Introduction
Tumor chemotherapy is an important part of the
comprehensive treatment for solid tumors (1). Paclitaxel, vincristine and other
spindle poisons are widely used as first-line chemotherapy for
various types of malignant tumor, such as breast cancer, cervical
cancer and melanoma (2,3). One of the important methods in treating
cancer is inducing tumor cell death. In the clinical setting, it
was observed that certain types of tumor are sensitive to this type
of drug, and in such cases, the tumor tissue disappears following
several cycles of chemotherapy (4,5).
However, more commonly, the tumor tissue is sensitive to the drug
at the beginning of chemotherapy, but drug resistance eventually
occurs, or the tumor tissue has no obvious response to this class
of drugs (6). The latter two
phenomena are the root cause of uncontrollable tumors and
recurrence. Previous studies demonstrated that spindle poisons play
a role in the induction of apoptosis in some cells (2,7).
However, certain cells do not undergo apoptosis when exposed to
such drugs, and instead form polyploids. Polyploid tumors are
extremely insensitive to common radiotherapy and chemotherapy
options, and patients who have tumors with polyploid subclones have
a worse prognosis (8,9). Therefore, it was speculated that the
formation of polyploids may be associated with tumor
resistance.
For a number of years, research has focused on
elucidating the molecular mechanism underlying tumor resistance,
and numerous theories and potential mechanisms have been proposed;
however, a resolution to this clinical issue has not yet been
identified (10,11). With continuous scientific
development, it has become apparent that the causes of tumor
resistance are multi-faceted. In addition to the current multi-drug
resistance genes that are known, it was speculated that the
formation of polyploids is likely to lead to resistance to spindle
poisons and is an important cause of tumor drug resistance after
use (10–12); however, a mechanism for this possible
drug resistance has not yet been proposed. The phenomenon of
tumor-forming polyploidy induced by spindle poison was used as a
basis for the design of the present study. MDA-MB-231 cells were
treated with the spindle poison nocodazole in order to establish a
stable polyploid cell model. Subsequently, the sensitivity of
chemotherapeutic drugs on polyploid and original tumors was
investigated with the aim of elucidating the association between
polyploid formation and tumor resistance, and to attempt to
determine the molecular mechanism underlying polyploidy occurrence
through in-depth analyses. A potential intervention point/method to
address such resistance was identified, and a new strategy was
proposed in the present study to prevent the occurrence of tumor
resistance by inhibiting the formation of polyploid subclones.
Materials and methods
Cell lines and cell culture
For routine culturing, human breast cancer HCC1806
and MDA-MB-231 cells were obtained from China Infrastructure of
Cell Line Resources, Institute of Basic Medical Sciences, Chinese
Academy of Medical Sciences, and MDA-MB-231 polyploid cells
(T-MDA-MB-231) were established in the laboratory. These cells were
cultured in RPMI-1640 medium (Corning Inc.) containing 10% newborn
calf serum (Corning, Inc.) at 37°C and 5% CO2. Cells in
the logarithmic growth phase were used in the subsequent
experiments.
Nocodazole drug induction assay
HCC1806 and MDA- MB-231 cells (1×106
cells/ml) were inoculated into 10-cm culture dishes for 24 h and
then the antibiotic-free RPMI 1640 medium was replaced fresh
antibiotic-free RPMI-1640 medium with 10% fetal bovine serum
(Corning, Inc.) containing nocodazole (Sigma-Aldrich; Merk KGaA).
The base volume was 10 ml, and the final concentration of
nocodazole was 100 µg/l; the cells were treated for 0–72 h at 37°C,
and 3 duplicate wells were examined for each group. The
morphological changes in the HCC1806 and MDA-MB-231 cells prior to
and following nocodazole treatment were observed under a light
microscope at ×200 magnification and images were captured at
randomly selected fields.
Cell cycle analysis
MDA-MB-231 and HCC1806 cells were treated with 100
µg/l nocodazole for 0, 6, 12, 24, 48 or 72 h, and the cells were
trypsinized by 0.25% trypsin, collected, fixed in 70% cold ethanol
and incubated overnight at 4°C. After centrifuging for 5 min at 300
× g and at room temperature, the ethanol was discarded, and the
pellets were washed with PBS and incubated with 100 ng/l RNase for
37 min at 37°C. A total of 500 µl propidium iodide (PI; 2 µg/ml)
stain solution was added, and the cells were incubated at room
temperature for 30 min in the dark prior to being counted
(1×106 cells/ml). The cell cycle (G0/S or
G2/M) was detected via flow cytometry (FACSCanto II; BD
Biosciences), and the percentages of diploid, tetraploid and
octoploid cells in the total cell population were determined by the
FACSDiva software (version 8.0.1; BD Biosciences).
Flow cytometry sorting of
polyploids
According to the results of the cell cycle assay,
100 µg/l nocodazole-treated MDA-MB231 cells were harvested at the
point of maximal polyploid formation (72 h), and the cells were
resuspended in RPMI 1640 medium at 4°C at a concentration of
1×106 cells/ml. Subsequently, Hoechst 33342 (5 µg/ml)
was added, and the cells were incubated at 37°C for 90 min. After
centrifuging at 300 × g for 10 min at 4°C, the supernatant was
discarded, and the cells were resuspended in pre-chilled Hanks'
Balanced Salt Solution (Beyotime Institute of Biotechnology). A
total of 500 µl PI (2 µg/ml) was then added to the solution prior
to sieving using a 200-mesh nylon cell sieve. Flow cytometry
(FACSCanto II) was used to obtain drug-treated polyploid cells
(T-MDA-MB-231) using the FACSDiva software (version 8.0.1). After
routine stable growth, polyploid cells in the logarithmic growth
phase were subjected to secondary sorting using the same method.
Subcloning of polyploid cells was performed by limiting
dilution.
Apoptosis experiment
The present study aimed to compare the sensitivity
of polyploid cells and their diploid parental cells to commonly
used chemotherapeutic drugs. Therefore, representative drugs from
different chemotherapeutic drug classes, paclitaxel and docetaxel
(Jiangsu Hengrui Medicine Co., Ltd.), were used for the apoptosis
experiments. MDA-MB-231 and T-MDA-MB-231 cells in the logarithmic
growth phase (5×105 cells/ml) were plated into 6-well
cell culture plates and cultured for 24 h at 37°C and 5%
CO2 using the 30% inhibitory concentration
(IC30) and the 60% inhibitory concentration
(IC60) of 0.66 and 2.50 µM for paclitaxel and 3.48 and
12.10 µM for docetaxel, respectively, to treat the MDA-MB-231 and
T-MDA-MB-231 cells. After incubating for 72 h at 37°C, the cells
were centrifuged at 300 × g for 4 min at 4°C, and 5 µl Annexin V
and 10 µl PI were added, followed by incubation for 15 min at room
temperature in the dark. Apoptosis was subsequently detected by
flow cytometry (FACSCanto II; excitation wavelength, 488 nm) with
the FACSDiva software (version 8.0.1).
MTT assay
T-MDA-MB-231 cells were seeded at 5×103
cells/well in 96-well plates using 2 and 4 µM of the multi-kinase
inhibitor sorafenib (Sigma-Aldrich; Merck KGaA) alone or in
combination with 100 µg/l nocodazole or 100 nmol/l paclitaxel for
48 and 72 h, with 5 replicate wells per group; 4 µl of a 5 g/l MTT
solution were added for 4 h. At termination, the medium was removed
from each well, and 200 µl of dimethyl sulfoxide was added per
well, followed by shaking until the crystals dissolved. The same
method was applied to compare differences in the cell cytotoxicity
of MDA-MB-231 and T-MDA-MB-231 cells treated with spindle poisons,
paclitaxel (0, 0.39, 0.78, 1.56, 3.125, 6.25, 12.5, 25 and 50 µM),
docetaxel (0, 0.39, 0.78, 1.56, 3.125, 6.25, 12.5, 25 and 50 µM),
epirubicin (0, 0.39, 0.78, 1.56, 3.125, 6.25, 12.5, 25 and 50 µM),
5-fluorouracil (FU) (0, 0.78, 1.56, 3.125, 6.25, 12.5, 25, 50 and
100 µM), oxaliplatin (0, 0.78, 1.56, 3.125, 6.25, 12.5, 25, 50, and
100 µM) or etoposide (0, 0.0195, 0.039, 0.078, 0.156, 0.3125,
0.625, 1.25 and 2.50 µM). Epirubicin was purchased from Pfizer Inc.
5-FU, oxaliplatin or etoposide were purchased from Jiangsu Hengrui
Medicine Co., Ltd. Differences in the cytotoxicity of paclitaxel,
docetaxel, epirubicin, 5-FU, oxaliplatin or etoposide on
T-MDA-MB-231 cells and MDA-MB-231 cells were also examined by MTT
assay. The absorbance value at 490 nm of each well was measured
using a microplate reader with the tecan i-control 2.0 software,
and the cytotoxicity rate was calculated as 1-A490 drug
group/A490 control group ×100%.
Western blotting
MDA-MB-231 and T-MDA-MB-231 cells (3×105)
were treated with 100 µg/l nocodazole for 48 h and then digested
and centrifuged at 300 × g for 5 min at 4°C and collected.
Subsequently, the cells were lysed using radioimmunoprecipitation
assay lysis buffer (Beyotime Institute of Biotechnology), and the
cell lysates (total protein) were collected by centrifugation at
16,000 × g for 10 min at 4°C. Protein concentration was determined
using a BCA kit (Beyotime Institute of Biotechnology). For each
sample, 100 µg of total cell protein per lane was separated via
SDS-PAGE (12% gel) and then transferred to a polyvinylidene
fluoride membrane. Following blocking with 5% skimmed milk at 4°C
for 1 h, the membrane was incubated with primary monoclonal
antibodies (all purchased from Abcam) against F-box and WD repeat
domain containing 7 (FBW7; 1:1,500; cat. no. ab109617), MCL1
apoptosis regulator BCL2 family member (MCL-1; 1:1,500; cat. no.
ab32087), Bcl-2 (1:1,500; cat. no. ab185002), Bax (1:1,500; cat.
no. ab232479), caspase-3 (1:1,500; cat. no. ab179517), caspase-9
(1:1,500; cat. no. ab2013) and β-actin (1:5,000; cat. no. ab179467)
overnight at 4°C. The membrane was washed three times to remove the
primary antibody, and then incubated with goat
anti-rabbit-horseradish peroxidase-labeled secondary antibody
(1:1,000; cat. no. ab6721; Abcam) for 1 h at room temperature.
After washing three times, Immobilon Western chemiluminescent
horseradish peroxidase substrate (EMD Millipore) was used to
develop the membranes. The experiment was performed three times and
visualized by Image J (National Institutes of Health; version
1.8.0).
Reverse transcription-quantitative PCR
(RT-qPCR)
MDA- MB-231 and T-MDA-MB-231 cells were digested,
centrifuged at 300 × g for 5 min at 4°C and collected. Total RNA
was extracted using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.), and cDNA was obtained via the BeyoRT™ RT kit
(Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. qPCR was subsequently performed using
BeyoFast™ SYBR Green qPCR Mix kit (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. The
following primer pairs were used: Bcl-2 forward,
5′-ATGTGTGTGGAGAGCGTCAA-3′ (140 bp) and reverse,
5′-GCCGTACAGTTCCACAAAGG-3′ (140 bp); and GAPDH forward,
5′-GAAGGTGAAGGTCGGAGTC-3′ (225 bp) and reverse,
5′-GAAGATGGTGATGGGATTTC-3′ (225 bp). For each reaction,
pre-denaturation was performed at 95°C for 7 min, followed by 35
cycles of denaturation at 95°C for 40 sec, annealing at 56°C for 40
sec, and extension at 72°C for 40 sec; and a final extension was
performed at 72°C for 10 min. GAPDH was used as an internal
reference to calculate the relative expression of the Bcl-2 gene
via the 2−∆∆Cq method (13).
RNA interference
T-MDA-MB231 cells were transfected using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol, and MDA-MB231 cells were
used as controls. Logarithmic growth phase T-MDA-MB231 cells at
1.5×105 cells per well were added to 6-well plates and
incubated for 24 h, after which Lipofectamine™ 2000 diluted in
Opti-MEMI (Corning; Thermo Fisher Scientific, Inc.) was added at 50
µl/well and mixed at room temperature for 5 min. After 5 min, 100
µl of liposomes (Invitrogen; Thermo Fisher Scientific Inc.) and
Bcl-2 small interfering (si) RNA (forward,
5′-UGUGGAUGACUGAGUACCUGA-3′; reverse,
5′-UCAGGUACUCAGUCAUCCACAGG-3′) or control siRNA
(AATTCTCCGAACGTGTCACGTCCTGTCTC) were purchase from Ambion (Thermo
Fisher Scientific, Inc.) and incubated at room temperature for 20
min. The liposome-siRNA complexes were added to the cells and then
incubated at 37°C for 6 h, at which time the media was replaced
with fresh RPMI-1640 medium with 10% fetal bovine serum to continue
the culture. The mRNA expression of Bcl-2 in T-MDA-MB-231 cells was
detected by RT-qPCR as aforementioned, 72 h following siRNA
transfection.
Statistical analysis
The statistical analyses were performed using SPSS
software (version 17.0; SPSS, Inc.). Data are expressed as the mean
± standard deviation from at least three separate experiments.
Experimental results were assessed using one-way analysis of
variance and SNK-q post hoc tests. P<0.05 was considered to
indicate a statistically significant difference. GraphPad Prism
software (version 5.0; GraphPad Software, Inc.) was used to
calculate the inhibitory concentrations.
Results
Nocodazole induces apoptosis in
HCC1806 cells and polyploidy in MDA-MB-231 cells
A final nocodazole concentration of 100 ng/ml was
used in the present study as it has been demonstrated that 100
ng/ml could induce polyploid (14,15)
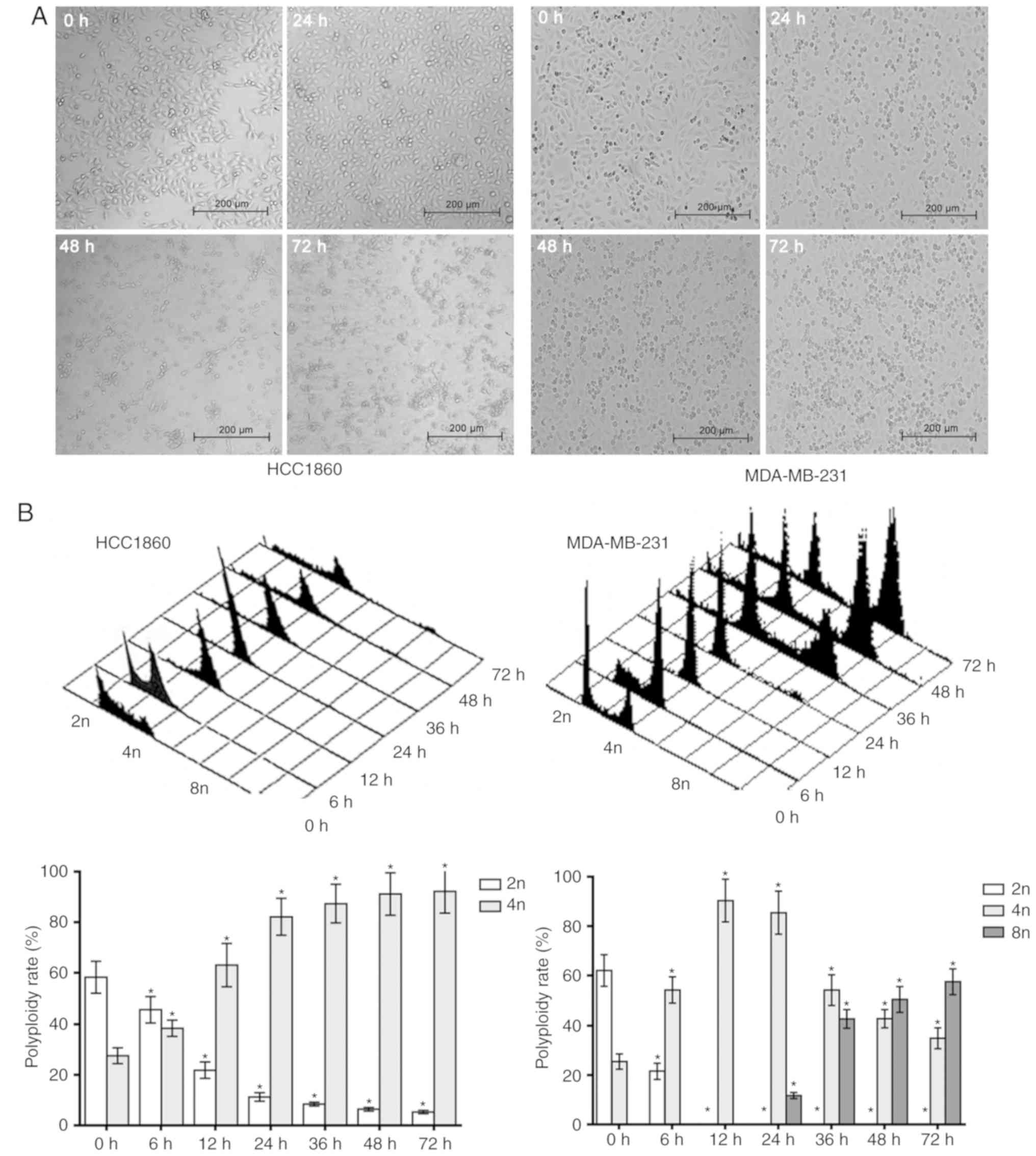
After nocodazole was applied to MDA-MB-231 and HCC1806 cells for 24
h, the cells began to appear rounded, and after 48 h, the cells
were all rounded. However, with prolonged exposure for 72 h to
nocodazole, the HCC1806 cells exhibited typical apoptotic
morphological changes; the cell volume gradually decreased, the
cells became deformed and floated, their nuclei shrank, and
apoptotic bodies formed. Fragmentation was gradually increased, and
after 72 h, apoptotic bodies were widespread. However, the
MDA-MB-231 cells exhibited increased cell volume over time with
drug exposure, and typical polyploid morphological changes were
apparent, such as gradually increasing nuclei. After 72 h, the
polyploid appearance was prevalent (Fig.
1A).
In order to further verify the responses of
MDA-MB-231 and HCC1806 breast cancer cells to nocodazole in the
present study, samples of the two cell lines were analyzed by flow
cytometry in the present study. After HCC1806 cells were treated
with nocodazole for 6 h, the percentage of G2/M phase
cells increased, and the proportion of sub-G1 phase
cells gradually increased. For MDA-MB-231 cells treated with
nocodazole for 6, 12 and 24 h, the percentages of cells in the
sub-G1 and G2/M phases were the same as those
of HCC1806 cells, but the percentage of cells in the
G1/S/G2/M phase increased after 36 h, and
body (tetraploid and octoploid) cell formation increased
significantly (Fig. 1B). These
experiments indicated that nocodazole can induce apoptosis in
HCC1806 cells but instead induces polyploidy in MDA-MB-231
cells.
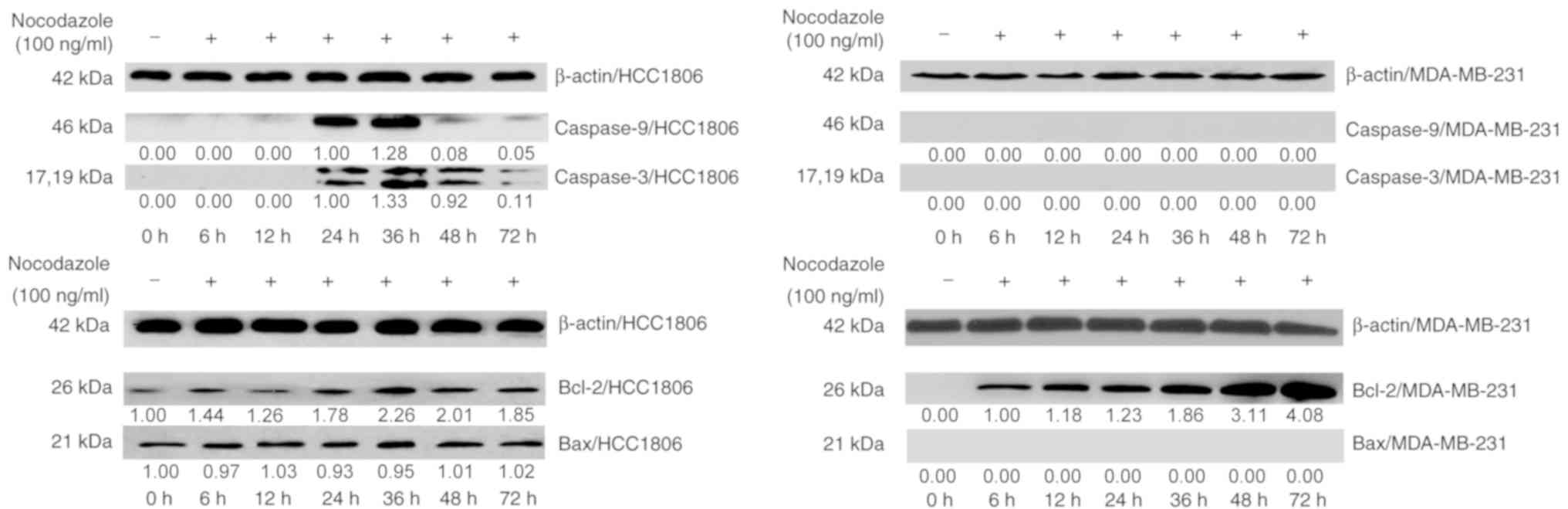
The western blot analysis revealed that, compared
with the control group, HCC1806 cells expressed caspase-9 and
caspase-3 after 24 h of incubation, indicating that nocodazole
induced apoptosis in HCC1806 cells. The expression of caspase-9,
caspase-3 and Bax was not detected in MDA-MB-231 cells. However,
the Bax protein was continuously expressed in HCC1806 cells. In
MDA-MB-231 cells, the expression of Bcl-2 increased significantly
until 36 h, and its expression levels were significantly different.
Bcl-2 was also expressed in HCC1806 cells, and its expression
increased after 36 h (Fig. 2). These
experiments indicated that an increasing Bcl-2 in HCC1806 cells
promoted apoptosis following treatment with nocodazole, while the
high expression of Bcl-2 in MDA-MB-231 cells inhibited apoptosis
and formed polyploid cells. High expression levels of Bcl-2 inhibit
nocodazole-induced apoptosis in breast cancer cells and may be one
of the primary molecular mechanisms underlying polyploid
formation.
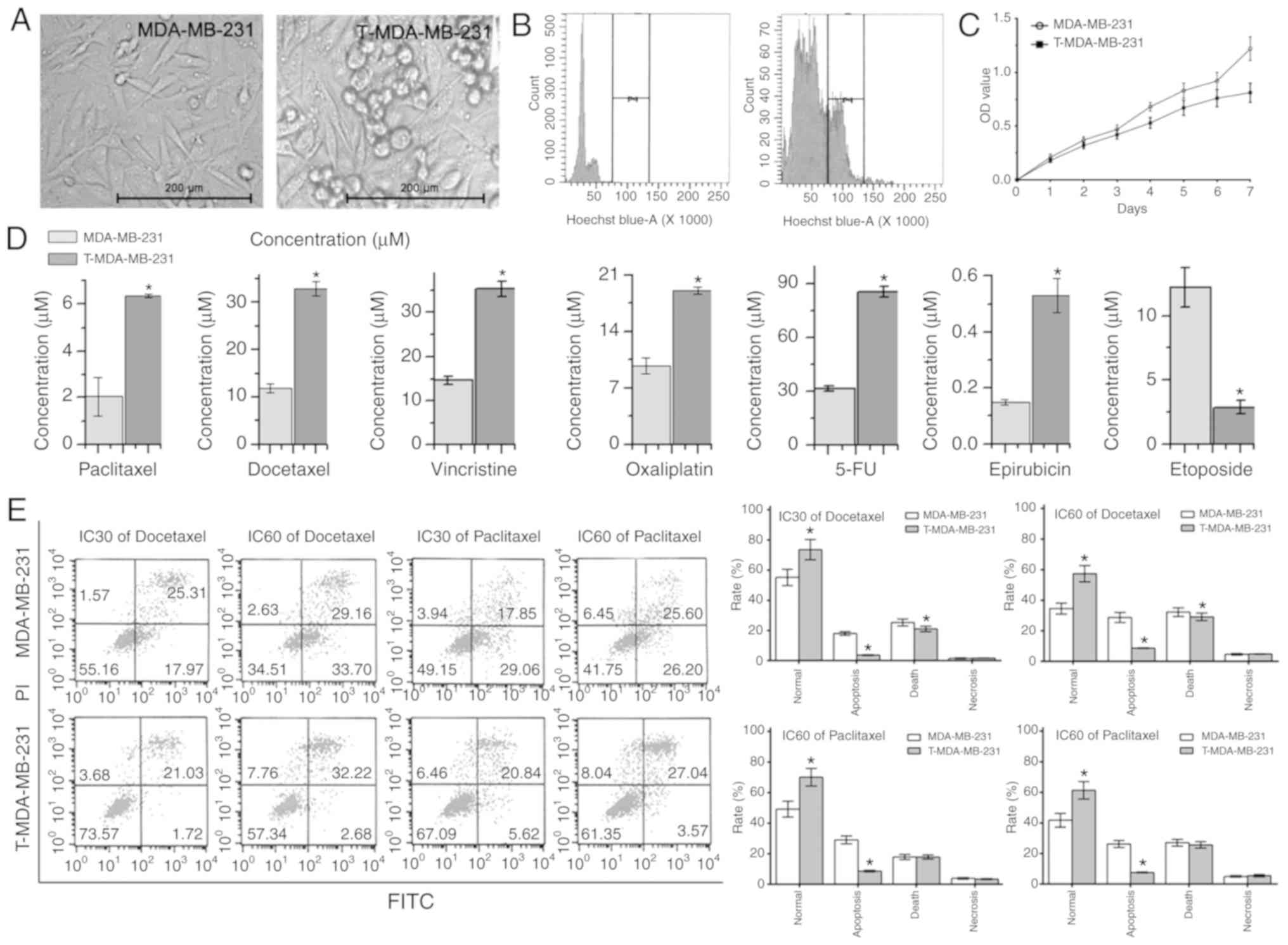
Effect of polyploid MDA-MB-231 on
chemosensitivity
The appearance and polyploid certification of
MDA-MB-231 parent cells and T-MDA-MB-231 polyploid cells is
presented in Fig. 3A and B, viewed
under a light microscope at ×200 magnification and flow cytometry.
Meanwhile, there was a slower rate of cytotoxicity in T-MDA-MB-231
cells, demonstrated by the MTT assay (Fig. 3C). The MTT assay results also
revealed that T-MDA-MB-231 cells were less sensitive to paclitaxel,
docetaxel, vincristine, oxaliplatin, 5-FU and epirubicin than
MDA-MB-231 cells (all P<0.05). However, T-MDA-MB-231 polyploid
cells were more sensitive to etoposide (Fig. 3D). The flow cytometry results
revealed that following treatment with paclitaxel and docetaxel at
IC30, IC50 and IC60 for 72 h, the
survival rates of the polyploid T-MDA-MB-231 cells were higher than
those of the parental MDA-MB-231 cells. The proportion of cells in
early apoptosis was also lower in polyploid cells (Fig. 3E), indicating that polyploid tumors
are more resistant to these anticancer drugs.
Role of the Bcl-2 protein family in
polyploidy-induced tumor resistance
The RT-qPCR results revealed that the relative
expression of Bcl-2 mRNA in T-MDA-MB-231 polyploid cells was
significantly higher than that of the MDA-MB-231 parental cells.
After silencing the expression of Bcl-2 mRNA in T-MDA-MB-231 cells
using siRNA technology, the relative expression decreased
significantly (P<0.05; Fig. 4A)
but was not significantly different to the expression levels of the
MDA-MB-231 parental cells. Following the silencing of Bcl-2
expression, mRNA was downregulated, the IC50 of
docetaxel for T-MDA-MB-231 polyploid cells was significantly
decreased, and the IC50 of etoposide was significantly
increased; however, Bcl-2 silencing had no significant effect on
the paclitaxel IC50 (Fig.
4B). These results indicated that the higher expression of
Bcl-2 in polyploidy cells plays a key role in the process by which
cells escape apoptosis and further form polyploid cells, but has
different effects on anti-tumor drugs with different
mechanisms.
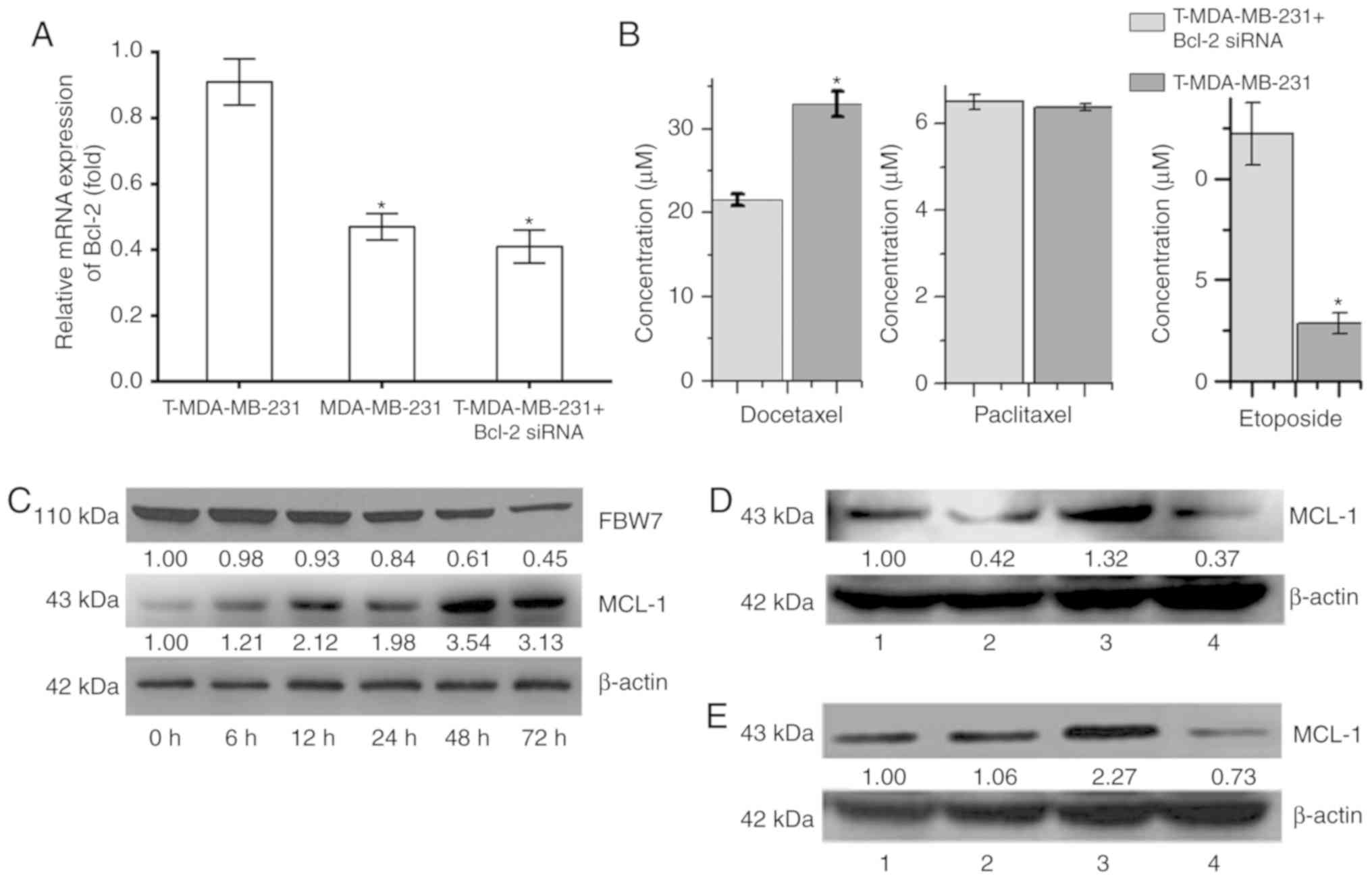 | Figure 4.Role of the Bcl-2 protein family in
polyploidy-induced tumor resistance in MDA-MB-231. (A) The relative
mRNA expression of Bcl-2 of T-MDA-MB-231 cells, MDA-MB-231 parental
cells and T-MDA-MB-231 cells following Bcl-2 silencing. (B) The
half maximal inhibitory concentration of docetaxel, paclitaxel and
etoposide for T-MDA-MB-231 cells with or without Bcl-2 silencing.
(C) The protein expression of FBW7 and MCL-1 in MDA-MB-231 cells at
different time points. (D) The protein expression of MCL-1 protein
in MDA-MB-231 cells following treatment with nocodazole and
sorafenib. β-actin was used as internal control. 1, control; 2, 2
µM sorafenib+ 100 µg/l nocodazole; 3, 100 µg/l nocodazole; 4, 2 µM
sorafenib. (E) The protein expression of MCL-1 protein in
MDA-MB-231 cells following treatment with paclitaxel and sorafenib.
β-actin was used as an internal control. 1, control; 2, 2 µM
sorafenib+ 100 nM paclitaxel; 3, 100 nM paclitaxel; 4, 2 µM
sorafenib. (F and G) The effect of ploidy on treatment with
nocodazole, sorafenib, paclitaxel and sorafenib in the MDA-MB-231
cells. (H) The effect of sorafenib on the cytotoxicity of
MDA-MB-231 at 48 and 72 h. *P<0.05 vs. 100 µg/l Nocodazole
group; ΔP<0.05 vs. 100 µg/l Nocodazole+ 2 µM
Sorafenib group; **P<0.05 vs. 100 nM Paclitaxel group;
#P<0.05 vs. 100 nM Paclitaxel+ 2 µM Sorafenib
group. |
MCL-1 is an important anti-apoptotic protein in the
Bcl-2 family, and it may play an important role in polyploidy
resistance (16). With the prolonged
nocodazole treatment time, expression of the FBW7 protein was
decreased in MDA-MB-231 cells, while expression of MCL-1 was
increased (Fig. 4C). The present
study observed that after 48 h of treatment, the expression of
MCL-1 was decreased and demonstrated by comparing the
nocodazole+sorafenib group with the nocodazole group, and the
paclitaxel+sorafenib group with the paclitaxel group in MDA-MB-231.
At 48 h, with sorafenib, expression of the MCL-1 protein was
decreased, the number of diploid cells was increased, and the
number of polyploid cells was decreased in MDA-MB-231 cells
(Fig. 4D and E). There was an
increase in diploid and decrease in polyploid cells in the
nocodazole+sorafenib and sorafenib groups compared with the
nocodazole group. There was a similar effect of ploidy in the
nocodazole+sorafenib, paclitaxel+sorafenib and sorafenib groups,
which polyploid was downregulated after sorafenib treating cells
for 48 and 72 h (Fig. 4F and G),
indicating that the combination of sorafenib and single-purpose
spindle poisons can increase drug sensitivity and be used to
reverse polyploid resistance, the occurrence of which may be
associated with the regulation of MCL-1 protein expression and the
cell cycle. The present study tested different concentrations of
nocodazole and sorafenib and it was concluded that 100 µg/l
nocodazole+4 µΜ sorafenib may be the best option by increasing
cytotoxicity (Fig. 4H).
Discussion
At present, paclitaxel, vincristine and other
spindle poisons are the first-line chemotherapy options for the
majority of solid tumors, such as breast and ovarian cancer
(17,18). These drugs affect the structure and
function of spindles during the process of mitosis in tumor cells,
destroying the kinetochore and affecting the adhesion of the tube
and the integrity of the mitotic apparatus to prevent normal
splitting of the chromosome centromere, which is originally placed
in the center of the equatorial plate (19,20).
Consequently, the chromosome cohort cannot be normally separated
from the two poles of the spindle, and the spindle assembly
checkpoint is activated. Under normal conditions, if the cell
damage cannot be repaired, then the cells are unable to undergo
mitosis, thereby initiating the apoptotic pathway and inducing
apoptosis to achieve the ultimate goal of tumor cell death
(21–24). However, certain cells ignore the
spindle structure that is formed during mitosis, and in the case of
functional disruption, the nucleus does not divide prior to
entering the next cell cycle, forming polyploid tumor cells, which
enhances resistance to chemotherapeutic drugs (25–27).
Nocodazole is responsible for inducing cell cycle
arrest in the G2/M phase (28–30).
Nocodazole interferes with the cell microtubules to form a spindle,
which blocks cell division (7). The
results of the present study demonstrated that nocodazole had a
significant effect on the cell cycle, and G2/M phase
arrest occurred following induction in MDA-MB-231 cells; however,
the cells did not completely exit mitosis or activate apoptosis and
instead entered the next cell cycle. The human breast cancer
MDA-MB-231 cells were polyploidized, with typical polyploid
morphological changes such as cell volume increase and nuclear
enlargement observed. At present, the occurrence of drug resistance
following the polyploidization of tumor cells is controversial.
Castedo et al (31) reported
that polyploid tumors exhibit significant resistance to cisplatin
and camptothecin. Havas et al (32) demonstrated that cells were more
sensitive to chemotherapeutic drugs following polyploidization. The
present study revealed that polyploid tumor cells induced by
spindle poisons were less sensitive to paclitaxel, docetaxel,
vincristine, oxaliplatin, 5-FU and epirubicin than the original
tumor cells. In addition, the results indicated that induced
polyploid cells are relatively resistant to the majority of
commonly used chemotherapeutic drugs and relatively sensitive to
the topoisomerase II inhibitor etoposide, which not only validates
the genetic instability of polyploid cells but also suggests that
induced polyploid cells are likely to be a key problem in drug
resistance during tumor treatment. It was hypothesized that there
may be an especial mechanism underlying topoisomerase II
inhibitors, for example, etoposide for polyploid breast cancer,
which is different with other drugs such as paclitaxel and
docetaxel. Therefore, the present study further investigated the
specific molecular mechanism underlying polyploid cell
resistance.
Nocodazole did not produce polyploidy in HCC1806
cells. MDA-MB-231 and HCC1806 cells have distinct p53 mutations
that occur in response to spindle poisons, and both have complete
spindle assembly checkpoints (33).
MDA-MB-231 cells are characterized by polypoloid formation, and
HCC1806 cells are characterized by apoptosis, meaning that their
differential responses to nocodazole may be associated with
inhibition of the apoptotic pathway. In order to further
investigate whether the apoptotic pathway is involved in the
formation of polyploid tumor cells induced by nocodazole,
expression of the apoptotic pathway proteins Bcl-2 and Bax in
nocodazole-induced polyploid tumors cells was investigated using
two strains treated with the drug. Human breast cancer cells with
different responses were studied: MDA-MB-231 cells were
characterized by the formation of polyploid cells, and HCC1806
cells were characterized by apoptosis. Whether key proteins of the
apoptosis pathway, Bcl-2 and Bax, were involved in the mechanism of
polyploid tumor formation was preliminarily investigated.
The flow cytometry analysis revealed that the
percentage of HCC1806 cells in the G2/M phase increased
following nocodazole treatment for 6 h, and the subdiploid peak
(sub-G1 phase), also called the ‘apoptosis peak’, was
increased at 36 and 48 h compared with 0 h. Changes in the
percentages of MDA-MB-231 cells in the G2/M phase and
sub-G1 phase following nocodazole treatment for 6, 12
and 24 h were the same as those of the HCC1806 cells. However, as
the cells were incubated with the drug for a prolonged period of
time, the two human breast cancer cells exhibited different
outcomes: HCC1806 cells exited mitosis and activated apoptosis,
which then occurred, while MDA-MB-231 cells re-entered mitosis in
the absence of a nucleus, with the next cell cycle forming
tetraploids. Therefore, it can be suggested that the two breast
cancer cells have a perfect spindle monitoring point, and the
effect of nocodazole on the cell cycle of MDA-MB-231 and HCC180
cells is noteworthy. MDA-MB-231 cells may be inhibiting the
apoptotic pathway in order to enter the next cell cycle to form
polyploids; thus, cell cycle regulation abnormalities may allow the
cells to escape the restriction point ‘lock’, a mitotic slip that
leads to polyploidy (tetraploid production).
Inhibition of the apoptotic pathway is one of the
major causes of polyploid tumor cell formation; the Bax-dependent
mitochondrial membrane permeabilization pathway may play an
important role in preventing polyploid cell formation, particularly
in polyploid p53 mutants (34).
Variations in p53 mutations in different cell lines are
particularly important (35). In the
present study, MDA-MB-231 and HCC1806 cells were selected as the
p53 mutants, and both cell lines had different responses to the
spindle poisons, despite both having had perfect spindle assembly
checkpoint functions. Western blotting was used to detect
associated proteins in the two cells. The expression of caspase-9
and caspase-3 was examined; both were observed in HCC1806 cells 24
h after drug treatment. Bcl-2 was also expressed in HCC1806 cells;
after 24 h, its expression increased. However, MDA-MB-231 cells
were primarily polyploidized following incubation with nocodazole.
The western blot analysis revealed that the overexpression of Bcl-2
with no expression of Bax, caspase-9 and caspase-3. From these
results, it can be suggested that nocodazole induces the mechanism
by which MDA-MB-231 cells form polyploid tumor cells. Bcl-2, which
is highly expressed in MDA-MB-231 cells, could possibly bind to
another Bcl-2 to form a homodimer. The pro-apoptotic effect of the
Bcl-2/Bax heterodimer would be counteracted, thereby preventing the
apoptotic effect of Bcl-2 and causing the formation of polyploid
tumor cells.
In the present study, the mRNA expression levels of
Bcl-2 were detected in MDA-MB-231 parental cells and T-MDA-MB-231
polyploid cells. The results revealed that the mRNA expression of
Bcl-2 in T-MDA-MB-231 polyploid cells was significantly increased,
suggesting that overexpression of Bcl-2 is likely to be the key to
polyploid cell resistance. siRNAs were also used to inhibit the
expression of Bcl-2 mRNA in T-MDA-MB-231 polyploid cells in the
present study. When Bcl-2 mRNA expression was downregulated, the
IC50 of docetaxel for T-MDA-MB-231 polyploid cells was
significantly decreased, and more polyploid cells were apoptotic,
which significantly restored the sensitivity of polyploid cells to
this chemotherapy drug. These results suggest that the resistance
of polyploid tumors to certain chemotherapeutic drugs, such as
docetaxel, may be significantly associated with high expression
levels of Bcl-2. However, it was also observed that downregulating
the expression of Bcl-2 mRNA in T-MDA-MB-231 polyploid cells caused
them to be more resistant to etoposide. The IC50 values
of the chemotherapy drugs evaluated revealed no significant changes
for MDA-MB-231 cells when the expression of Bcl-2 mRNA was
downregulated in paclitaxel. The mechanism by which polyploids
cause tumor resistance is complex and may involve a number of genes
and their interactions. There may be different roles that Bcl-2
plays in different types of breast cancer, which further research
will elucidate.
Studies have reported that MCL-1, an important anti-
apoptotic protein in the Bcl-2 family, is a key regulator of
apoptosis that is induced by spindle poisons (16,36). The
results of the present study also revealed that expression of the
FBW7 protein was decreased, and that of the MCL-1 protein was
significantly increased following treatment with nocodazole for 48
h, suggesting they may affect the formation of polyploidy in
MDA-MB-231 cells. FBW7 degrades the downstream oncogenic protein
MCL-1 by recognizing phosphorylation groups, promoting apoptosis
and exerting anticancer effects (37–38).
Therefore, it could be speculated that the lack of FBW7 due to low
expression may cause decreased ubiquitination and degradation of
MCL-1, resulting in the high levels of MCL-1 in MDA-MB-231cells,
which may play a key role in the formation of polyploid tumors.
MCL-1 is a potential target for reversing polyploid resistance. In
the present study, it was observed that the polyploid and
proliferation of MDA-MB-231 cells was decreased following treatment
with the MCL-1 inhibitor sorafenib in, indicating the key role of
the Bcl-2 family in breast cancer polyploidy.
The present study revealed that Bcl-2 protein likely
inhibits apoptosis induced by spindle poisons in breast cancer
MDA-MB-231 cells, which leads to polyploid cell tumor formation.
Furthermore, polyploid tumor cell formation and tumor resistance
are multifactorial, with numerous important molecular mechanisms.
The present study investigated the molecular mechanisms underlying
polyploidy and the association between polyploidy and tumor
resistance in an attempt to ascertain potential interventional
methods, which may lead to an improved understanding of polyploid
tumor cell formation.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant nos. 31501159 and 30872969), the
Tianjin Public Health Key Research Project (grant no. 15KG108), the
Tianjin Science and Technology Key Project on Chronic Disease
Prevention and Treatment (grant no. 16ZXMJSY00020), the Special
Program of Talent Development for Excellent Youth Scholars in
Tianjin (grant no. TJTZJH-QNBJRC-2-9) and the Tianjin 131 Creative
Talents Cultivation Project (1st Class, 2016), the Science and
Technology Development Fund of Bengbu Medical College (grant no.
BYKF1783) and The Fund of Tianjin People's Hospital (grant no.
2017YJ026).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
BY and YZ contributed to the study conception and
design. YZ wrote and revised the manuscript. JH, QZ and YW were
responsible for the acquisition and analysis of data. All authors
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fajka-Boja R, Marton A, Tóth A, Blazsó P,
Tubak V, Bálint B, Nagy I, Hegedűs Z, Vizler C and Katona RL:
Increased insulin-like growth factor 1 production by polyploid
adipose stem cells promotes growth of breast cancer cells. BMC
Cancer. 18:8722018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yasuhira S, Shibazaki M, Nishiya M and
Maesawa C: Paclitaxel-induced aberrant mitosis and mitotic slippage
efficiently lead to proliferative death irrespective of canonical
apoptosis and p53. Cell Cycle. 15:3268–3277. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stojanovic N, Dekanic A, Paradzik M,
Majhen D, Ferencak K, Ruscic J, Bardak I, Supina C, Tomicic MT,
Christmann M, et al: Differential effects of integrin αv knockdown
and cilengitide on sensitisation of triple-negative breast cancer
and melanoma cells to microtubule poisons. Mol Pharmacol.
94:1334–1351. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang W, Xu J, Ji D, Li Z, He W, Yang F,
Lan H, Wang Y, Wu Z, Liu X, et al: CyclinG1 amplification enhances
aurora kinase inhibitor-induced polyploid resistance and inhibition
of Bcl-2 pathway reverses the resistance. Cell Physiol Biochem.
43:94–107. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharma S, Zeng JY, Zhuang CM, Zhou YQ, Yao
HP, Hu X, Zhang R and Wang MH: Small-molecule inhibitor BMS-777607
induces breast cancer cell polyploidy with increased resistance to
cytotoxic chemotherapy agents. Mol Cancer Ther. 12:725–736. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gerashchenko BI, Salmina K, Eglitis J,
Huna A, Grjunberga V and Erenpreisa J: Disentangling the aneuploidy
and senescence paradoxes: A study of triploid breast cancers
non-responsive to neoadjuvant therapy. Histochem Cell Biol.
145:497–508. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han J, Wang QC, Zhu CC, Liu J, Zhang Y,
Cui XS, Kim NH and Sun SC: Deoxynivalenol exposure induces
autophagy/apoptosis and epigenetic modification changes during
porcine oocyte maturation. Toxicol Appl Pharmacol. 300:70–76. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li P, Zhou L, Liu X, Jin X, Zhao T, Ye F,
Liu X, Hirayama R and Li Q: Mitotic DNA damages induced by
carbon-ion radiation incur additional chromosomal breaks in
polyploidy. Toxicol Lett. 230:36–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nano M, Gemble S, Simon A, Pennetier C,
Fraisier V, Marthiens V and Basto R: Cell-cycle asynchrony
generates DNA damage at mitotic entry in polyploid cells. Curr
Biol. 29:3937–3945.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hussain S, Singh A, Nazir SU, Tulsyan S,
Khan A, Kumar R, Bashir N, Tanwar P and Mehrotra R: Cancer drug
resistance: A fleet to conquer. J Cell Biochem. 120:14213–14225.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Müller A, Gillissen B, Richter A, Richter
A, Chumduri C, Daniel PT and Scholz CW: Pan-class I PI3-kinase
inhibitor BKM120 induces MEK1/2-dependent mitotic catastrophe in
non-Hodgkin lymphoma leading to apoptosis or polyploidy determined
by Bax/Bak and p53. Cell Death Dis. 9:3842018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mittal K, Donthamsetty S, Kaur R, Yang C,
Gupta MV, Reid MD, Choi DH, Rida PCG and Aneja R: Multinucleated
polyploidy drives resistance to Docetaxel chemotherapy in prostate
cancer. Br J Cancer. 116:1186–1194. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hao J, Yuan BB, Xu YF, Yu J, Liu GY and
Wang DH: Sensitivity to chemotherapeutic drugs of polyploid tumor
cells induced by a spindle poison nocodazole. Zhonghua Zhong Liu Za
Zhi. 34:419–424. 2012.(In Chinese). PubMed/NCBI
|
|
15
|
Inuzuka H, Shaik S, Onoyama I, Gao D,
Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al:
SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for
ubiquitylation and destruction. Nature. 471:104–109. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Srivastava S and Panda D: A centrosomal
protein STARD9 promotes microtubule stability and regulates spindle
microtubule dynamics. Cell Cycle. 17:2052–2068. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chowdhury P, Nagesh PKB, Hatami E, Wagh S,
Dan N, Tripathi MK, Khan S, Hafeez BB, Meibohm B, Chauhan SC, et
al: Tannic acid-inspired paclitaxel nanoparticles for enhanced
anticancer effects in breast cancer cells. J Colloid Interface Sci.
535:133–148. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zeng F, Ju RJ, Liu L, Xie HJ, Mu LM and Lu
WL: Efficacy in treating lung metastasis of invasive breast cancer
with functional vincristine plus dasatinib liposomes. Pharmacology.
101:43–53. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bosco B, Defant A, Messina A, Incitti T,
Sighel D, Bozza A, Ciribilli Y, Inga A, Casarosa S and Mancini I:
Synthesis of 2,6-diamino-substituted purine derivatives and
evaluation of cell cycle arrest in breast and colorectal cancer
cells. Molecules. 23:E19962018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jemaà M, Galluzzi L, Kepp O, Senovilla L,
Brands M, Boemer U, Koppitz M, Lienau P, Prechtl S, Schulze V, et
al: Characterization of novel MPS1 inhibitors with preclinical
anticancer activity. Cell Death Differ. 20:1532–1545. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krisenko MO, Cartagena A, Raman A and
Geahlen RL: Nanomechanical property maps of breast cancer cells as
determined by multiharmonic atomic force microscopy reveal
Syk-dependent changes in microtubule stability mediated by MAP1B.
Biochemistry. 54:60–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choi HJ and Zhu BT: Role of cyclin B1/Cdc2
in mediating Bcl-XL phosphorylation and apoptotic cell death
following nocodazole-induced mitotic arrest. Mol Carcinog.
53:125–137. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Z, Chen X, Zhong MZ, Yang S, Zhou J,
Klinkebiel DL, Karpf AR, Chen Y and Dong J: Cyclin-dependent kinase
1-mediated phosphorylation of YES links mitotic arrest and
apoptosis during antitubulin chemotherapy. Cell Signal. 52:137–146.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hsu CT, Huang YF, Hsieh CP, Wu CC and Shen
TS: JNK inactivation induces polyploidy and drug-resistance in
coronarin D-treated osteosarcoma cells. Molecules. 23:E21212018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Horiuchi M, Kuga T, Saito Y, Nagano M,
Adachi J, Tomonaga T, Yamaguchi N and Nakayama Y: The tyrosine
kinase v-Src causes mitotic slippage by phosphorylating an
inhibitory tyrosine residue of Cdk1. J Biol Chem. 293:15524–15537.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vitale I, Manic G, Castedo M and Kroemer
G: Caspase 2 in mitotic catastrophe: The terminator of aneuploid
and tetraploid cells. Mol Cell Oncol. 4:e12992742017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou W, Xu J, Gelston E, Wu X, Zou Z, Wang
B, Zeng Y, Wang H, Liu A, Xu L and Liu Q: Inhibition of Bcl-xL
overcomes polyploidy resistance and leads to apoptotic cell death
in acute myeloid leukemia cells. Oncotarget. 6:21557–21571.
2015.PubMed/NCBI
|
|
28
|
Romanova LY, Mushinski F and Kovalchuk AL:
Transcriptional activation of p21Waf1 contributes to suppression of
HR by p53 in response to replication arrest induced by
camptothecin. Oncotarget. 9:25427–25440. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hsu CY, Lecland N, Pendaries V, Viodé C,
Redoulès D, Paul C, Merdes A, Simon M and Bierkamp C: Stabilization
of microtubules restores barrier function after cytokine-induced
defects in reconstructed human epidermis. J Dermatol Sci. 91:87–96.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dinić J, Ríos-Luci C, Karpaviciene I,
Cikotiene I, Fernandes MX, Pešić M and Padrón JM: CKT0353, a novel
microtubule targeting agent, overcomes paclitaxel induced
resistance in cancer cells. Invest New Drugs. Jun 8–2019.(Epub
ahead of print). View Article : Google Scholar
|
|
31
|
Castedo M, Coquelle A, Vivet S, Vitale I,
Kauffmann A, Dessen P, Pequignot MO, Casares N, Valent A, Mouhamad
S, et al: Apoptosis regulation in tetraploid cancer cells. EMBO J.
25:2584–2595. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Havas AP, Rodrigues KB, Bhakta A,
Demirjian JA, Hahn S, Tran J, Scavello M, Tula-Sanchez AA, Zeng Y,
Schmelz M and Smith CL: Belinostat and vincristine demonstrate
mutually synergistic cytotoxicity associated with mitotic arrest
and inhibition of polyploidy in a preclinical model of aggressive
diffuse large B cell lymphoma. Cancer Biol Ther. 17:1240–1252.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ferraz da Costa DC, Campos NPC, Santos RA,
Guedes-da-Silva FH, Martins-Dinis MMDC, Zanphorlin L, Ramos C,
Rangel LP and Silva JL: Resveratrol prevents p53 aggregation in
vitro and in breast cancer cells. Oncotarget. 9:29112–29122.
2018.PubMed/NCBI
|
|
34
|
Zou X, Qu M, Fang F, Fan Z, Chen L, Yue W,
Xie X and Pei X: Small molecule supplements improve cultured
megakaryocyte polyploidization by modulating multiple cell cycle
regulators. Biomed Res Int. 2017:23205192017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dabiri Y, Abu El Maaty MA, Chan HY, Wölker
J, Ott I, Wölfl S and Cheng X: p53-Dependent anti-proliferative and
pro-apoptotic effects of a gold(I) N-heterocyclic carbene (NHC)
complex in colorectal cancer cells. Front Oncol. 9:4382019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maity A, Majumdar S and Ghosh Dastidar S:
Flexibility enables to discriminate between ligands: Lessons from
structural ensembles of Bcl-xl and Mcl-1. Comput Biol Chem.
77:17–27. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tong J, Tan S, Nikolovska-Coleska Z, Yu J,
Zou F and Zhang L: FBW7-dependent Mcl-1 degradation mediates the
anticancer effect of Hsp90 inhibitors. Mol Cancer Ther.
16:1979–1988. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tong J, Tan S, Zou F, Yu J and Zhang L:
FBW7 mutations mediate resistance of colorectal cancer to targeted
therapies by blocking Mcl-1 degradation. Oncogene. 36:787–796.
2017. View Article : Google Scholar : PubMed/NCBI
|