Introduction
Breast cancer (BC) is the most frequent type of
cancer in female patients, with high morbidity and mortality rates
worldwide. It has been estimated that there will be 27,1270 new
cases of BC and 42,260 deaths associated with BC in the USA in 2019
(1). Previous epidemiological
studies have reported that obesity (2), utilization of estrogen and progestin
(3), advanced maternal age at first
birth (4) and alcohol abuse
(5) increase the risk of developing
BC. In addition, genetic mutations and epigenetic modifications are
regarded as potential causes of tumorigenesis in BC (6,7). In the
past decades, there has been considerable progress in preventing,
diagnosing and treating BC. However, the prognosis for BC remains
poor. In consequence, understanding the molecular and genetic
mechanisms associated with the pathogenesis of BC is essential.
Circular RNAs (circRNAs), a new type of 3′-5′
head-to-tail covalently closed non-coding RNA, were identified in
1976 (8,9). However, in subsequent decades, circRNAs
were considered to be the product of incorrect splicing (10). Recently, it was recognized that
circRNAs are normal co-products of numerous eukaryotic
protein-coding genes. Furthermore, circRNAs have extensive
functions, including: i) Sponging microRNAs (miRNAs/miRs), thus
inhibiting the expression of target genes (11); ii) binding to proteins in order to
form a complex and consequently serving a role in transcription
(12); iii) post-transcriptional
regulation via a variety of mechanisms (13); and iv) translational functions
involving the modification of the internal ribosome entry site or
N6-methyladenosine (14,15). However, the majority of functions and
mechanisms of circRNAs remain unknown, which suggests that circRNAs
may be a promising avenue to explore in medical research (16).
miRNAs are a type of non-coding RNAs with a length
of ~22 nucleotides (17). miRNAs
perform a repressive function on the expression of their target
genes through miRNA response elements (MREs), which exist in their
target RNA transcripts (18).
Previous studies have demonstrated that circRNAs sponge miRNAs via
MREs in numerous diseases. This mechanism has also been reported in
cancer initiation and progression (19). Accumulating evidence has demonstrated
that circRNAs regulate the growth, development and metastasis of BC
by acting as miRNA sponges. For instance, ciRS-7 sponges miR-7 and
inhibits the expression of its target gene in numerous tumors
(20). In addition, circHIPK3 may
function as a miR-124 sponge and inhibit its antineoplastic
function, thus inducing the proliferation of BC cells (21). However, further studies are required
to explore the potential mechanisms of tumorigenesis, which may aid
in the diagnosis and treatment of BC.
The application of microarray technology has enabled
the extensive study of gene expression and has facilitated the
research of disease susceptibility in favor of treating diseases at
the molecular level. Several studies have indicated that microarray
technology and in silico analysis have broad application in
identifying pathogenetic mechanisms and novel diagnostic and
therapeutic targets (22,23).
The present study identified novel circRNAs and
revealed their underlying mechanisms in BC via the combined
application of in silico analysis and microarray technology.
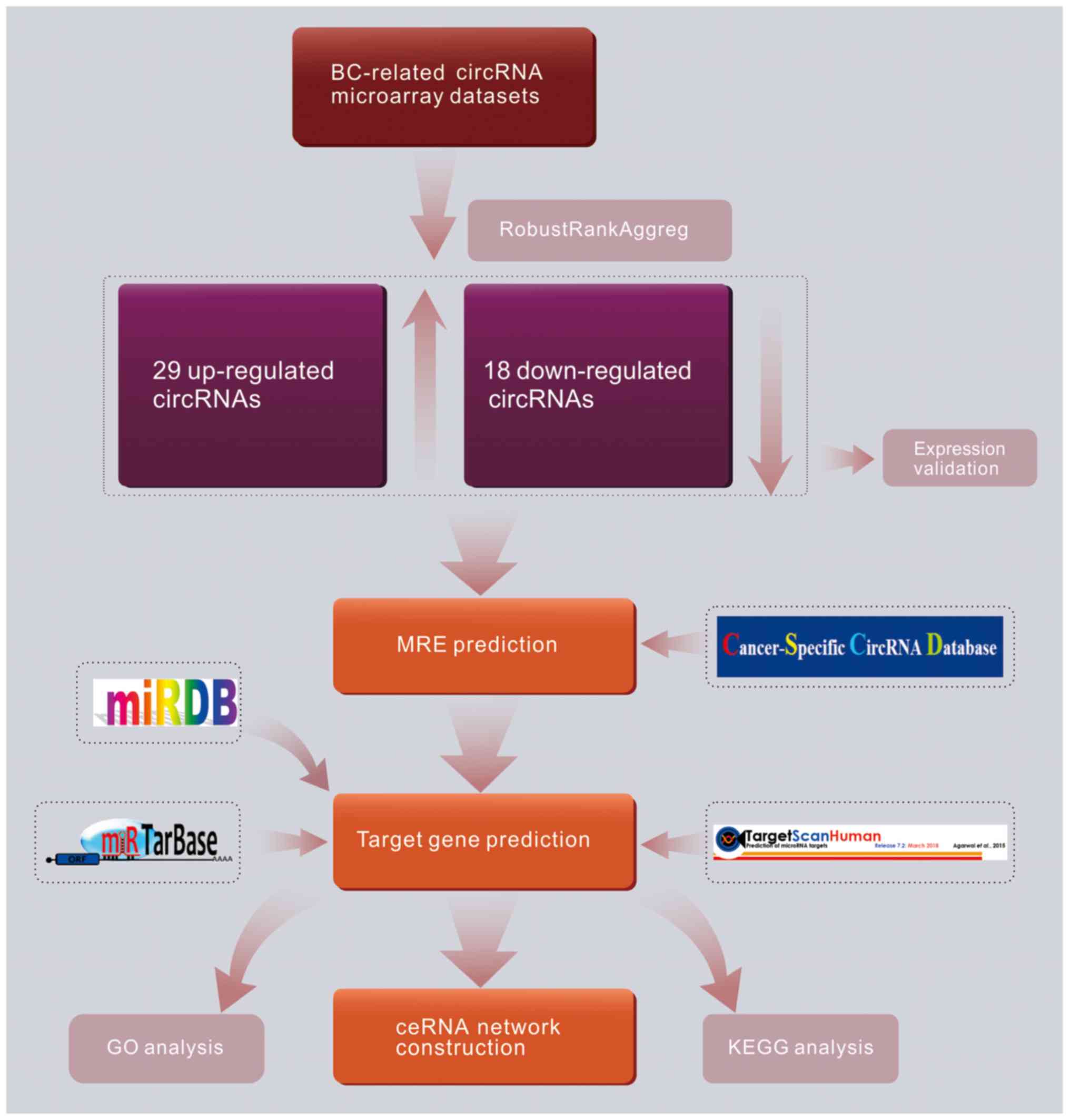
A diagram representing the overall study design is presented in
Fig. 1. First, BC-associated
microarray datasets from the Gene Expression Omnibus (GEO) database
were screened, the bioinformatic data were analyzed and
differentially expressed circRNAs (DECs) were obtained. Next, the
potential miRNAs sponged by DECs were identified through the
cancer-specific circRNA database (CSCD) database. Moreover, a
bioinformatics prediction of target mRNAs was performed, and a
circRNA-miRNA-mRNA competing endoegenous network was constructed.
Gene enrichment analyses of the candidate mRNAs were performed with
the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes
(KEGG) databases, which resulted in the prediction of the signaling
pathways involved in BC. The identification of DECs and potential
mechanisms reported in the present study may facilitate future
research in BC treatment and diagnosis.
Materials and methods
Screening of DECs in BC from the GEO
database
The microarray datasets analyzed in the present
study, including the circRNA expression profile in BC, were
downloaded from the GEO database [National Center for Biotechnology
Information (NCBI); http://www.ncbi.nlm.nih.gov/geo/] (24). The GSE101124 dataset was derived from
the GEO database, and contained gene expression data from 15
samples, including 4 breast cancer cell lines, 8 cancerous breast
tissues and 3 healthy mammary gland tissue samples (25). The GPL19978 platform (Agilent-069978
Arraystar Human circRNA microarray V1; Agilent Technologies, Inc.)
was used to identify the GSE101124 dataset. The raw data were
pretreated and homogenized, followed by detection, screening and
annotation of circRNAs. The Limma package of R/Bioconductor
software (version 3.9) (26) was
used for the analysis of raw microarray data and the identification
of DECs in the dataset. The following filtering criteria were
applied: |logFC|>2 and adjusted P<0.05.
Prediction of miRNAs and competing
endogenous (ceRNA) network construction
The CSCD database (http://gb.whu.edu.cn/CSCD) can calculate predictions
of MREs, RNA-binding proteins (RBPs) and open reading frames (ORFs)
(27). miRNAs sponged to circRNAs
were identified from the GSE101124 dataset. Furthermore, potential
ORFs in DECs and RBPs combined with DECs were examined. A ceRNA
network was constructed to predict the association between miRNAs
and the identified DECs or mRNAs. Using the sequences and
annotations of miRNAs obtained from miRBase (http://www.mirbase.org/), the mRNA-miRNA interactions
were predicted by miRTarbase (28),
TargetScan (29) and miRDB (30). Target genes were selected from the
overlapping genes for the construction of the ceRNA network.
The ceRNA network was constructed as a graphical
representation by Cytoscape software (version 3.6.1) (31) with the miRNA target genes and
DECs.
Functional term and signaling pathway
enrichment analyses
GO (http://www.geneontology.org) annotates genes to
biological process (BP), molecular function (MF) and cellular
component (CC) (32). The
identification and annotation of homologous genes and protein
sequences in various organisms helps to elucidate the specific
roles of certain genes.
The KEGG (http://www.genome.jp/kegg/) database consists of 16
main databases and is comprised of disease information organized in
computable forms (33). Among these
databases, the KEGG pathway database provides molecular networks
for molecular systems by annotating genes to pathways (33). The R (version 3.9) package
(http://www.bioconductor.org/)
clusterProfiler (34) allows gene
classification and calculation of enrichment for GO terms and KEGG
pathways. In the present study, GO annotation and KEGG pathway
analyses were conducted with clusterProfiler to explore the
potential biological roles of target genes. The analysis results
were visualized with the ggplot2 package of the R software.
Adjusted P<0.05 and q<0.05 were considered to indicate
statistically significant enriched function annotations.
Patient samples
For the validation of DECs in clinical samples, RNA
was extracted from 20 BC tissue specimens and their paired adjacent
normal tissue specimens to perform reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis. These samples were collected at The Affiliated Hospital
of Qingdao University between April 2018 and December 2018.
Inclusion criteria of the patients were as follows: i) Age ≥18
years; ii) BC was confirmed via pathological examination; iii)
routine blood count, liver and kidney function and
electrocardiogram examination met the treatment requirements before
operation; and iv) no serious complications prior to surgery. The
exclusion criteria were: i) No pathological diagnosis and unknown
TNM stage; ii) received chemotherapy or radiotherapy prior to
surgery; iii) presence of other tumors; and iv) poor compliance of
patients. Tissue specimens located ≥5 cm from the tumor margin were
obtained via excision surgery. Patients were all female. The median
age of patients was 45 years (range, 30–56). Immediately after
obtaining the specimen, tissues were frozen in liquid nitrogen and
stored at −80°C for preservation. Ethical approval was obtained
from the Ethics Committee of The Affiliated Hospital of Qingdao
University. The study participants approved the use of clinical
samples by providing written informed consent.
Cell culture
BC cell lines (MCF-7 and MDA-MB-231) and a human
normal mammary epithelial cell line (MCF-10A) were obtained from
the American Type Culture Collection. MCF-10A cells were used as
the control. BC cell lines were cultured in Dulbecco's modified
Eagle's medium (DMEM) with 10% FBS (both Gibco; Thermo Fisher
Scientific, Inc.). MCF-10A cells were cultured in Roswell Park
Memorial Institute (RPMI)-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS. All cell lines were
used within 6 months and cultured in a 5% CO2 incubator
at 37°C. RNA was extracted when the cell confluence reached ~80%.
All experiments were repeated in triplicate.
RT-qPCR
TRIzol reagent (Takara Bio, Inc.) was used for the
extraction of total RNA from tissues or cells following the
manufacturer's protocols. After treatment with DNAse I (Takara Bio,
Inc.), reverse transcription of 1 µg total RNA was conducted with
reverse transcriptase (RT reagent Kit; cat. no. RR037A, Takara Bio,
Inc.).
qPCR was subsequently performed using SYBR Premix Ex
Taq (Takara Bio, Inc.) on a CFX96 Real-Time PCR Detection system
(Bio-Rad Laboratories, Inc.). The conditions of the amplification
reaction procedure were set as follows: Pre-denaturation at 95°C
for 30 sec; 95°C for 10 sec; and 60°C for 40 sec for a total of 40
cycles. The expression of circRNAs was normalized to that of
internal control GAPDH, using the 2−ΔΔCq method
(35). The following primer pairs
were used for qPCR: GAPDH forward, 5′-CGCTCTCTGCTCCTCCTGTTC-3′ and
reverse, 5′-ATCCGTTGACTCCGACCTTCAC-3′; Homo sapiens
(hsa)_circ_0000520 forward, 5′-AGACTAGGGCCAGAGGCG-3′ and reverse,
5′-GAGCTTCCCTCCCCGAAG-3′; hsa_circ_0006220 forward,
5′-ATTCCATTTCACTGCAGGATGTAGC-3′ and reverse,
5′-ACATCCTGCAGTGAAATG-3′; hsa_circ_0000977 forward,
5′-ATGTGGAATAAGAACTCC-3′ and reverse,
5′-AACCTATAAACTTCAGAATGGAATG-3′; and hsa_circ_0043278 forward,
5′-GCATTTCATCAATAACCCTC-3′ and reverse, 5′-TAGTGAAATGGAATGGCTGT-3′.
The expression of DECs was normalized to that of GAPDH.
Validation of circRNAs
The expression levels of DECs were verified in
clinical samples and cells by RT-qPCR. In addition, to demonstrate
the circularized junction of DECs, divergent (circular) primers
were designed for the spliced junction of circRNAs and PCR was
performed as previously described (36). The cDNA of MCF-7 cells was reverse
transcribed (RT reagent Kit, Code No. RR037A, Takara Bio, Inc.), as
above. The thermocycling conditions were as follows: Initial
denaturation at 95°C for 5 min, and 38 cycles at 95°C for 30 sec,
61°C (annealing temperature) for 30 sec and 72°C for 30 sec,
followed by a 7-min extension at 72°C (36). Convergent (linear) primers were used
as the control. The spliced junction in the PCR product of DECs was
then confirmed by Sanger sequencing (37). Combined with agarose gel
electrophoresis, these results validated the existence of circRNAs.
The primer sequences used in the present study were synthesized by
the Beijing Genomics Institute and are listed in Table I.
 | Table I.Divergent and convergent primers used
for PCR. |
Table I.
Divergent and convergent primers used
for PCR.
| circRNA ID | Primer type | Sequence
(5′-3′) |
|---|
|
hsa_circ_0000520 | Div | F:
GGTCTGAGACTAGGGCCAGAGG |
|
|
| R:
GGAGTGGAGTGACAGGACGCAC |
|
| Con | F:
ACGAGCTGAGTGCGTCCTGT |
|
|
| R:
AAGCTCAGGGAGAGCCCTGT |
|
hsa_circ_0006220 | Div | F:
CTACCCTGCTGAACCTGAAACA |
|
|
| R:
TCACACTCCTCCTTGGTCTTGG |
|
| Con | F:
GCAGGATGTAGCCAATCAAATGT |
|
|
| R:
TTTTTGCTTCCTCTGCTTGTTTC |
|
hsa_circ_0000977 | Div | F:
TTTACTTCCTTGGAGCCAGAGC |
|
|
| R:
CAAACATTATTCTCCGCAGCAT |
|
| Con | F:
GGAACAACCACAGGGCAGGT |
|
|
| R:
ATGCTCTGGCTCCAAGGAAGTAA |
|
hsa_circ_0043278 | Div | F:
GAAACAAGCAGAGGAAGCAAAA |
|
|
| R:
CATTTGATTGGCTACATCCTGC |
|
| Con | F:
GCAGGATGTAGCCAATCAAATGT |
|
|
| R:
TTTTTGCTTCCTCTGCTTGTTTC |
Statistical analysis
Data are presented as the mean ± standard error of
the mean of ≥3 independent experiments. For the comparison of two
groups, a two-tailed paired Student's t-test was performed. One-way
analysis of variance (ANOVA) was used to analyze more than two
groups. Dunnett's test was performed as the post hoc test.
P<0.05 was considered to indicate a statistically significant
difference. All statistical analyses were performed with GraphPad
Prism software (version 6; GraphPad Software, Inc.).
Results
Identification of DECs in BC
GEO is an international public database containing
high-throughput functional genomic data (24). GEO may be used for searching,
analyzing and visualizing data. The present study screened the
microarray datasets of BC from the GEO database in NCBI. The
retrieval terms used in GEO were as follows: (‘breast’ OR ‘mammary’
OR ‘HBL’) and (‘tumor’ OR ‘cancer’ OR ‘carcinoma’ OR ‘neoplasm’ OR
‘malignant’). ‘Homosapiens’ in the ‘Top Organisms’ and ‘Non-coding
RNA profiling by array’ were selected in the ‘Study type’.
circRNA microarray datasets were selected as the
subject of analysis. Data consisting only microarray samples of
tissues or cells were excluded. As a result, the final search
identified GSE101124, which included information from 15 microarray
samples of BC cells, and tissues and paired adjacent normal
tissues, and was consequently selected for further analaysis.
Subsequently, the CSCD was used to identify the DECs in the
GSE101124 microarray dataset. Limma, an R/Bioconductor software
package, offers an integrated solution for processing large
datasets with advanced computational algorithms (26). Pretreatment and homogenization of the
raw data was performed with the Llimma package of R software. After
filtering the data based on the inclusion criteria (|logFC|>2.0
and adjusted P<0.05), 47 circRNAs were revealed to be
differentially expressed between BC and paired adjacent normal
tissues. As demonstrated in Table
SI, 29 upregulated and 18 downregulated circRNAs were screened
out in the GSE101124 dataset. The circRNAs that ranked in the top
four according to the |logFC| were hsa_circ_0000520,
hsa_circ_0006220, hsa_circ_0000977 and hsa_circ_0043278. The
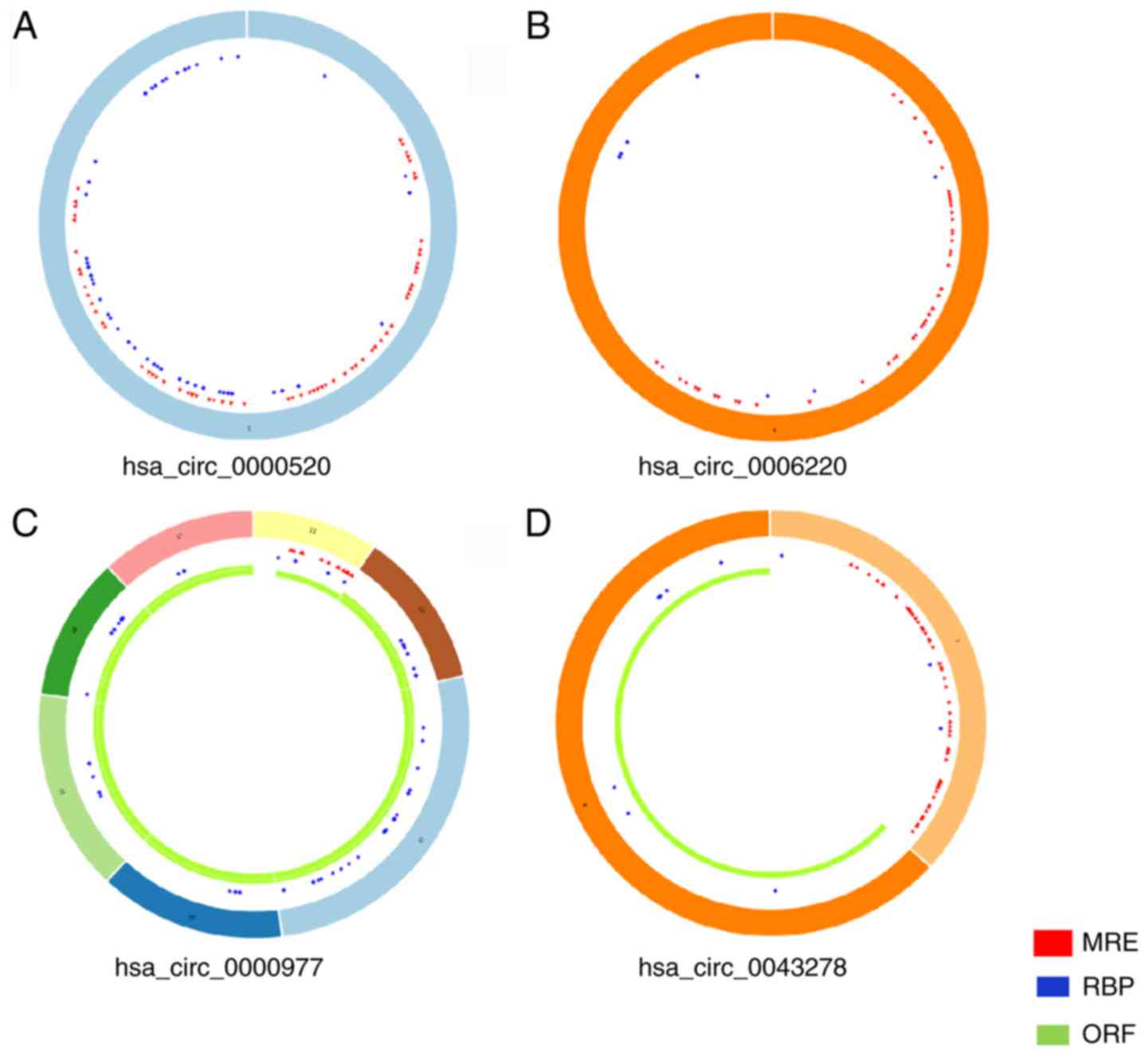
essential characteristics and basic structural patterns of these
four circRNAs are presented in Table
II and Fig. 2, respectively.
Furthermore, a Circos plot (http://circos.ca/) demonstrating DECs in human
chromosomes was generated to exhibit the expression of DECs
(Fig. 3). The results revealed that
the DECs were extensively distributed in all chromosomes, including
chromosome X.
| Table II.Essential characteristics of
hsa_circ_0000520, hsa_circ_0006220, hsa_circ_0000977 and
hsa_circ_0043278. |
Table II.
Essential characteristics of
hsa_circ_0000520, hsa_circ_0006220, hsa_circ_0000977 and
hsa_circ_0043278.
| circRNA ID | circRNA type | Genomic length,
bp | Spliced length,
bp | Best
transcript | Gene symbol | Type of
regulation |
|---|
|
hsa_circ_0043278 | Exonic | 2,925 | 250 | NM_001488 | TADA2A | Downregulation |
|
hsa_circ_0000977 | Exonic | 24,404 | 562 | NM_024894 | NOL10 | Downregulation |
|
hsa_circ_0006220 | Exonic | 158 | 158 | NM_001488 | TADA2A | Downregulation |
|
hsa_circ_0000520 | Exonic | 123 | 123 | NR_002312 | RPPH1 | Upregulation |
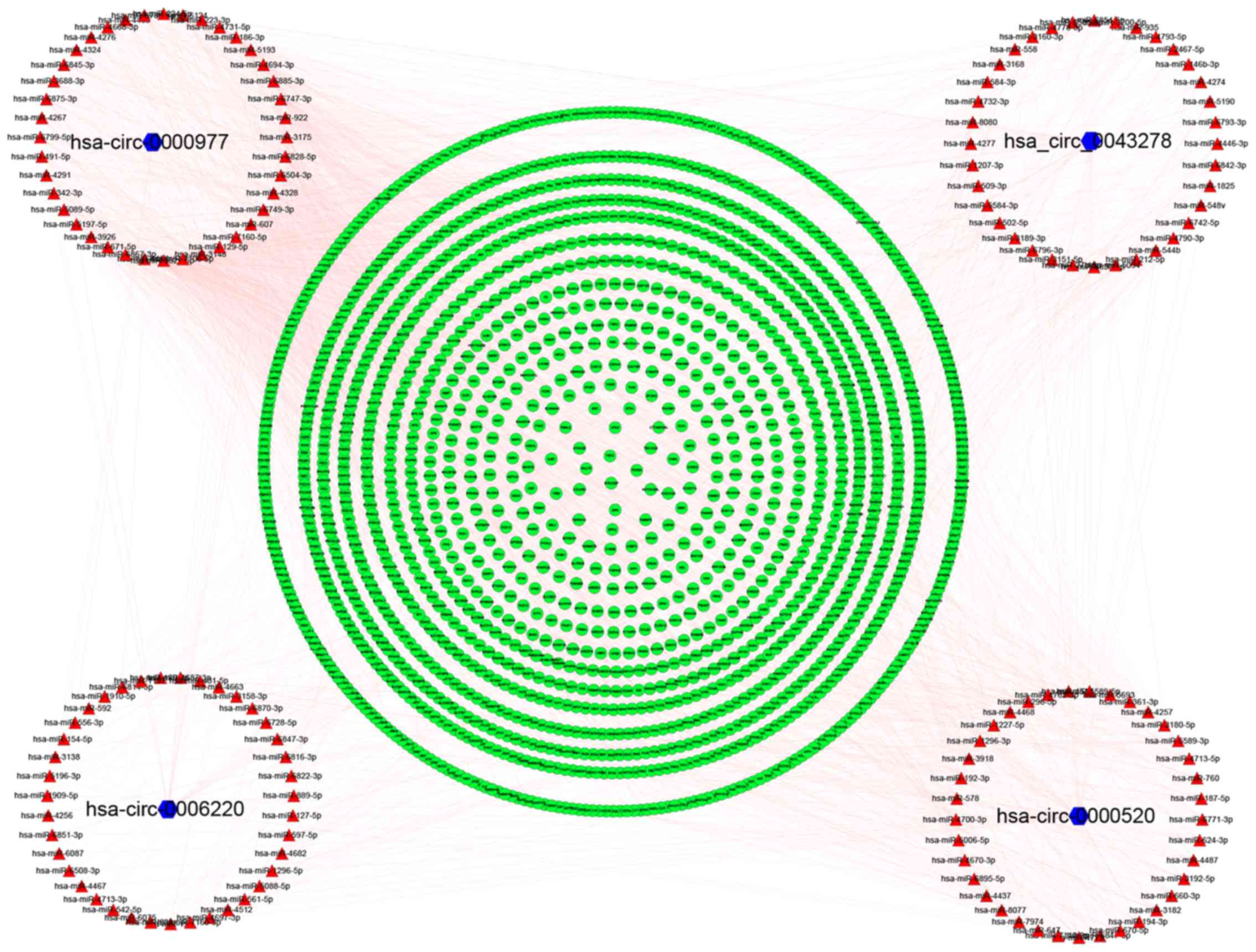
Prediction of a circRNA-miRNA-mRNA
network
RNA transcripts with the same MREs can competitively
sponge miRNAs and subsequently inhibit their downstream functions
(38). circRNAs are able to
participate in miRNA-mediated post-transcriptional regulation by
sponging miRNAs (36).
circRNAs are emerging as essential modulators of
ceRNA networks (39). For example,
circGFRA1 may sponge miR-34a to serve its regulatory role in
triple-negative BC (TNBC) (40).
Furthermore, circEPSTI1 may affect the proliferation and apoptosis
of TNBC cells by sponging miR-4753 and miR-6809 (41). The next step in the present study was
to explore whether the identified DECs acted as miRNA sponges.
CSCD supports the prediction of MREs, RBPs and ORFs
for circRNAs (27). The predictions
are integrated from TargetScan (29), starBase (http://starbase.sysu.edu.cn/index.php) and ORFfinder
(https://indra.mullins.microbiol.washington.edu/sms2/orf_find.html)
making CSCD the first comprehensive database specific for
cancer-associated circRNAs. This database serves a vital role in
the research of the mechanism of action of these molecules. The
present study predicted the MREs, RBPs and ORFs of DECs using CSCD
(Tables SII–SIV). Moreover, the target genes of miRNAs
were predicted and a ceRNA network was constructed to describe the
overall functions of DECs. The predicted target genes are
demonstrated in Tables SV–SVIII. The specific mRNAs that bound the
target miRNAs were identified using miRTarBase, TargetScan and
miRDB. The regulatory network was constructed by Cytoscape and
consists of DECs, miRNAs and mRNAs (Fig.
4).
GO annotation and KEGG pathway
analysis
In cases where the functional role of the
aforementioned DECs has not yet been determined, the roles of mRNAs
have been explored to understand the circRNAs. In particular, GO
functional term and KEGG pathway enrichment analyses of
significantly differentially expressed mRNAs facilitate the
understanding of circRNAs (42).
The clusterProfiler software of the R package was
used in the present study in order to perform the GO analysis of
target genes of the identified DECs. The results for the four
aforementioned circRNAs are presented in Fig. 5. For hsa_circ_0000520, the most
enriched GO term for target genes in BP was ‘transcriptional
activator activity, RNA polymerase II transcription regulatory
region sequence-specific DNA binding’ (Fig. 5A and Table SIX). For hsa_circ_0006220, the most
enriched GO term in BP for target genes was ‘proximal promoter
sequence-specific DNA binding’ (Fig.
5B and Table SX). For
hsa_circ_0000977 and hsa_circ_0043278, the most enriched GO term
for target genes in BP was ‘transcription factor activity, RNA
polymerase II proximal promoter sequence-specific DNA binding’
(Fig. 5C and D; Tables SXI and SXII). In summary, GO enrichment analysis
revealed the potential regulatory role of circRNAs in
transcription. Recently, Ding et al (43) verified that circ-DONSON is capable of
activating the transcription of SOX4, a transcription promoter, to
promote gastric cancer progression. Thus, the results of the
present study suggest the potential use of circRNAs as therapeutic
biomarkers for patients with BC.
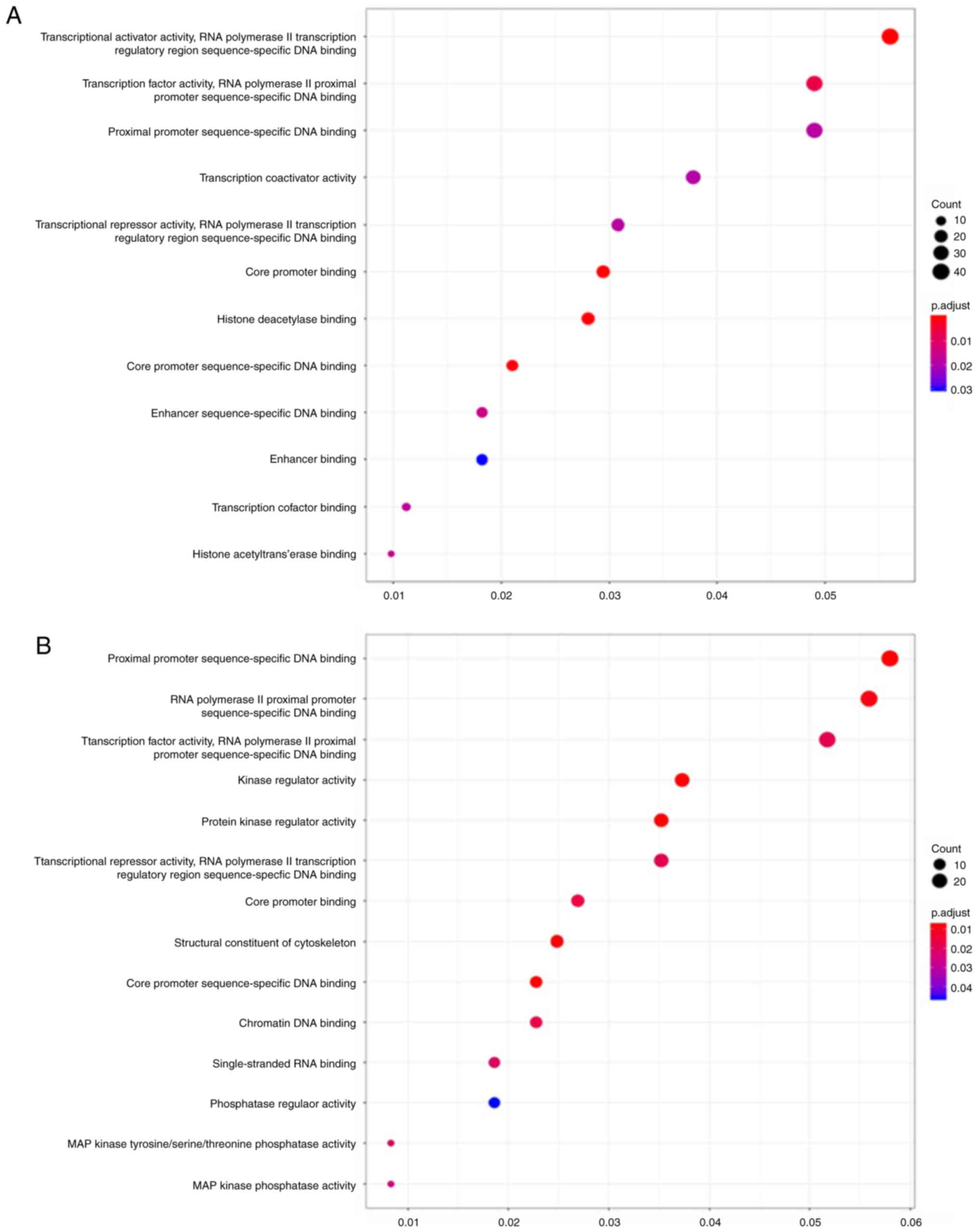 | Figure 5.GO enrichment annotations for the
target mRNAs of the competing endogenous RNA network. GO analysis
of (A) hsa_circ_0000520, (B) hsa_circ_0006220 on the basis of the
regulatory network. GO analysis was performed with the
clusterProfiler package of the R software and visualized with the
ggplot2 package of the R software (version 3.9). Adjusted P<0.05
was considered to indicate significantly enriched GO annotations.
p.adjust, adjusted P-value; MAP, mitogen-activated protein; UTR,
untranslated region; GO, Gene Ontology; circ, circular; hsa,
Homo sapiens. GO enrichment annotations for the target mRNAs
of the competing endogenous RNA network. GO analysis of (C)
hsa_circ_0000977 and (D) hsa_circ_0043278 on the basis of the
regulatory network. GO analysis was performed with the
clusterProfiler package of the R software and visualized with the
ggplot2 package of the R software (version 3.9). Adjusted P<0.05
was considered to indicate significantly enriched GO annotations.
p.adjust, adjusted P-value; MAP, mitogen-activated protein; UTR,
untranslated region; GO, Gene Ontology; circ, circular; hsa,
Homo sapiens. |
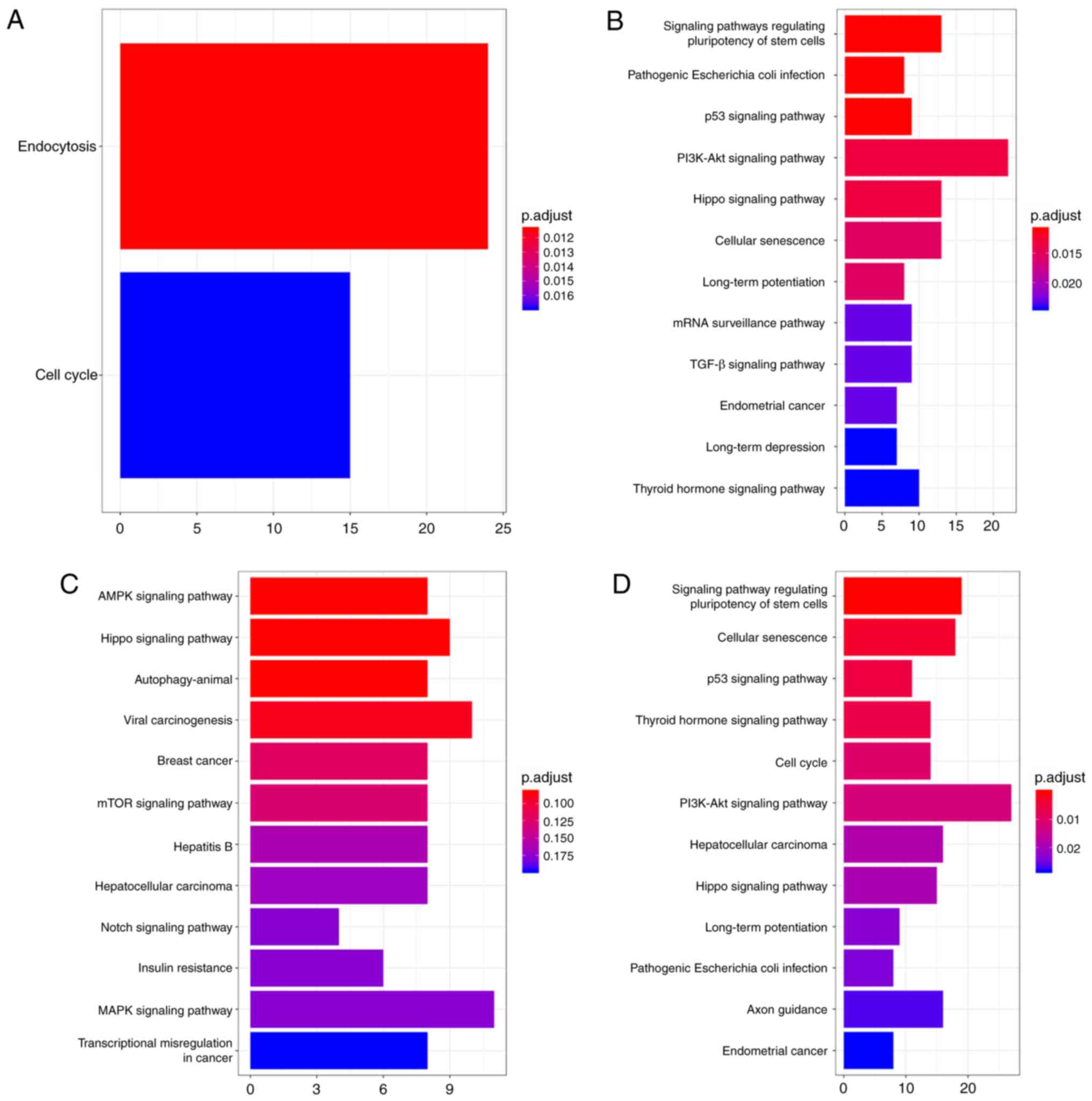
The present study also performed KEGG pathway
analysis (Fig. 6). For
hsa_circ_0000520, the main enriched pathways for target genes were
‘endocytosis’ and the ‘cell cycle’ (Fig.
6A). For hsa_circ_0006220, the most enriched pathway for target
genes was the ‘PI3K-AKT signaling pathway’ (Fig. 6B). For target genes of
hsa_circ_0000977, the ‘MAPK signaling pathway’ was the most
enriched pathway (Fig. 6C). For
target genes of hsa_circ_0043278, KEGG pathway analysis
demonstrated that the main association was with the ‘PI3K-AKT
signaling pathway’ and the ‘signaling pathway regulating
pluripotency of stem cells’ (Fig.
6D). These results suggested that these DECs may be involved in
tumor development.
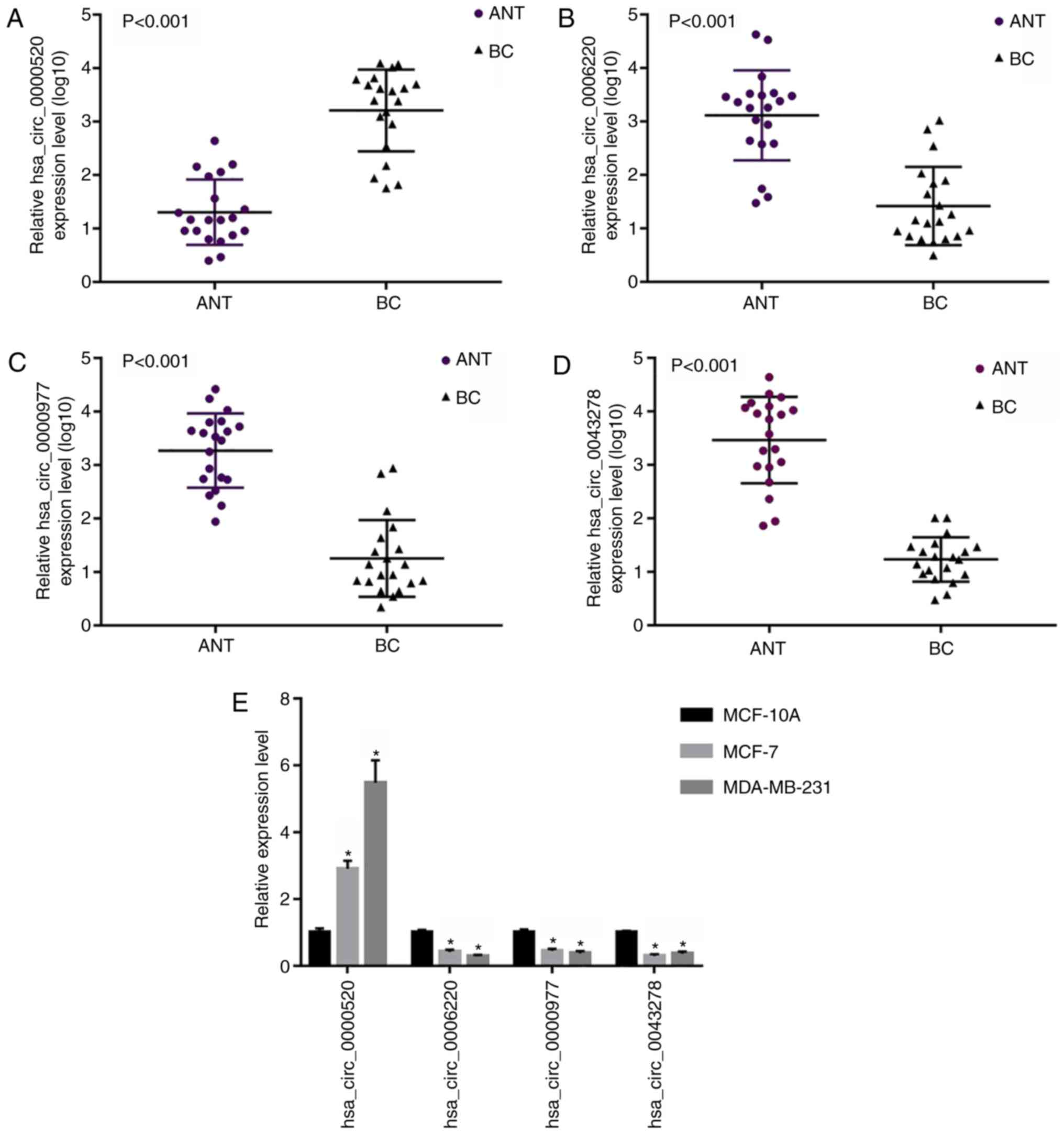
Validation of candidate circRNAs
The circRNAs (hsa_circ_0000520, hsa_circ_0006220,
hsa_circ_0000977 and hsa_circ_0043278) that ranked in the top four
according to |logFC| (>2.0) were selected for validation
experiments. First, the expression levels of these four DECs were
evaluated in 20 BC tissue specimens and their paired adjacent
normal tissue specimens. Subsequently, the expression levels of
these four DECs were also examined in BC cells (MCF-7 and
MDA-MB-231) and a human normal mammary epithelial cell line
(MCF-10A). The relative expression of hsa_circ_0000520 was
significantly higher in BC than that in the adjacent normal tissues
and MCF-10A cells (Fig. 7A and E).
The relative expression levels of hsa_circ_0006220,
hsa_circ_0000977 and hsa_circ_0043278 were significantly lower in
BC than in the adjacent normal tissues and MCF-10A cells (Fig. 7B-E). To validate whether these
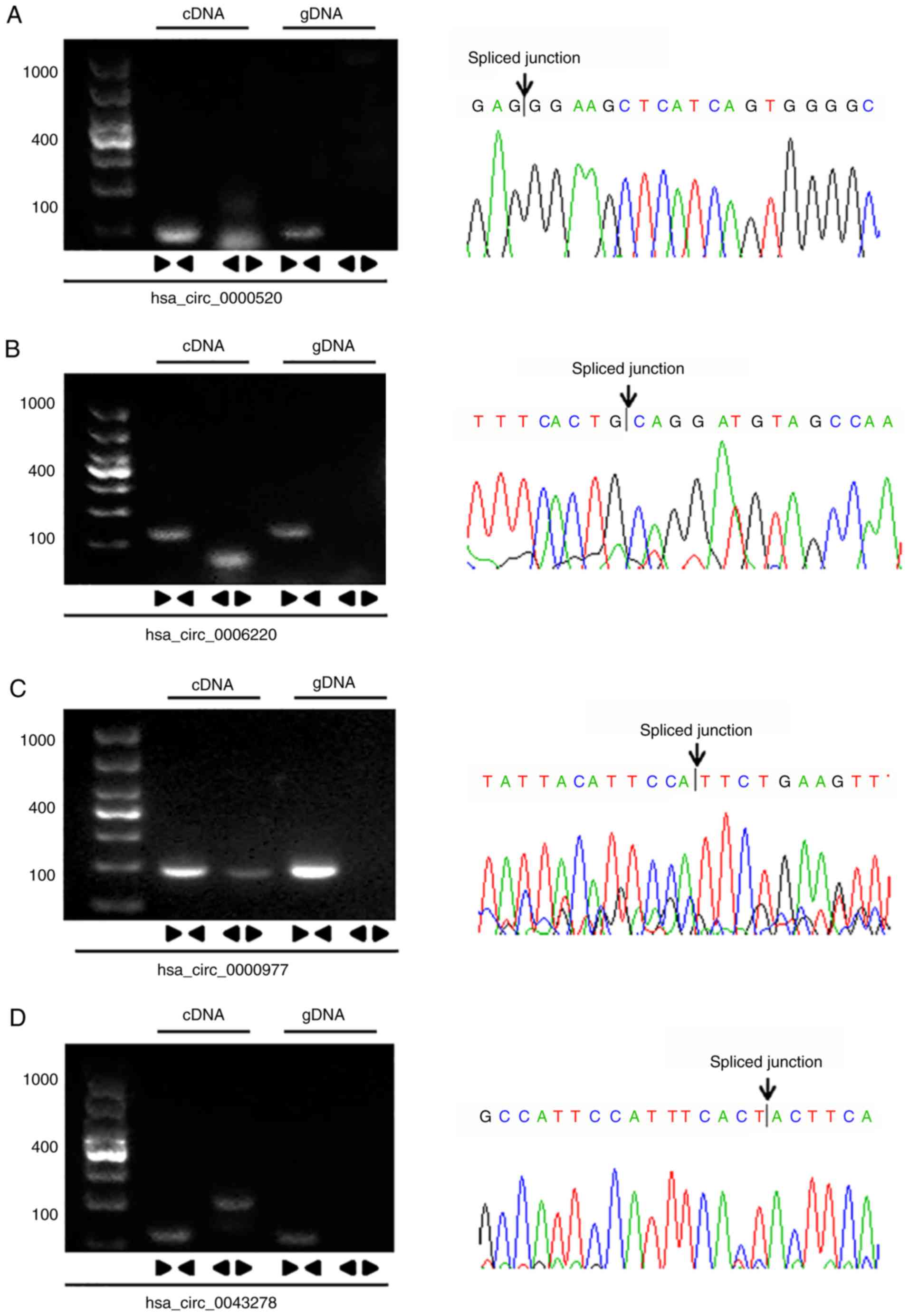
circRNAs were actually circular transcripts, PCR was performed
using divergent and convergent primers specific for spliced
junctions of circRNAs. The cDNA and genomic DNA (gDNA) extracted
from MCF-7 cells were used as templates. The results revealed that
these circRNAs were amplified with the divergent primers on cDNA
instead of gDNA. Furthermore, the sequences of the spliced
junctions were confirmed by Sanger sequencing (Fig. 8A-D).
Discussion
As the most common type of cancer among females, BC
has high morbidity and mortality rates, and its incidence is
gradually increasing worldwide. A previous study in the USA
estimated 27,1270 new cases and 42,260 deaths caused by BC in 2019
(1). The main reasons for morbidity
and mortality in patients with BC include cancer cell invasion,
migration and metastasis. The mechanisms of tumorigenesis remain
unclear due to the complex genetic mutations and epigenetic
alterations involved in this process.
The development of effective therapies for patients
with BC requires the identification of therapeutic and prognostic
targets. Recent studies have focused on the identification of
differentially expressed non-coding RNAs, including long non-coding
RNAs, miRNAs and circRNAs, by gene chip and high-throughput
sequencing (44–46). It is essential to identify early
clinical diagnostic markers in order to understand the molecular
mechanism underlying, and facilitate the diagnosis of, BC.
As naturally occurring RNAs, circRNAs are a subclass
of non-coding RNAs widely expressed in mammalian cells (47). It was recently demonstrated that
circRNAs may serve a vital role in the progression of various human
cancer types (48–51). For example, circCEP128 promotes
bladder cancer progression by sponging miR-145-5p, which inhibits
the function of SRY-box transcription factor 11 (48). However, their roles in breast
tumorigenesis are not yet well understood. Further research of the
molecular mechanisms of circRNAs may provide insight into the
diagnosis and treatment of BC.
There are two primary methods of circRNA detection:
RNA sequencing (RNA-seq) and high-throughput circRNA microarray.
RNA-seq is a common method for genome-wide analysis of circRNAs,
though limited by the low reads that cover the specific splicing
junction (52). In previous studies,
Lan et al (53) investigated
circRNA expression profiles via high-throughput RNA-seq in
papillary thyroid carcinoma, while Coscujuela et al
(45) used RNA-seq to profile and
characterize circRNAs in luminal-like BC. Nair et al
(44) created a comprehensive
workflow termed Circ-Seq based on existing bioinformatics
approaches to screen expressed circRNAs in BC subtypes.
For the detection of known circRNAs, high-throughput
circRNA microarray assays are able to efficiently target the
reported back-splice sites in the samples of interest. Microarray
data have been largely examined to identify pathogenetic pathways
and therapeutic targets in several diseases, including cancer and
autoimmune, fibrotic, neurodegenerative and infectious diseases
(22,23,54–59).
Presti et al (22) utilized
RNA-seq data of 281 and 283 DNA-sequenced glioblastoma multiforme
and low-grade glioma patients, respectively, to evaluate the
expression levels of migration inhibitory factor (MIF) and related
genes in glioma. After screening for differentially expressed
genes, a correlation analysis was performed. Moreover, microarray
transcriptomic data were used to assess the mean difference in MIF
levels between neoadjuvant and non-neoadjuvant therapy. In uveal
melanoma (UM), Petralia et al (54) characterized the pathophysiological
role of CD47. CD47 expression levels were found to affect the tumor
microenvironment, immune capacity and stroma in UM. Microarray
datasets were used to identify 64 differentially expressed genes in
metastatic UM. These genes, which are associated with the
metastatic properties of UM, may be useful for the verification of
potential chemotherapeutic methods.
In colorectal cancer (CRC), KCNMA1 has been reported
to be downregulated, and assessment of the GSE35834 microarray
dataset has suggested that mir-17-5p may be a target for KCNMA1.
Therefore, KCNMA1 has been indicated as a therapeutic target in the
early stages of CRC (60). For the
study of multiple sclerosis (MS), Fagone et al (23) utilized microarray dataset analysis to
obtain 45 genes that were differentially expressed between low and
high responder CD4+ T cells. The identification of a
specific gene signature for natalizumab responsiveness may provide
a theoretical basis for suitable treatments for patients with MS,
indicating the potential of microarray dataset analysis in
improving the understanding of multiple disease types. Fagone et
al (57) analyzed 254
upregulated genes in activated hepatic stellate cells by screening
microarray datasets in order to investigate liver fibrosis.
Regarding BC, a recent study systematically identified DECs by
circRNA microarray, and identified the
hsa_circ0087378/miR-1260b/secreted frizzled-related protein 1 axis
as a potential regulatory mechanism in estrogen receptor-positive
BC (61).
In the present study, circRNA microarray was
utilized to construct a general expression profile of BC. Specific
circRNAs were analyzed, and a ceRNA network was constructed with
the aim of exploring the potential mechanisms of these circRNAs as
diagnostic biomarkers.
First, the GSE101124 dataset from the GEO database
was screened and 47 DECs were detected using Limma. Among them,
hsa_circ_0000520 was found to be upregulated, whereas
hsa_circ_0006220, hsa_circ_0000977 and hsa_circ_0043278 were found
to be downregulated. hsa_circ_0000520 was spliced from ribonuclease
PRNA component H1 gene, the RNA component of the RNase P
ribonucleoprotein, which is an endoribonuclease that assists in the
formation of the mature 5′ termini of tRNA sequences by cleaving
tRNA precursor molecules (62).
hsa_circ_0006220 and hsa_circ_0043278 were found to be encoded by
transcriptional adaptor 2A (TADA2A), which encodes proteins as a
transcriptional activator adaptor and as part of the PCAF (human
Gcn5 homologue) histone acetylase complex (63). circTADA2A, also known as
hsa_circ_0043278, has been demonstrated to serve a role in
promoting osteosarcoma progression and metastasis by sponging
miR-203a-3p (64). hsa_circ_0000977
was derived from the nucleolar protein 10 (NOL10) gene. Bammert
et al (65) hypothesized that
NOL10 formed a salt-stable protein complex in conjunction with
neuroguidin (NGDN) and apoptosis-antagonizing transcription factor
(AATF/Che-1/TRB) and demonstrated that the AATF-NGDN-NOL10 complex
is involved in ribosome biogenesis. In the present study, the
presence and expression level of the four circRNAs were validated
in both 20 BC tissues and their paired adjacent normal tissues,
using qPCR and RT-qPCR, respectively. hsa_circ_000052 was
upregulated in BC, whereas hsa_circ_0006220, hsa_circ_0000977 and
hsa_circ_0043278 were downregulated. Back-splicing in the RT-qPCR
products of hsa_circ_000052, hsa_circ_0006220, hsa_circ_0000977 and
hsa_circ_0043278 was confirmed by Sanger sequencing.
circRNAs have been demonstrated to serve a vital
role in cancer cell proliferation (66), migration and invasion (67), anchorage-independent cell growth
(68) and cell cycle progression
(69). It has been reported that
exonic circRNAs are involved in miRNA or protein sponging and are
able to inhibit their functional activities in the cytoplasm
(70). hsa_circ_0000520,
hsa_circ_0006220, hsa_circ_0000977 and hsa_circ_0043278 were both
cyclized from exons. To identify whether the identified circRNAs
act as miRNA sponges, the bioinformatics database CSCD was employed
to analyze the presence of potential miRNA binding sites. In
addition, CSCD can also be used for the prediction of RBPs and ORFs
for circRNAs. hsa_circ_0000977 and hsa_circ_0043278 were revealed
to have ORFs, which implies that these circRNAs may be translated.
The results demonstrated that hsa_circ_0000520 has 87 potential
MREs, hsa_circ_0006220 has 56 potential MREs, hsa_circ_0000977 has
37 potential MREs and hsa_circ_0043278 has 68 potential MREs. To
elucidate the molecular mechanism behind the functions of miRNAs,
their hypothetical target genes were analyzed using TargetScan,
miRDB and miRTarBase. The results of the analysis were used to
construct the ceRNA network.
Currently, there is no direct method of predicting
the function of circRNAs. However, the functions of mRNAs in the
ceRNA network may be analyzed to identify the potential mechanisms
of circRNAs. GO, as a bioinformatics tool, consists of three
separate ontologies: BP, MF and CC. The clusterProfiler software of
the R package was used to perform GO analysis, and the results
demonstrated that the four circRNAs may function as transcriptional
regulatory factors. For example, the most enriched GO term for
hsa_circ_0000520 in BP was ‘transcriptional activator activity, RNA
polymerase II transcription regulatory region sequence-specific DNA
binding’. Ding et al (43)
reported that circDONSON recruited the nucleosome remodeling factor
complex to the SOX4 promoter and initiated its transcription in
gastric cancer. Huang et al (71) confirmed that circNfix, as a super
enhancer-associated circRNA, may be the key regulator of cardiac
regeneration. The results of the GO analysis in the present study
provided information on the mechanism of circRNA regulation in BC
tumorigenesis.
KEGG analysis revealed that the aforementioned DECs
were related to tumor-associated pathways. For hsa_circ_0000520,
the main enriched pathways were ‘endocytosis’ and the ‘cell cycle’.
Cell cycle arrest results in decreased cell proliferation and
increased cell apoptosis. For example, upregulation of
circRNA-BARD1 has been identified to inhibit BC cell proliferation,
lead to cell cycle arrest and stimulate apoptosis (72). For hsa_circ_0006220, the most
enriched pathway was the ‘PI3K-AKT signaling pathway’. The PI3K-AKT
pathway is a ubiquitous signaling pathway that triggers a series of
responses, including cell survival, growth and proliferation
(73). Imbalance in the components
of this pathway may cause cells to proliferate abnormally, which
may facilitate tumor formation and progression (74). Recently, Zhen et al (75) revealed that circHMGCS1 serves an
important role in hepatoblastoma cell proliferation by regulating
IGF2 and IGF1R expression and upregulating the downstream effectors
of the PI3K-AKT signaling pathway. Regarding hsa_circ_0000977, the
‘MAPK signaling pathway’ was observed as the most enriched pathway.
Once activated, mitogen-activated protein kinases (MAPKs)
phosphorylate other protein kinases and numerous transcription
factors in various cellular processes, including cell survival,
differentiation, proliferation and migration (76). A previous study has reported that the
circSETD3/miR-421/MAPK14 signaling axis prevents the proliferation
of hepatocellular carcinoma (77).
KEGG pathway analysis demonstrated that hsa_circ_0043278 was mainly
associated with the ‘PI3K-AKT signaling pathway’ and ‘signaling
pathways regulating pluripotency of stem cells’. One study
demonstrated that the combined inhibition of PI3K/Akt/mTOR and
sonic hedgehog pathways modulates the activity of pluripotency
promoting factors and inhibits the survival, self-renewal and
tumorigenic potential of glioblastoma-initiating cells (78).
However, there are various limitations in the
present study due to the small sample size of the dataset employed.
In the future, integrated analysis of additional datasets may
reduce the likelihood of false-positives. In addition, the DECs
identified in the present study should be verified in a larger
number of clinical samples and other experiments should be
conducted, including in vivo studies and western blotting.
The present study provides a theoretical basis for the future study
of circRNAs-miRNAs-mRNAs in BC.
In conclusion, the present study revealed the
expression profiles of specific DECs in BC. Specifically, 47 DECs
were identified by circRNA microarray analysis of the GSE101124
dataset. Four specific DECs (hsa_circ_0000520, hsa_circ_0006220,
hsa_circ_0000977 and hsa_circ_0043278) were selected for further
validation. Following the construction of ceRNA networks and
function enrichment analysis, it was concluded that these four
circRNAs may serve a role in BC tumorigenesis as transcriptional
regulators. In summary, the present study provides further
knowledge on the mechanism by which circRNAs may serve as
biomarkers in BC.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Mr. Tong Lu and Mr.
Yuanyong Wang (Department of Thoracic Surgery, Affiliated Hospital
of Qingdao University) for their suggestions on the design of the
present study.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81770900) and the
Science and Technology Development Foundation of Shandong Province
(grant no. 2014GHY115025).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding authors on reasonable
request.
Authors' contributions
XW contributed to the conception of the present
study and completed the draft of the manuscript. All authors
participated in the design of the study and conducted the research.
YD and WL processed and analyzed the data from the dataset. QW, TL
and YW were responsible for the clinical data collection. CL and WX
contributed to the experimental design and were responsible for
revising the manuscript and approving the version to be published.
All authors reviewed all the data and approved the final version of
the manuscript.
Ethics approval and consent to
participate
The present study was conducted in accordance with
the Declaration of Helsinki. The Ethics Committee of the Affiliated
Hospital of Qingdao University approved the study. The participants
approved the use of clinical samples by providing written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guo Y, Warren Andersen S, Shu XO,
Michailidou K, Bolla MK, Wang Q, Garcia-Closas M, Milne RL, Schmidt
MK, Chang-Claude J, et al: Genetically predicted body mass index
and breast cancer risk: Mendelian randomization analyses of data
from 145,000 women of european descent. PLoS Med. 13:e10021052016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chlebowski RT, Manson JE, Anderson GL,
Cauley JA, Aragaki AK, Stefanick ML, Lane DS, Johnson KC,
Wactawski-Wende J, Chen C, et al: Estrogen plus progestin and
breast cancer incidence and mortality in the women's health
initiative observational study. J Natl Cancer Inst. 105:526–535.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lambertini M, Santoro L, Del Mastro L,
Nguyen B, Livraghi L, Ugolini D, Peccatori FA and Azim HA Jr:
Reproductive behaviors and risk of developing breast cancer
according to tumor subtype: A systematic review and meta-analysis
of epidemiological studies. Cancer Treat Rev. 49:65–76. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rice MS, Eliassen AH, Hankinson SE, Lenart
EB, Willett WC and Tamimi RM: Breast cancer research in the Nurses'
health studies: Exposures across the life course. Am J Public
Health. 106:1592–1598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu L, Shen Y, Peng X, Zhang S, Wang M, Xu
G, Zheng X, Wang J and Lu C: Aberrant promoter methylation of
cancer-related genes in human breast cancer. Oncol Lett.
12:5145–5155. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang SM, Dowhan DH and Muscat G:
Epigenetic Arginine Methylation in Breast Cancer: Emerging
therapeutic strategies. J Mol Endocrinol. 62:R223–R237. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sanger HL, Klotz G, Riesner D, Gross HJ
and Kleinschmidt AK: Viroids are single-stranded covalently closed
circular RNA molecules existing as highly base-paired rod-like
structures. Proc Natl Acad Sci USA. 73:3852–3856. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer
M, et al: Circular RNAs are a large class of animal RNAs with
regulatory potency. Nature. 495:333–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Salzman J: Circular RNA expression: Its
potential regulation and function. Trends Genet. 32:309–316. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu H, Guo S, Li W and Yu P: The circular
RNA Cdr1as, via miR-7 and its targets, regulates insulin
transcription and secretion in islet cells. Sci Rep. 5:124532015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P
and Yang BB: Foxo3 circular RNA retards cell cycle progression via
forming ternary complexes with p21 and CDK2. Nucleic Acids Res.
44:2846–2858. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Z, Huang C, Bao C, Chen L, Lin M, Wang
X, Zhong G, Yu B, Hu W, Dai L, et al: Exon-intron circular RNAs
regulate transcription in the nucleus. Nat Struct Mol Biol.
22:256–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang M, Zhao K, Xu X, Yang Y, Yan S, Wei
P, Liu H, Xu J, Xiao F, Zhou H, et al: A peptide encoded by
circular form of LINC-PINT suppresses oncogenic transcriptional
elongation in glioblastoma. Nat Commun. 9:44752018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang
Y, Jin Y, Yang Y, Chen LL, Wang Y, et al: Extensive translation of
circular RNAs driven by N6-methyladenosine. Cell Res.
27:626–641. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shao Y, Li J, Lu R, Li T, Yang Y, Xiao B
and Guo J: Global circular RNA expression profile of human gastric
cancer and its clinical significance. Cancer Med. 6:1173–1180.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thomas M, Lieberman J and Lal A:
Desperately seeking microRNA targets. Nat Struct Mol Biol.
17:1169–1174. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Swain AC and Mallick B: miRNA-mediated
‘tug-of-war’ model reveals ceRNA propensity of genes in cancers.
Mol Oncol. 12:855–868. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Peng L, Yuan XQ and Li GC: The emerging
landscape of circular RNA ciRS-7 in cancer (Review). Oncol Rep.
33:2669–2674. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen
B, Luo Y, Lyu D, Li Y, Shi G, et al: Circular RNA profiling reveals
an abundant circHIPK3 that regulates cell growth by sponging
multiple miRNAs. Nat Commun. 7:112152016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Presti M, Mazzon E, Basile MS, Petralia
MC, Bramanti A, Colletti G, Bramanti P, Nicoletti F and Fagone P:
Overexpression of macrophage migration inhibitory factor and
functionally-related genes, D-DT, CD74, CD44, CXCR2 and CXCR4, in
glioblastoma. Oncol Lett. 16:2881–2886. 2018.PubMed/NCBI
|
|
23
|
Fagone P, Mazzon E, Mammana S, Di Marco R,
Spinasanta F, Basile MS, Petralia MC, Bramanti P, Nicoletti F and
Mangano K: Identification of CD4+ T cell biomarkers for
predicting the response of patients with relapsing-remitting
multiple sclerosis to natalizumab treatment. Mol Med Rep.
20:678–684. 2019.PubMed/NCBI
|
|
24
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:D991–D995. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu JZ, Shao CC, Wang XJ, Zhao X, Chen JQ,
Ouyang YX, Feng J, Zhang F, Huang WH, Ying Q, et al: circTADA2As
suppress breast cancer progression and metastasis via targeting
miR-203a-3p/SOCS3 axis. Cell Death Dis. 10:1752019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xia S, Feng J, Chen K, Ma Y, Gong J, Cai
F, Jin Y, Gao Y, Xia L, Chang H, et al: CSCD: A database for
cancer-specific circular RNAs. Nucleic Acids Res. 46:D925–D929.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chou CH, Shrestha S, Yang CD, Chang NW,
Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, et al: miRTarBase
update 2018: A resource for experimentally validated
microRNA-target interactions. Nucleic Acids Res. 46:D296–D302.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:2015. View Article : Google Scholar
|
|
30
|
Wong N and Wang X: miRDB: An online
resource for microRNA target prediction and functional annotations.
Nucleic Acids Res. 43:D146–D152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Su G, Morris JH, Demchak B and Bader GD:
Biological network exploration with Cytoscape 3. Curr Protoc
Bioinformatics. 47:8.13.1–24. 2014. View Article : Google Scholar
|
|
32
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kanehisa M, Goto S, Furumichi M, Tanabe M
and Hirakawa M: KEGG for representation and analysis of molecular
networks involving diseases and drugs. Nucleic Acids Res.
38:D355–D360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li M, Ding W, Tariq MA, Chang W, Zhang X,
Xu W, Hou L, Wang Y and Wang J: A circular transcript of ncx1 gene
mediates ischemic myocardial injury by targeting miR-133a-3p.
Theranostics. 8:5855–5869. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ludwig NF, Sperb-Ludwig F, Velho RV and
Schwartz IV: Next-generation sequencing corroborates a probable de
novo GNPTG variation previously detected by Sanger sequencing. Mol
Genet Metab Rep. 11:92–93. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhong Y, Du Y, Yang X, Mo Y, Fan C, Xiong
F, Ren D, Ye X, Li C, Wang Y, et al: Circular RNAs function as
ceRNAs to regulate and control human cancer progression. Mol
Cancer. 17:792018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
He R, Liu P, Xie X, Zhou Y, Liao Q, Xiong
W, Li X, Li G, Zeng Z and Tang H: circGFRA1 and GFRA1 act as ceRNAs
in triple negative breast cancer by regulating miR-34a. J Exp Clin
Cancer Res. 36:1452017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen B, Wei W, Huang X and Xie X, Kong Y,
Dai D, Yang L, Wang J, Tang H and Xie X: circEPSTI1 as a prognostic
marker and mediator of triple-negative breast cancer progression.
Theranostics. 8:4003–4015. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang R, Xing L, Wang M, Chi H, Zhang L and
Chen J: Comprehensive analysis of differentially expressed profiles
of lncRNAs/mRNAs and miRNAs with associated ceRNA networks in
triple-negative breast cancer. Cell Physiol Biochem. 50:473–488.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ding L, Zhao Y, Dang S, Wang Y, Li X, Yu
X, Li Z, Wei J, Liu M and Li G: Circular RNA circ-DONSON
facilitates gastric cancer growth and invasion via NURF complex
dependent activation of transcription factor SOX4. Mol Cancer.
18:452019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nair AA, Niu N, Tang X, Thompson KJ, Wang
L, Kocher JP, Subramanian S and Kalari KR: Circular RNAs and their
associations with breast cancer subtypes. Oncotarget.
7:80967–80979. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Coscujuela Tarrero L, Ferrero G, Miano V,
De Intinis C, Ricci L, Arigoni M, Riccardo F, Annaratone L,
Castellano I, Calogero RA, et al: Luminal breast cancer-specific
circular RNAs uncovered by a novel tool for data analysis.
Oncotarget. 9:14580–14596. 2018.PubMed/NCBI
|
|
46
|
Zhang HD, Jiang LH, Hou JC, Zhou SY, Zhong
SL, Zhu LP, Wang DD, Yang SJ, He YJ, Mao CF, et al: Circular RNA
hsa_circ_0072995 promotes breast cancer cell migration and invasion
through sponge for miR-30c-2-3p. Epigenomics. 10:1229–1242. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Du WW, Yang W, Chen Y, Wu ZK, Foster FS,
Yang Z, Li X and Yang BB: Foxo3 circular RNA promotes cardiac
senescence by modulating multiple factors associated with stress
and senescence responses. Eur Heart J. 38:1402–1412.
2017.PubMed/NCBI
|
|
48
|
Wu Z, Huang W, Wang X, Wang T, Chen Y,
Chen B, Liu R, Bai P and Xing J: Circular RNA CEP128 acts as a
sponge of miR-145-5p in promoting the bladder cancer progression
via regulating SOX11. Mol Med. 24:402018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wei H, Pan L, Tao D and Li R: Circular RNA
circZFR contributes to papillary thyroid cancer cell proliferation
and invasion by sponging miR-1261 and facilitating C8orf4
expression. Biochem Biophys Res Commun. 503:56–61. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang L, Tong X, Zhou Z, Wang S, Lei Z,
Zhang T, Liu Z, Zeng Y, Li C, Zhao J, et al: Circular RNA
hsa_circ_0008305 (circPTK2) inhibits TGF-β-induced
epithelial-mesenchymal transition and metastasis by controlling
TIF1γ in non-small cell lung cancer. Mol Cancer. 17:1402018.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Qu S, Liu Z, Yang X, Zhou J, Yu H, Zhang R
and Li H: The emerging functions and roles of circular RNAs in
cancer. Cancer Lett. 414:301–309. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li S, Teng S, Xu J, Su G, Zhang Y, Zhao J,
Zhang S, Wang H, Qin W, Lu ZJ, et al: Microarray is an efficient
tool for circRNA profiling. Brief Bioinform. 20:1420–1433. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lan X, Xu J, Chen C, Zheng C, Wang J, Cao
J, Zhu X and Ge M: the landscape of circular RNA expression
profiles in papillary thyroid carcinoma based on RNA sequencing.
Cell Physiol Biochem. 47:1122–1132. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Petralia MC, Mazzon E, Fagone P, Russo A,
Longo A, Avitabile T, Nicoletti F, Reibaldi M and Basile MS:
Characterization of the pathophysiological role of CD47 in uveal
melanoma. Molecules. 24(pii): E24502019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Basile MS, Mazzon E, Russo A, Mammana S,
Longo A, Bonfiglio V, Fallico M, Caltabiano R, Fagone P, Nicoletti
F, et al: Differential modulation and prognostic values of
immune-escape genes in uveal melanoma. PLoS One. 14:e02102762019.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fagone P, Caltabiano R, Russo A, Lupo G,
Anfuso CD, Basile MS, Longo A, Nicoletti F, De Pasquale R, Libra M
and Reibaldi M: Identification of novel chemotherapeutic strategies
for metastatic uveal melanoma. Sci Rep. 7:445642017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fagone P, Mangano K, Mammana S, Pesce A,
Pesce A, Caltabiano R, Giorlandino A, Portale TR, Cavalli E,
Lombardo GA, et al: Identification of novel targets for the
diagnosis and treatment of liver fibrosis. Int J Mol Med.
36:747–752. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fagone P, Mazzon E, Cavalli E, Bramanti A,
Petralia MC, Mangano K, Al-Abed Y, Bramati P and Nicoletti F:
Contribution of the macrophage migration inhibitory factor
superfamily of cytokines in the pathogenesis of preclinical and
human multiple sclerosis: In silico and in vivo evidences. J
Neuroimmunol. 322:46–56. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fagone P, Nunnari G, Lazzara F, Longo A,
Cambria D, Distefano G, Palumbo M, Nicoletti F, Malaguarnera L and
Di Rosa M: Induction of OAS gene family in HIV monocyte infected
patients with high and low viral load. Antiviral Res. 131:66–73.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Basile MS, Fagone P, Mangano K, Mammana S,
Magro G, Salvatorelli L, Li Destri G, La Greca G, Nicoletti F,
Puleo S and Pesce A: KCNMA1 expression is downregulated in
colorectal cancer via epigenetic mechanisms. Cancers (Basel).
11(pii): E2452019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yuan C, Zhou L, Zhang L, Yin K, Peng J,
Sha R, Zhang S, Xu Y, Sheng X, Wang Y, et al: Identification and
integrated analysis of key differentially expressed circular RNAs
in ER-positive subtype breast cancer. Epigenomics. 11:297–321.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Baer M, Nilsen TW, Costigan C and Altman
S: Structure and transcription of a human gene for H1 RNA, the RNA
component of human RNase P. Nucleic Acids Res. 18:97–103. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Barlev NA, Emelyanov AV, Castagnino P,
Zegerman P, Bannister AJ, Sepulveda MA, Robert F, Tora L,
Kouzarides T, Birshtein BK and Berger SL: A novel human Ada2
homologue functions with Gcn5 or Brg1 to coactivate transcription.
Mol Cell Biol. 23:6944–6957. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wu Y, Xie Z, Chen J, Chen J, Ni W, Ma Y,
Huang K, Wang G, Wang J, Ma J, et al: Circular RNA circTADA2A
promotes osteosarcoma progression and metastasis by sponging
miR-203a-3p and regulating CREB3 expression. Mol Cancer. 18:732019.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bammert L, Jonas S, Ungricht R and Kutay
U: Human AATF/Che-1 forms a nucleolar protein complex with NGDN and
NOL10 required for 40S ribosomal subunit synthesis. Nucleic Acids
Res. 44:9803–9820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhao F, Han Y, Liu Z, Zhao Z, Li Z and Jia
K: circFADS2 regulates lung cancer cells proliferation and invasion
via acting as a sponge of miR-498. Biosci Rep. 38(pii):
BSR201805702018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
He JH, Li YG, Han ZP, Zhou JB, Chen WM, Lv
YB, He ML, Zuo JD and Zheng L: The CircRNA-ACAP2/Hsa-miR-21-
5p/Tiam1 regulatory feedback circuit affects the proliferation,
migration, and invasion of colon cancer SW480 cells. Cell Physiol
Biochem. 49:1539–1550. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hsiao KY, Lin YC, Gupta SK, Chang N, Yen
L, Sun HS and Tsai SJ: Noncoding effects of circular RNA CCDC66
promote colon cancer growth and metastasis. Cancer Res.
77:2339–2350. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Xue J, Liu Y, Luo F, Lu X, Xu H, Liu X, Lu
L, Yang Q, Chen C, Fan W and Liu Q: Circ100284, via miR-217
regulation of EZH2, is involved in the arsenite-accelerated cell
cycle of human keratinocytes in carcinogenesis. Biochim Biophys
Acta Mol Basis Dis. 1863:753–763. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chen LL: The biogenesis and emerging roles
of circular RNAs. Nat Rev Mol Cell Biol. 17:205–211. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huang S, Li X, Zheng H, Si X, Li B, Wei G,
Li C, Chen Y, Chen Y, Liao W, et al: Loss of
super-enhancer-regulated CircRNA Nfix induces cardiac regeneration
after myocardial infarction in adult mice. Circulation.
139:2857–2876. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhao J, Zou H, Han C, Ma J, Zhao J and
Tang J: Circlular RNA BARD1 (Hsa_circ_0001098) overexpression in
breast cancer cells with TCDD treatment could promote cell
apoptosis via miR-3942/BARD1 axis. Cell Cycle. 17:2731–2744. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mehrian-Shai R, Chen CD, Shi T, Horvath S,
Nelson SF, Reichardt JK and Sawyers CL: Insulin growth
factor-binding protein 2 is a candidate biomarker for PTEN status
and PI3K/Akt pathway activation in glioblastoma and prostate
cancer. Proc Natl Acad Sci USA. 104:5563–5568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhen N, Gu S, Ma J, Zhu J, Yin M, Xu M,
Wang J, Huang N, Cui Z, Bian Z, et al: CircHMGCS1 promotes
hepatoblastoma cell proliferation by regulating the IGF signaling
pathway and glutaminolysis. Theranostics. 9:900–919. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Xu L, Feng X, Hao X, Wang P, Zhang Y,
Zheng X, Li L, Ren S, Zhang M and Xu M: CircSETD3
(Hsa_circ_0000567) acts as a sponge for microRNA-421 inhibiting
hepatocellular carcinoma growth. J Exp Clin Cancer Res. 38:982019.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Nanta R, Shrivastava A, Sharma J, Shankar
S and Srivastava RK: Inhibition of sonic hedgehog and PI3K/Akt/mTOR
pathways cooperate in suppressing survival, self-renewal and
tumorigenic potential of glioblastoma-initiating cells. Mol Cell
Biochem. 454:11–23. 2019. View Article : Google Scholar : PubMed/NCBI
|