2- and 3-year survival times in the test set
(Fig. 4B, middle panel),
respectively, and 0.756, 0.830, 0.829 at 1-, 2- and 3-year survival
times in the entire set (Fig. 4C,
middle panel), respectively.
Stratified analysis and independence
analysis in the entire set
The results from subgroup analysis based on age,
sex, height, weight, ethnicity, alcohol consumption history,
Barrett's disease and TNM stage suggested that patients with
high-risk of mortality may present with poor clinical outcomes. The
results were all significant, except for women, non-white, no
alcohol consumption history, tumor size I+II and stage I+II
(Fig. S1). Furthermore, the
mRNA-lncRNA signature was considered as an independent prognostic
factor in univariate and multivariate Cox regression analyses in
the entire set following adjustment for various clinicopathological
characteristics [hazard ratio (HR), 2.5, 95% confidence interval
(CI), 1.55–4.06, P<0.001 in the univariate Cox regression
analysis; HR, 2.41, 95% CI, 1.47–3.96, P<0.001 in the
multivariate Cox regression analysis; Table II].
| Table II.Univariate and multivariate Cox
regression analysis of risk score and clinical variables. |
Table II.
Univariate and multivariate Cox
regression analysis of risk score and clinical variables.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
|
Characteristics | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Risk score | 2.50 | 1.55–4.06 | <0.001 | 2.41 | 1.47–3.96 | <0.001 |
| Age (≥60 years vs.
<60 years) | 0.68 | 0.36–1.29 | 0.238 |
|
|
|
| Sex (male vs.
female) | 1.70 | 0.52–5.54 | 0.382 |
|
|
|
| Height (≥175 cm vs.
<175 cm) | 1.15 | 0.60–2.19 | 0.681 |
|
|
|
| Weight (≥85 kg vs.
<85 kg) | 0.79 | 0.41–1.51 | 0.471 |
|
|
|
| Pathology (squamous
vs. adenocarcinoma) | 1.04 | 0.51–2.12 | 0.903 |
|
|
|
| Alcohol consumption
(yes vs. no) | 0.57 | 0.30–1.08 | 0.083 |
|
|
|
| Barrett's disease
(yes vs. no) | 1.46 | 0.74–2.88 | 0.271 |
|
|
|
| Stage (III+IV vs.
I+II) | 2.09 | 1.29–3.41 | 0.003 | 1.98 | 1.20–3.27 | 0.007 |
Construction of a novel nomogram for
predicting prognosis in ESCA
Risk score and TNM stage derived from the previous
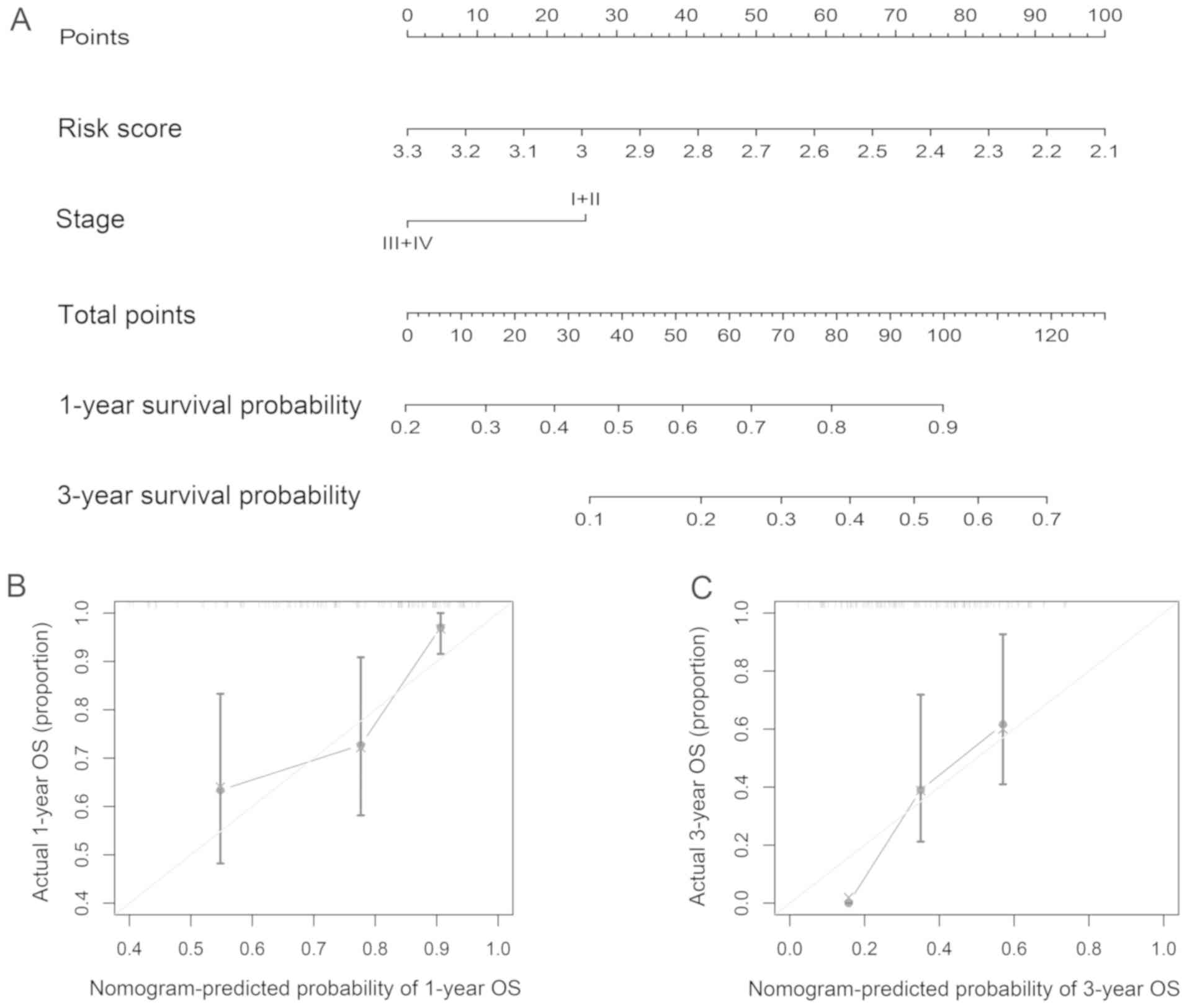
analyses were developed and presented as a nomogram (Fig. 5A). A high score of the nomogram
indicated a high probability of 1- and 3-year survival. In the
primary cohort, the nomogram C-index was 0.717 (95% CI,
0.543–0.891). The established nomogram calibration curve for the
probability of survival outcome indicated good agreement between
observation and prediction (Fig. 5B and
C).
Functional analysis of the mRNA-lncRNA
signature
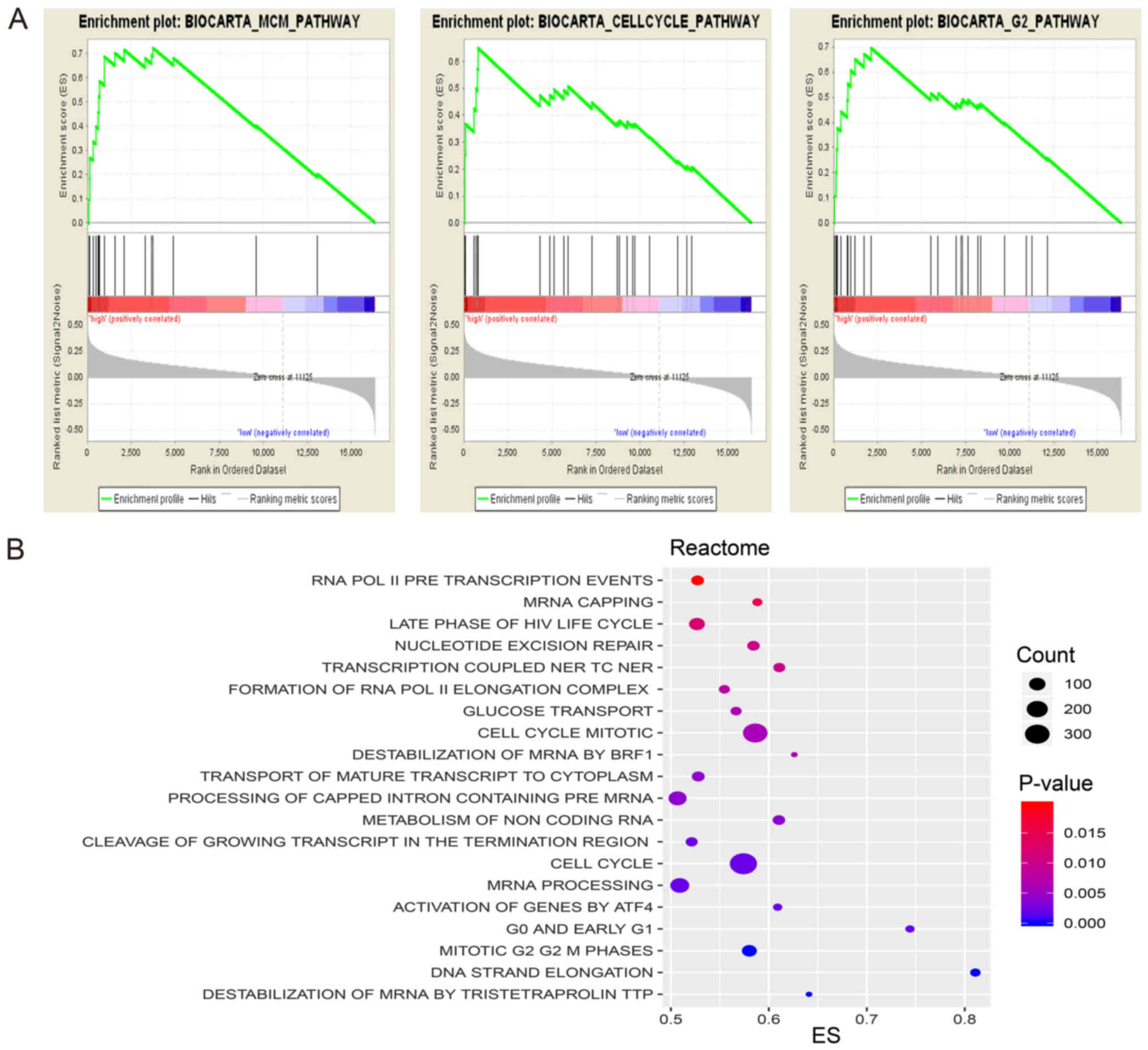
In order to determine the possible signature
mechanism, BioCarta and Reactome pathway enrichment analysis was
performed using GSEA. The results demonstrated that compared with
genes in the low-risk group, genes in the high-risk group were
significantly enriched with numerous biological processes, in
particular cell cycle signaling pathway, minichromosome maintenance
pathway and mRNA capping and processing pathway (Fig. 6).
Discussion
Clinicopathological characteristics of patients with
ESCA, including TNM stage, have been widely used to adapt
individual treatment and predict prognosis for patients. However,
due to the heterogeneity of ESCA, it is difficult to precisely
predict prognosis by using clinicopathological characteristics
(16). Therefore, application of
gene-based biomarkers represents a promising alternative to predict
prognosis.
Numerous studies have identified molecular markers
for prognosis in ESCA. Peters et al (17) generated a 4-mRNA prognostic signature
for patients with esophageal adenocarcinoma. Recently, a reliable
6-immunohistochemical-based signature has been determined for
patients with esophageal squamous cell cancer (18). Furthermore, a 3-lncRNA signature
associated with OS has been built for patients with esophageal
squamous cell carcinoma (19). Fan
et al (20) developed a novel
8-lncRNA signature for the prediction of prognosis in patients with
ESCA. Additionally, a previous study reported that the dysregulated
expression of certain micro (mi)RNAs (hsa-mir-425, hsa-let-7b,
hsa-mir-23a, hsa-mir-3074, hsa-mir-424 and hsa-mir-505) could be
used for the prognosis of esophageal adenocarcinoma (6). To the best of our knowledge, previous
studies have only focused on mRNAs, lncRNAs or miRNAs, without
investigating multi-RNA-based data. It is therefore crucial to
assess whether multi-RNA-based signatures could predict prognosis
in ESCA.
In the present study, a mRNA-lncRNA prognostic
signature was established by conducting LASSO Cox regression model
analysis. Based on the risk score of the signature, patients were
divided into high- and low-risk groups. Patients in the low-risk
group exhibited improved clinical outcomes. Furthermore, the
mRNA-lncRNA signature was independent of the clinicopathological
characteristics in univariate and multivariate Cox regression
analyses.
The prognostic signature for patients with ESCA
contained five genes, three mRNAs, PCNA, TNS4, SLC26A9, and two
lncRNAs, ZFAS1, AC104041.1.
Among them, the genes proliferating cell nuclear
antigen (PCNA), ZNFX1 antisense RNA 1 (ZFAS1) and AC10401.1 were
negatively associated with OS, whereas the two other genes tensin 4
(TNS4) and solute carrier family 26 member 9 (SLC26A9) were
associated with an improved survival outcome. PCNA has been
identified as a proliferating cell nuclear antigen that can serve a
role in DNA replication and repair (21). Furthermore, previous studies have
identified PCNA as a cell growth marker and a predictive indicator
of various types of cancer, including colorectal cancer, gastric
carcinoma and parotid gland cancer (22–24). In
addition, it has been reported that upregulated PCNA is associated
with poor survival in patients with esophagus squamous cell cancer
(25), which is consistent with the
results from our previous study on ESCA. Furthermore, Kimos et
al (26) identified PCNA as a
biomarker of esophageal neoplastic progression, suggesting that
PCNA could be a target for ESCA management. TNS4, which is
identified as a COOH-terminus tensin-like molecule, has been
reported to be a cell adhesion factor that is associated with
cancer cell mobility and migration (27). Previous studies have demonstrated
that TNS4 can promote cancer cell migration, and can be considered
as an oncogene in hepatocellular cancer (28), pancreatic cancer (29) and colon carcinoma (30). Furthermore, none or lowly expressed
TNS4 has been reported in kidney and prostate cancer (31,32).
These findings suggest the biological functions of TNS4 are
dependent on the type of cancer. In the present study, reduced
expression of TNS4 in ESCA samples was associated with short
survival in patients. Regarding ZFAS1, its overexpression has been
reported to be associated with metastasis and poor prognosis in
gastric cancer (33), colorectal
cancer (34) and hepatocellular
carcinoma (35), suggesting that
ZFAS1 may be considered as a potential prognostic marker in cancer.
Consistently, the present study demonstrated that ZFAS1 expression
was inversely associated with the OS of patients with ESCA.
Regarding SLC26A9 and AC104041.1, there was no correlative
literature in cancer. The biological role of these genes in ESCA
should be further investigated.
The present study presented some limitations.
Firstly, only 106 ESCA samples were available in the TCGA dataset,
hence the sub-group sample size was too small. Secondly, the
identified signature should be further validated in an external
dataset. Thirdly, in vitro and in vivo experiments
should be performed in order to better understand the potential
mechanism of this signature.
In conclusion, the present study established and
validated a mRNA-lncRNA integrated signature for the prognosis of
patients with ESCA by using a TCGA dataset. Although further
investigation is required to confirm the importance of this
signature, the present study provided valuable information
regarding ESCA pathology and its clinical management.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TL and YL conceived, designed and performed the
experiments. TL and YL analyzed the data, prepared figures and/or
tables and wrote and revised the manuscript. ZX, KS and OY
performed the experiments. HL and CZ analyzed the data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dy GW, Gore JL, Forouzanfar MH, Naghavi M
and Fitzmaurice C: Global burden of urologic cancers, 1990–2013.
Eur Urol. 71:437–446. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arnold M, Soerjomataram I, Ferlay J and
Forman D: Global incidence of oesophageal cancer by histological
subtype in 2012. Gut. 64:381–387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alsop BR and Sharma P: Esophageal cancer.
Gastroenterol Clin North Am. 45:399–412. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rice TW, Ishwaran H, Blackstone EH,
Hofstetter WL, Kelsen DP and Apperson-Hansen C; Worldwide
Esophageal Cancer Collaboration Investigators, : Recommendations
for clinical staging (cTNM) of cancer of the esophagus and
esophagogastric junction for the 8th edition AJCC/UICC staging
manuals. Dis Esophagus. 29:913–919. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rice TW, Gress DM, Patil DT, Hofstetter
WL, Kelsen DP and Blackstone EH: Cancer of the esophagus and
esophagogastric junction-major changes in the American joint
committee on cancer eighth edition cancer staging manual. CA Cancer
J Clin. 67:304–317. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lan T, Lu Y, Xiao Z, Xu H, He J, Hu Z and
Mao W: A six-microRNA signature can better predict overall survival
of patients with esophagus adenocarcinoma. PeerJ. 7:e73532019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yuan LY, Qin X, Li L, Zhou J, Zhou M, Li
X, Xu Y, Wang XJ and Xing H: The transcriptome profiles and
methylation status revealed the potential cancer-related lncRNAs in
patients with cervical cancer. J Cell Physiol. 234:9756–9763. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huarte M: The emerging role of lncRNAs in
cancer. Nat Med. 21:1253–1261. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang YZ, Liu YR, Xu XE, Jin X, Hu X, Yu
KD and Shao ZM: Transcriptome Analysis of triple-negative breast
cancer reveals an integrated mRNA-lncRNA signature with predictive
and prognostic value. Cancer Res. 76:2105–2114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi YM, Li YY, Lin JY, Zheng L, Zhu YM and
Huang J: The discovery of a novel eight-mRNA-lncRNA signature
predicting survival of hepatocellular carcinoma patients. J Cell
Biochem. Nov. 28–2018.(Epub ahead of print).
|
|
12
|
Cancer Genome Atlas Research Network;
Analysis Working Group: Asan University; BC Cancer Agency; Brigham
and Women's Hospital; Broad Institute; Brown University; Case
Western Reserve University; Dana-Farber Cancer Institute; Duke
University et al, . Integrated genomic characterization of
oesophageal carcinoma. Nature. 541:169–175. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gu J, Zhang X, Miao R, Ma X, Xiang X, Fu
Y, Liu C, Niu W and Qu K: A three-long non-coding
RNA-expression-based risk score system can better predict both
overall and recurrence-free survival in patients with small
hepatocellular carcinoma. Aging (Albany NY). 10:1627–1639. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu JH, Zuo ZX, Wang W, Zhao Q, Qiu MZ, Luo
HY, Chen ZH, Mo HY, Wang F, Yang DD, et al: A two-microRNA-based
signature predicts first-line chemotherapy outcomes in advanced
colorectal cancer patients. Cell Death Discov. 4:1162018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao J, Kwan PW and Shi D: Sparse kernel
learning with LASSO and Bayesian inference algorithm. Neural Netw.
23:257–264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Allison KH and Sledge GW: Heterogeneity
and cancer. Oncology (Williston Park). 28:772–778. 2014.PubMed/NCBI
|
|
17
|
Peters CJ, Rees JR, Hardwick RH, Hardwick
JS, Vowler SL, Ong CA, Zhang C, Save V, O'Donovan M, Rassl D, et
al: A 4-gene signature predicts survival of patients with resected
adenocarcinoma of the esophagus, junction, and gastric cardia.
Gastroenterology. 139:1995–2004 e15. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Meng J, Zhang J, Xiu Y, Jin Y, Xiang J,
Nie Y, Fu S and Zhao K: Prognostic value of an immunohistochemical
signature in patients with esophageal squamous cell carcinoma
undergoing radical esophagectomy. Mol Oncol. 12:196–207. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Chen Z, Tian L, Zhou C, He MY, Gao
Y, Wang S, Zhou F, Shi S, Feng X, et al: LncRNA profile study
reveals a three-lncRNA signature associated with the survival of
patients with oesophageal squamous cell carcinoma. Gut.
63:1700–1710. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fan Q and Liu B: Identification of a
RNA-Seq based 8-long non-coding RNA signature predicting survival
in esophageal cancer. Med Sci Monit. 22:5163–5172. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang SC: PCNA: A silent housekeeper or a
potential therapeutic target? Trends Pharmacol Sci. 35:178–186.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou H, Huang T, Xiong Y, Peng L, Wang R
and Zhang GJ: The prognostic value of proliferating cell nuclear
antigen expression in colorectal cancer: A meta-analysis. Medicine
(Baltimore). 97:e137522018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu L, Li HL, Li WF, Chen JM, Yang JT, Gu
JJ and Xin L: Clinical significance of expression of proliferating
cell nuclear antigen and E-cadherin in gastric carcinoma. World J
Gastroenterol. 23:3721–3729. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stenner M, Demgensky A, Molls C, Hardt A,
Luers JC, Grosheva M, Huebbers CU and Klussmann JP: Prognostic
value of proliferating cell nuclear antigen in parotid gland
cancer. Eur Arch Otorhinolaryngol. 269:1225–1232. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kinugasa S, Tachibana M, Hishikawa Y, Abe
S, Yoshimura H, Monden N, Dhar DK and Nagasue N: Prognostic
significance of proliferating cell nuclear antigen (PCNA) in
squamous cell carcinoma of the esophagus. Jpn J Clin Oncol.
26:405–410. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kimos MC, Wang S, Borkowski A, Yang GY,
Yang CS, Perry K, Olaru A, Deacu E, Sterian A, Cottrell J, et al:
Esophagin and proliferating cell nuclear antigen (PCNA) are
biomarkers of human esophageal neoplastic progression. Int J
Cancer. 111:415–417. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Al-Ghamdi S, Albasri A, Cachat J, Ibrahem
S, Muhammad BA, Jackson D, Nateri AS, Kindle KB and Ilyas M: Cten
is targeted by kras signalling to regulate cell motility in the
colon and pancreas. PLoS One. 6:e209192011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chan LK, Chiu YT, Sze KM and Ng IO:
Tensin4 is up-regulated by EGF-induced ERK1/2 activity and promotes
cell proliferation and migration in hepatocellular carcinoma.
Oncotarget. 6:20964–20976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Al-Ghamdi S, Cachat J, Albasri A, Ahmed M,
Jackson D, Zaitoun A, Guppy N, Otto WR, Alison MR, Kindle KB and
Ilyas M: C-terminal tensin-like gene functions as an oncogene and
promotes cell motility in pancreatic cancer. Pancreas. 42:135–140.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liao YC, Chen NT, Shih YP, Dong Y and Lo
SH: Up-regulation of C-terminal tensin-like molecule promotes the
tumorigenicity of colon cancer through beta-catenin. Cancer Res.
69:4563–4566. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Martuszewska D, Ljungberg B, Johansson M,
Landberg G, Oslakovic C, Dahlbäck B and Hafizi S: Tensin3 is a
negative regulator of cell migration and all four tensin family
members are downregulated in human kidney cancer. PLoS One.
4:e43502009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu WM and Liao YC: Downregulation of
C-terminal tensin-like protein (CTEN) suppresses prostate cell
proliferation and contributes to acinar morphogenesis. Int J Mol
Sci. 19:E31902018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nie F, Yu X, Huang M, Wang Y, Xie M, Ma H,
Wang Z, De W and Sun M: Long noncoding RNA ZFAS1 promotes gastric
cancer cells proliferation by epigenetically repressing KLF2 and
NKD2 expression. Oncotarget. 8:38227–38238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang W and Xing C: Upregulation of long
noncoding RNA ZFAS1 predicts poor prognosis and prompts invasion
and metastasis in colorectal cancer. Pathol Res Pract. 212:690–695.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li T, Xie J, Shen C, Cheng D, Shi Y, Wu Z,
Deng X, Chen H, Shen B, Peng C, et al: Amplification of long
noncoding RNA ZFAS1 promotes metastasis in hepatocellular
carcinoma. Cancer Res. 75:3181–3191. 2015. View Article : Google Scholar : PubMed/NCBI
|