Introduction
Renal cell carcinoma (RCC) is one of the top 10 most
common types of cancer worldwide, whereby its incidence increased
by 8.1% from 1975–2016 (1,2). RCC is primarily composed of three
subtypes: Clear cell RCC (ccRCC), papillary RCC and chromophobe
RCC. ccRCC accounts for 75–80% of these tumors and is the most
common RCC subtype with the highest degree of local invasion,
metastasis and mortality (3,4). At present, the primary treatment of
early stage, localized RCC is surgical resection. However, despite
tumor removal, 20–40% of patients still experience tumor recurrence
(5). It is generally well accepted
that renal cell carcinogenesis is the result of multiple factors
(6–8); however, a consensus in the field has
not yet been reached and the precise underlying molecular
mechanisms remain unclear.
Cell division cycle-associated protein 2 (CDCA2)
belongs to a class of cyclin-associated proteins (9,10).
Previous studies have demonstrated that CDCA2 can form a complex
with protein phosphatase 1 (PP1) γ and control the PP1γ-dependent
DNA damage response (DDR) (11,12).
Furthermore, CDCA2 promotes major mitotic histone H3
dephosphorylation in a PP1-dependent manner (13). CDCA2 is highly expressed in a number
of different types of tumors. Previous studies have demonstrated
that CDCA2 protein expression is associated with tumor volume and
Tumor-Node-Metastasis (TNM) stage of oral squamous cell carcinoma
(14–16). Furthermore, silencing the CDCA2 gene
can lead to cell cycle arrest, inhibition of cell proliferation and
apoptosis (16,17) However, the expression of CDCA2 and
its function in ccRCC remain unclear.
The present study demonstrated that CDCA2 expression
in ccRCC tissue is upregulated compared with normal healthy tissue.
Furthermore, silencing CDCA2 induced G1 arrest and
promoted apoptosis in 786-O and CAKI-1 cells. These results
indicated that CDCA2 regulates ccRCC carcinogenesis and may serve
as a potential therapeutic target for the disease.
Materials and methods
Bioinformatics analysis
The Cancer Genome Atlas (TCGA) dataset, TCGA kidney
Clear Cell Carcinoma (KIRC) (18),
was downloaded from the University of California Santa Cruz Xena
website (xena.ucsc.edu) and includes 534 ccRCC
cases and 72 normal controls (Table
SI). The tumor samples were matched to TNM stage and G stage
(19) in order to obtain data on
CDCA2 expression and clinical progression.
Cell culture and RNA transfection
The two human ccRCC cell lines (786-O and CAKI-1)
and a human tubular epithelial cell line (HK-2) were sourced from
the Key Laboratory of Environment and Genes Related to Diseases at
Xi'an Jiaotong University (Xi'an, China). 786-O and HK-2 cells were
cultured in RPMI-1640 medium (Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Biological Industries) and 1%
penicillin/streptomycin (PS). CAKI-1 cells were cultured in McCoy's
5a Modified Medium (Thermo Fisher Scientific, Inc.) supplemented
with 15% FBS and 1% PS. All cell lines were cultured in an
incubator with 5% CO2 at 37°C, until they reached 80%
confluence. Small interfering (si)RNA duplexes targeting human
CDCA2 were synthesized and purified by Shanghai GenePharma Co.,
Ltd. Non-specific siRNA sequences, purchased from Shanghai
GenePharma Co., Ltd., were used as a negative control. A total of
two siRNAs were used, and the sequences were as follows: CDCA2
siRNA-1; Forward, 5′-CACCUGCCUUUCUAAAUAUTT-3′ and reverse,
5′-AUAUUUAGAAAGGCAGGUGTT-3′; and CDCA2 siRNA-2; Forward,
5′GGGCAAAGGAUCAAGUGAUTT-3′ and reverse,
5′-AUCACUUGAUCCUUUGCCCTT-3′. The non-specific siRNA sequence was as
follows: Forward, 5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′. A total of 20 µM of siRNA was used,
and 3 µl was added to each well of the six-well plate. Transfection
of siRNAs was performed using jetPRIME reagent
(Polyplus-transfection SA), according to the manufacturer's
protocol.
RNA extraction, cDNA synthesis and
reverse transcription-quantitative (RT-q)PCR
Total RNA was extracted from transfected ccRCC cells
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol and
expression levels were quantified using a NanoDrop
spectrophotometer (Thermo Fisher Scientific, Inc.). Total RNA was
reverse transcribed into cDNA using the PrimeScript™ RT Reagent kit
(Takara Bio, Inc.), according to the manufacturer's protocol. qPCR
was subsequently performed using the iQ5 Optical real-time PCR
system (Bio-Rad Laboratories, Inc.) with SYBR Green Ex Taq™ II
(Takara Bio, Inc.). The following primer sequences were used for
the qPCR: CDCA2; Forward, 5′-ATGACCGGCTGTCTGGAAT-3′ and reverse,
5′-GCTGAGACCTTCCTTTCTGGT-3′ and GAPDH; Forward,
5′-TGAAGGTCGGAGTCAACGGATT-3′ and reverse,
5′-CCTGGAAGATGGTGATGGGATT-3′. The following thermocycling
conditions were used for the qPCR: Initial denaturation at 95°C for
10 min; 40 cycles of 95°C for 15 sec, annealing at 60°C for 60 sec
and extension at 72°C for 30 sec. CDCA2 mRNA levels were quantified
using the 2−ΔΔCq method (20) and normalized to the internal
reference gene GAPDH.
Cell proliferation assay
The effect of CDCA2 silencing on the proliferation
of 786-O and CAKI-1 cells was assessed using an MTT assay. Cells
were seeded into 96-well plates at 3,000 cells per 100 µl culture
media per well. Transfections were performed the following day. A
total of 10 µl MTT reagent (5 mg/ml) was added to every well at
various time points following transfection (24, 48 and 72 h), and
150 µl dimethyl sulfoxide was added 4 h later. The absorbance of
samples was measured at 490 nm using a high-throughput universal
micro plate reader.
Colony forming assays
ccRCC cells were seeded into 12-well plates at a
density of 5×104 cells/well and transfection was
performed the following day. 24 h post-transfection, cells were
reseeded into 6-well plates at a density of 1,000 cells/well in
triplicate and incubated at 37°C with 5% CO2 for 7–10
days until they reached 80% confluence. Cells were then fixed with
4% paraformaldehyde and stained with 0.5% crystal violet (Sigma
Aldrich; Merck KGaA) for 30 min at room temperature. Photos were
captured and colonies were counted using the Quantity
One® software (version 4.3.1; Bio-Rad Laboratories,
Inc.).
Cell cycle assay
786-O and CAKI-1 cells were seeded into 6-well
plates at a density of 1.5×105 cells/well in triplicate
and transfected 24 h later. Cells were then trypsinized 24 h
post-transfection, washed with cold PBS twice, and fixed in
ice-cold 75% alcohol at 4°C overnight. Fixed cells were washed with
PBS and then resuspended in 150 µl RNase A (0.1 mg/ml) and 150 µl
propidium iodide (PI; 0.05 mg/ml) for 30 min at room temperature.
Cell cycle distributions were measured using a flow cytometer.
Cell apoptosis analysis
In order to determine the effects of CDCA2 on ccRCC
cell apoptosis, Annexin V-FITC Apoptosis Detection kits (7Sea
PharmTech Shanghai, China) were used according to the
manufacturer's protocol. Cells were seeded into 12-well plates at a
density of 5×104 cells/well and transfected 24 h later.
Cells were then trypsinized 48 h post-transfection and stained with
5 µl of FITC Annexin V for 15 min at room temperature in the dark,
prior to incubating with 10 µl of PI on ice for 5 min in the dark.
Cell apoptosis was measured using a flow cytometer and the
percentage of apoptotic cells was calculated using ModFit software
(version 3.3.11; Verity Software House, Inc.). Cells stained with
Annexin V-FITC were considered to be early apoptotic cells, and
Annexin V-FITC and PI double stained cells were considered to be
late apoptotic cells.
Western blot analysis
Total protein was extracted from ccRCC cells 48 h
post- transfection using radioimmunoprecipitation assay buffer
(http://www.xfbio.com) supplemented with protease
inhibitor cocktail (100X) and 1 mM phenylmethylsulfonyl protease
inhibitor (both from MedChemExpress). Protein concentrations were
determined using a BCA protein assay kit (Takara Bio, Inc.) and 20
µg protein/lane was separated via SDS-PAGE on a 7.5–12.5% gel. The
separated proteins were subsequently transferred onto a
methanol-activated polyvinylidene membrane (EMD Millipore) and
blocked with 5% non-fat milk in Tris-buffered saline (pH 7.4)
containing 0.1% Tween, for 1 h at room temperature. The membranes
were incubated with primary antibodies against CDCA2 (cat. no.
14976), BAX (cat. no. 2774), Bcl-2 (cat. no. 15071), cyclin
dependent kinase 4 (CDK4; cat. no. 12790) and cyclin D1 (cat. no.
2922) (all 1:1,000; all from Cell Signaling Technology, Inc.)
overnight at 4°C. Following the primary incubation, membranes were
incubated with horseradish peroxidase-labeled secondary antibodies
(cat. no. 7076 and cat. no. 7074; all 1:5,000; all from Cell
Signaling Technology, Inc.) for 1 h at room temperature. Protein
bands were visualized using the chemiluminescence detection Syngene
GBox (Syngene Europe). The optical density of the image was
analyzed using ImageJ software (version 1.4.3.67; National
Institutes of Health) and protein levels were normalized to β-actin
(1:5,000, cat. no. ab822, Abcam).
Immunohistochemistry
ccRCC tissues were fixed in 4% paraformaldehyde for
24 h at room temperature and embedded in paraffin.
Paraffin-embedded samples were cut into 4-µm thick sections. Sample
information is presented in Table
SII. Sections were deparaffinized in xylene and rehydrated in a
descending ethanol series at room temperature. Deparaffinized
sections were blocked with 10% goat serum working solution and
incubated with 50 µl endogenous peroxidase inhibitor (both from
OriGene Technologies, Inc.), according to the manufacturer's
protocol, both at room temperature for 30 min. Antigen retrieval
and blocking was subsequently performed. Tissue sections were
incubated with primary antibody directed against CDCA2 (1:100; cat.
no. 14976; Cell Signaling Technology, Inc.) overnight at 4°C,
followed by incubation with 100 µl of horseradish
peroxidase-labeled secondary antibodies (cat. no. SP-9001; OriGene
Technologies, Inc.) for 15 min at room temperature. Chromogenic
development was performed using 3,3′-diaminobenzidine and
hematoxylin staining for 15 sec at room temperature. Positive
staining was analyzed by measuring the gray pixels using Image-pro
Plus (version 6.0; Media Cybernetics, Inc.).
Statistical analysis
All statistical analyses were performed using SPSS
software (version 22.0). All experiments were performed in
triplicate. Unpaired Student's t-test and one-way ANOVA analysis
followed by Dunnett's post-hoc test were performed for multiple
comparison between the groups. Data are presented as the mean ±
standard error of the mean. P<0.05 was considered to indicate a
statistically significant difference.
Results
CDCA2 is upregulated in ccRCC and is
associated with clinical stage in TCGA dataset
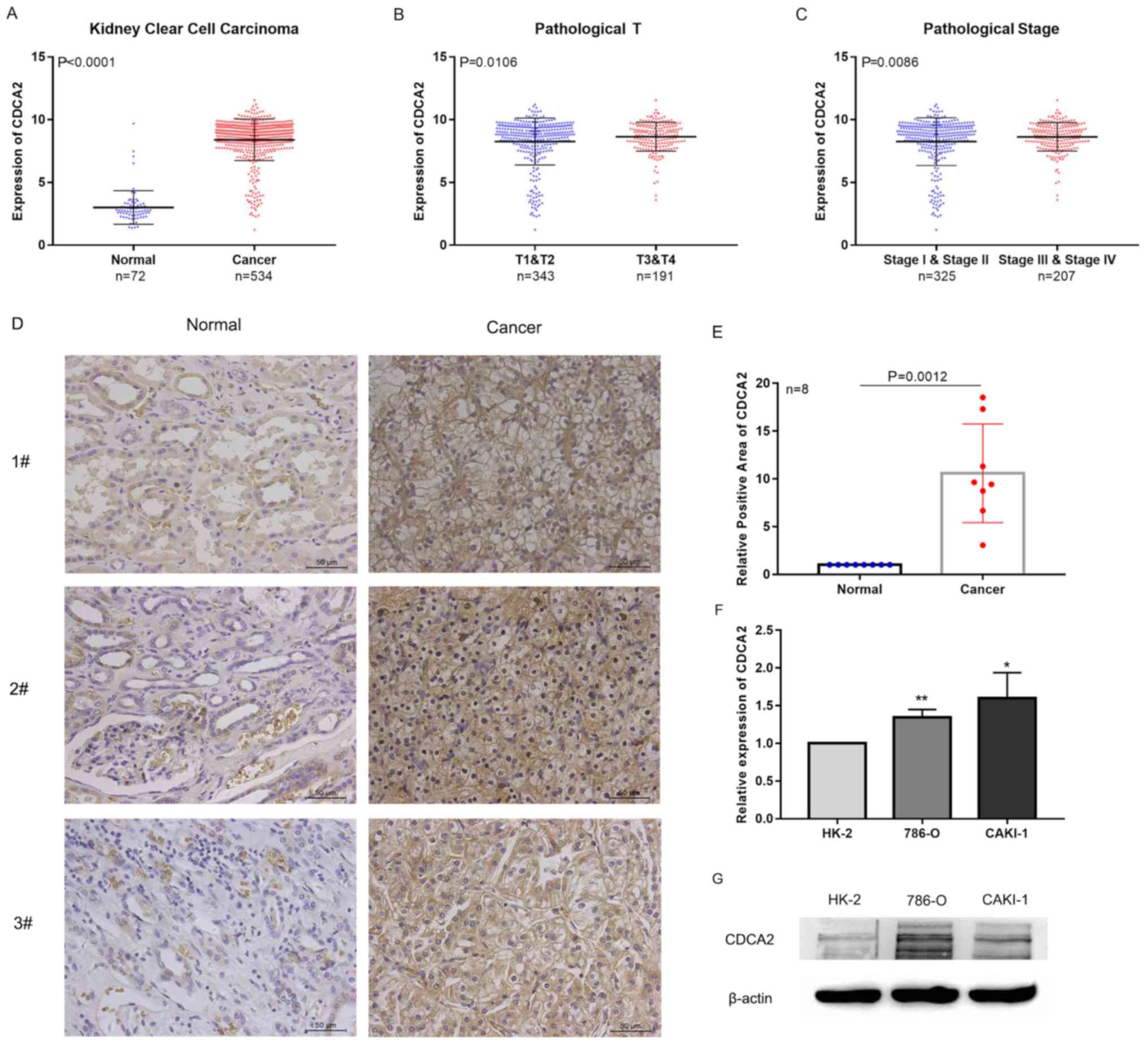
Tumor samples had significantly higher CDCA2
expression compared with normal samples (P<0.0001) in the TCGA
dataset (Fig. 1A). The present study
then assessed CDCA2 protein levels in ccRCC paired tissue samples
(n=8) using immunohistochemistry. As presented in Fig. 1D and E, positive staining of CDCA2
was higher in ccRCC tissue than normal, suggesting that CDCA2
protein expression is upregulated in ccRCC tissues compared with
normal tissue controls (P=0.0012). In order to assess the
expression of CDCA2 in the ccRCC cell lines 786-O and CAKI-1, the
present study used RT-qPCR and western blotting. CDCA2 was highly
expressed in 786-O and CAKI-1 cells compared with the normal renal
epithelial cell line HK-2 (both P<0.05; Fig. 1F and G).
The present study then assessed whether CDCA2
expression was associated with the clinical stage of ccRCC tumors.
The statistical analysis indicated that CDCA2 expression was
increased in tumors that had a high degree of malignancy compared
with samples with a lower degree of malignancy (both P<0.05;
Fig. 1B and C). These data suggest
that CDCA2 may be associated with tumor cell proliferation.
CDCA2 knockdown inhibits cell
viability and proliferation in 786-O and CAKI-1 cells
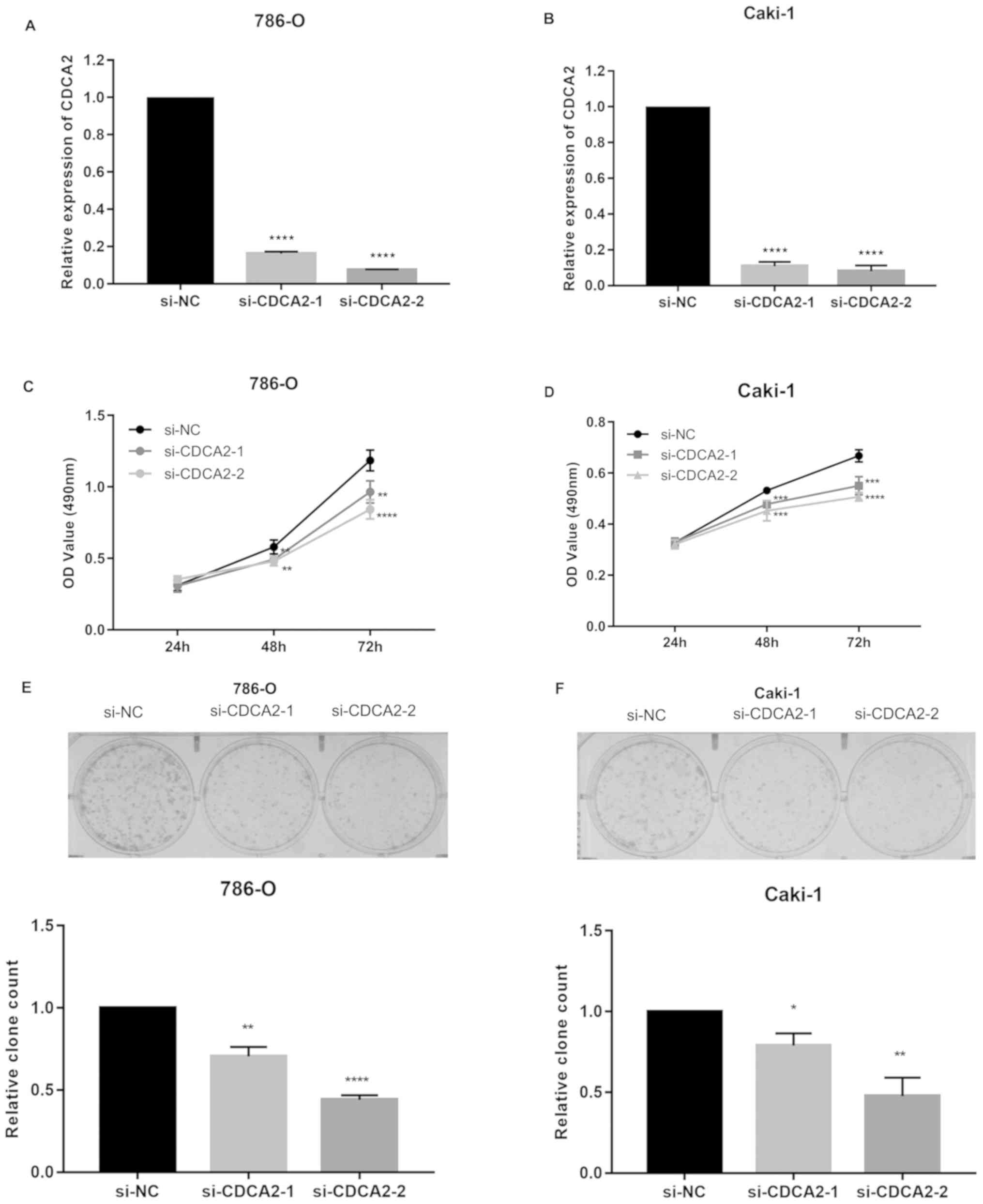
In order to investigate the effect of CDCA2 on ccRCC
cell proliferation, the present study designed siRNAs that target
human CDCA2. It was demonstrated that these CDCA2 siRNAs decreased
CDCA2 mRNA levels in 786-O and CAKI-1 cells by >60% (both
P<0.0001; Fig. 2A and B). MTT
assays were used to measure the effect of CDCA2 on ccRCC cell
viability. CDCA2 knockdown significantly inhibited ccRCC cell
viability compared to the control group (both P<0.05; Fig. 2C and D). In order to further
investigate the effect of CDCA2 on ccRCC cell proliferation, a
colony formation assay was performed. Fewer and smaller colonies
were observed in cells with CDCA2 knockdown compared with control
cells (both P<0.05; Fig. 2E and
F). These data demonstrated that silencing of CDCA2 inhibits
the growth of ccRCC cells, and that CDCA2 promotes ccRCC cell
viability and proliferation.
CDCA2 knockdown inhibits the
expression of cell cycle proteins and promotes G1 arrest
in ccRCC cells
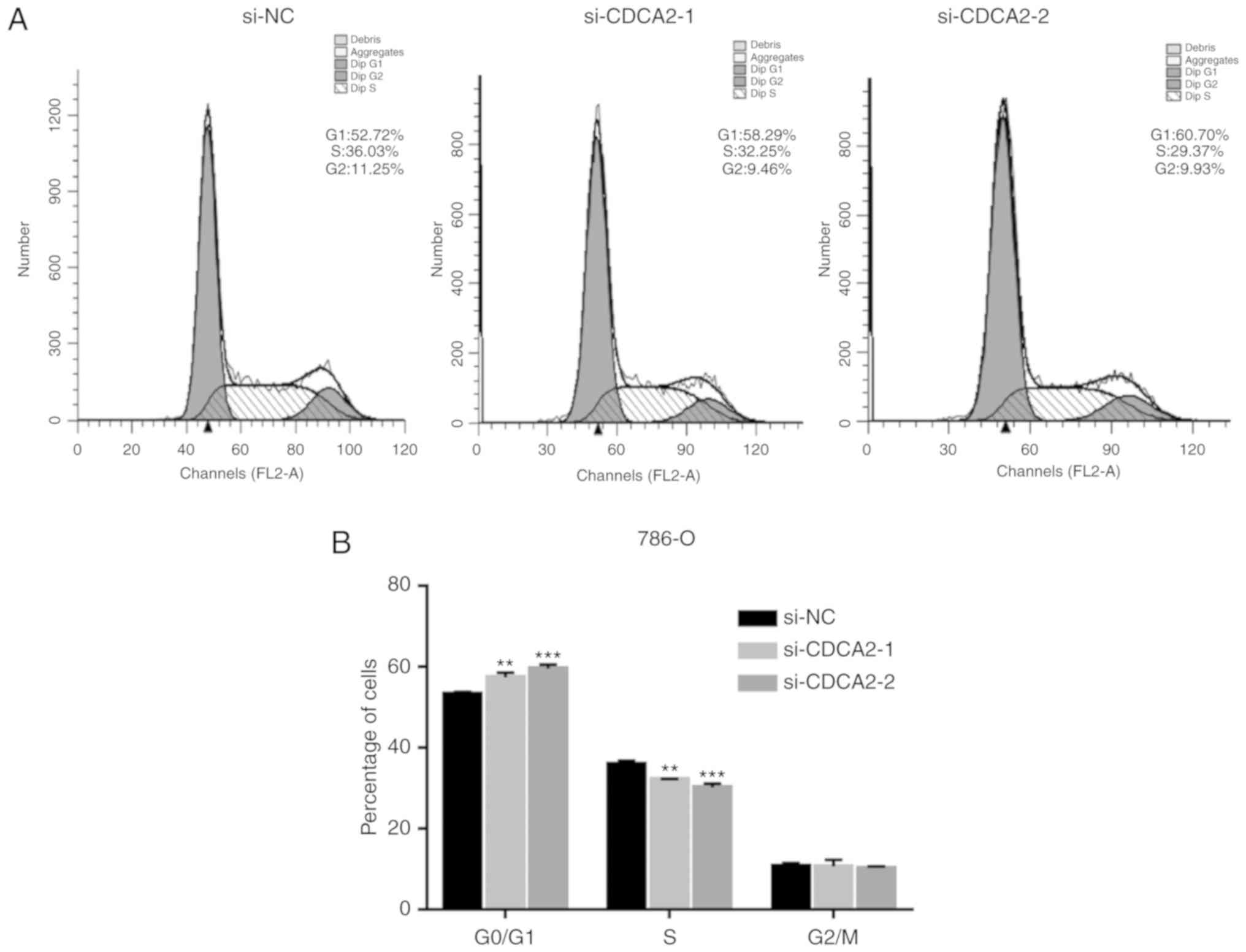
In order to determine whether cell cycle arrest
drove the inhibition of cell proliferation that was observed with
CDCA2 knockdown, flow cytometry was used to analyze cell cycle
distribution 24 h post-transfection. Compared with controls, the
percentage of siCDCA2 transfected 786-O and CAKI-1 cells in the
G1 phase increased, while the percentage of cells in S
phase decreased (Fig. 3).
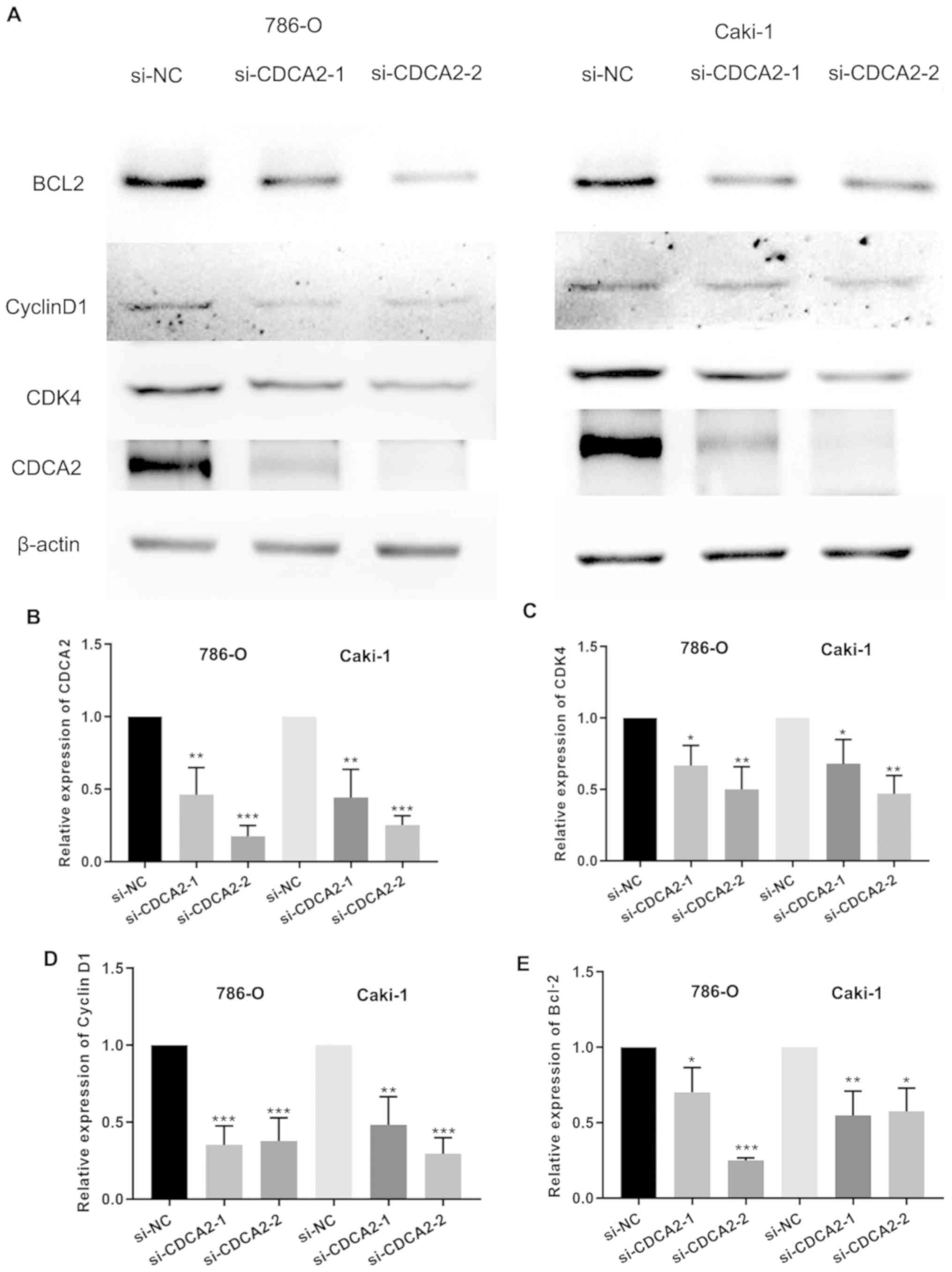
Furthermore, CDK4 and cyclin D1 expression was significantly
decreased in ccRCC cells transfected with siCDCA2 (both P<0.05;
Fig. 4A, C and D), and the silencing
efficiency of siCDCA2 was significant (P<0.05; Fig. 4B). These results demonstrated that
silencing CDCA2 inhibits the expression of cell cycle proteins in
ccRCC cells, causing G1 arrest.
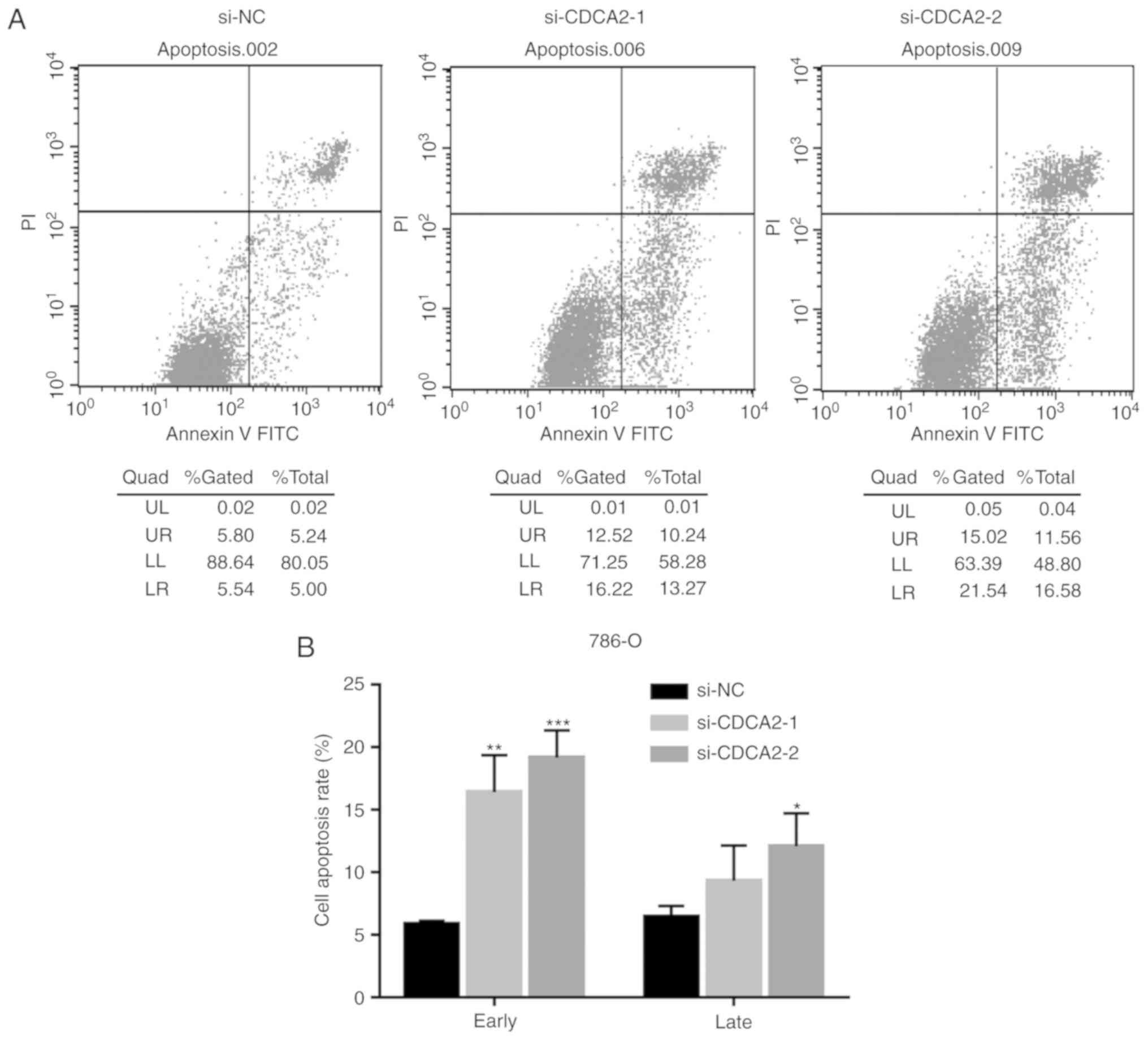
CDCA2 knockdown induces apoptosis of
ccRCC cells
Dysfunction in apoptosis caused by the dysregulation
of apoptosis-associated proteins plays an important role in the
development of cancer. An apoptosis assay and western blot analysis
were performed in order to determine whether CDCA2 affects
apoptosis in ccRCC cells. CDCA2 knockdown increased the proportion
of 786-O and CAKI-1 apoptotic cells (Fig. 5). Furthermore, B-cell lymphoma 2
(Bcl-2) expression significantly decreased in ccRCC cells
transfected with siCDCA2 (Fig. 4A and
E). These results revealed that silencing CDCA2 induces
apoptosis in ccRCC cells.
Discussion
Trinkle-Mulcahy et al (21) first identified CDCA2 as a binding
protein for PP1. Peng et al (12) reported that CDCA2 inhibits the
activation of Ataxia-telangiectasia mutated-dependent signaling by
promoting the binding of PP1c to chromatin. Peng et al
(12) also demonstrated that CDCA2
upregulation during cancer progression enhances CDCA2-dependent DDR
regulation, resulting in decreased DDR sensitivity. DNA damage
delays cell cycle entry by affecting cell cycle checkpoints,
causing cell cycle arrest at specific stages (22,23).
Genomic stability is maintained by offsetting DNA damage through a
series of pathways such as DNA repair, damage tolerance and
checkpoint pathways. DDR defects can lead to apoptosis, genomic
instability, dysregulation of cells and an increased risk of cancer
(24,25). The aforementioned studies indicate
that CDCA2 plays an important role in cell cycle progression and
apoptosis. Studies have reported that CDCA2 is upregulated in
neuroblastoma, melanoma and oral squamous cell carcinoma (15,16,18);
however, to the best of our knowledge, the expression and function
of CDCA2 in ccRCC has not been previously reported. The present
study demonstrated that CDCA2 is widely upregulated in ccRCC, and
the experiments in ccRCC cell lines revealed that CDCA2 knockdown
can significantly inhibit cell proliferation by promoting
G1 phase arrest and apoptosis. This is consistent with
previous findings in lung adenocarcinoma and oral squamous cell
carcinoma (16,18). Since CDCA2 knockdown can cause
G1 arrest in ccRCC cells, the present study assessed
changes in cyclin D1 and CDK4 protein levels, key downstream
regulators of the G1 to S transition. CDK4 and cyclin D1
expression levels were demonstrated to be decreased in 786-O and
CAKI-1 cells with CDCA2 knockdown. Similarly, it was observed that
silencing of CDCA2 significantly downregulated the
apoptosis-associated protein Bcl-2 in 786-O and CAKI-1 cells,
consistent with the results of the apoptosis assays.
Overall, the results of the present study
demonstrated that CDCA2 is upregulated in ccRCC, and knockdown of
CDCA2 promotes G1 arrest by inhibiting the expression of
CDK4 and cyclin D1. In addition, CDCA2 knockdown promoted apoptosis
by inhibiting Bcl-2 expression. This indicates that CDCA2 is
involved in the proliferation of human ccRCC cells and may play an
important role in the progression of the disease. The present study
investigated the role of CDCA2 in ccRCC development; however, its
underlying molecular mechanisms remain unclear. Future studies are
required on CDCA2 regulation of ccRCC and further research of its
targeted drugs, in order to improve the treatment of ccRCC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by The Scientific
Research and Sharing Platform Construction Project of Shaanxi
Province (grant no. 2018PT-09).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
CH designed the present study. YW, ZW and XW
collected the cancer tissues and interpreted the bioinformatics
data. FL, HZ, QL and FW performed the experiments. CH and FL
interpreted the data. FL and HZ drafted the initial manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
SEER Cancer Stat Facts: Kidney and Renal
Pelvis Cancer. National Cancer Institute; Bethesda, MD: https://seer.cancer.gov/statfacts/html/kidrp.htmlNovember
8–2019
|
|
3
|
Yan BC, Mackinnon AC and Al-Ahmadie HA:
Recent developments in the pathology of renal tumors: Morphology
and molecular characteristics of select entities. Arch Pathol Lab
Med. 133:1026–1032. 2009.PubMed/NCBI
|
|
4
|
Protzel C, Maruschke M and Hakenberg OW:
Epidemiology, aetiology, and pathogenesis of renal cell carcinoma.
Eur Urol. (Suppl 11):52–59. 2012. View Article : Google Scholar
|
|
5
|
Posadas EM, Limvorasak S and Figlin RA:
Targeted therapies for renal cell carcinoma. Nat Rev Nephrol.
13:496–511. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chow WH, Dong LM and Devesa SS:
Epidemiology and risk factors for kidney cancer. Nat Rev Urol.
7:245–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hofmann JN, Corley DA, Zhao WK, Colt JS,
Shuch B, Chow WH and Purdue MP: Chronic kidney disease and risk of
renal cell carcinoma: Differences by race. Epidemiology. 26:59–67.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song JK, Luo H, Yin XH, Huang GL, Luo SY,
Lin Du R, Yuan DB, Zhang W and Zhu JG: Association between cadmium
exposure and renal cancer risk: A meta-analysis of observational
studies. Sci Rep. 5:179762015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wurzenberger C, Held M, Lampson MA, Poser
I, Hyman AA and Gerlich DW: Sds22 and Repo-Man stabilize chromosome
segregation by counteracting aurora B on anaphase kinetochores. J
Cell Biol. 198:173–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taylor CM, Wang Q, Rosa BA, Huang SC,
Powell K, Schedl T, Pearce EJ, Abubucker S and Mitreva M: Discovery
of anthelmintic drug targets and drugs using chokepoints in
nematode metabolic pathways. PLoS Pathog. 9:e10035052013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thadani R, Uhlmann F and Heeger S:
Condensin, chromatin crossbarring and chromosome condensation. Curr
Biol. 22:R1012–R1021. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peng A, Lewellyn AL, Schiemann WP and
Maller JL: Repo-man controld a protein phosphatase 1-dependent
threshold for DNA damage checkpoint activation. Curr Biol.
20:387–396. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qian J, Lesage B, Beullens M, Van Eynde A
and Bollen M: PP1/Repo-man dephosphorylates mitotic histone H3 at
T3 and regulates chromosomal aurora B targeting. Curr Biol.
21:766–773. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi R, Zhang C, Wu Y, Wang X, Sun Q, Sun
J, Xia W, Dong G, Wang A, Jiang F and Xu L: CDCA2 promotes lung
adenocarcinoma cell proliferation and predicts poor survival in
lung adenocarcinoma patients. Oncotarget. 8:19768–19779.
2017.PubMed/NCBI
|
|
15
|
Krasnoselsky AL, Whiteford CC, Wei JS,
Bilke S, Westermann F, Chen QR and Khan J: Altered expression of
cell cycle genes distinguishes aggressive neuroblastoma. Oncogene.
24:1533–1541. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Uchida F, Uzawa K, Kasamatsu A, Takatori
H, Sakamoto Y, Ogawara K, Shiiba M, Bukawa H and Tanzawa H:
Overexpression of CDCA2 in human squamous cell carcinoma:
Correlation with prevention of G1 phase arrest and apoptosis. PLoS
One. 8:e563812013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ryu B, Kim DS, Deluca AM and Alani RM:
Comprehensive expression profiling of tumor cell line identifies
molecular signatures of melanoma progression. PLoS One. 2:e5942007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goldman M, Craft B, Hastie M, Repečka K,
Kamath A, McDade F, Rogers D, Brooks AN, Zhu J and Haussler D: The
UCSC Xena platform for public and private cancer genomics data
visualization and interpretation bioRxiv 326470. doi:
https://doi.org/10.1101/326470.
|
|
19
|
Telloni SM: Tumor staging and grading: A
primer. Methods Mol Biol. 1606:1–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Trinkle-Mulcahy L, Andersen J, Lam YW,
Moorhead G, Mann M and Lamond AI: Repo-Man recruits PP1 gamma to
chromatin and is essential for cell viability. J Cell Biol.
172:679–692. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ward JF: Complexity of damage produced by
ionizing radiation. Cold Spring Harb Symp Quant Biol. 65:377–382.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Khanna KK and Jackson SP: DNA
double-strand breaks: Signaling, repair and the cancer connection.
Nat Genet. 27:247–254. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hoeijmaker JH: DNA damage, aging, and
cancer. N Engl J Mcd. 361:1475–1485. 2009. View Article : Google Scholar
|
|
25
|
Meyn RE, Munshi A, Haymach JV, Milas L and
Ang KK: Receptor signaling as a regulatory mechanism of DNA repair.
Radiother Oncol. 92:316–322. 2009. View Article : Google Scholar : PubMed/NCBI
|