Introduction
Breast cancer (BC) is the most common cancer in
women worldwide with an estimated 2.4 million cases in 2015
(1). Standard treatments of patients
with BC include loco-regional therapy (surgery and radiation),
hormonal therapy, chemotherapy and molecular targeted therapy. The
afore-mentioned treatment regimens have existed for a number of
years, however, drug resistance and cancer recurrence still pose a
problem for the treatment of patients with BC (2,3). A
subpopulation of cells in tumors exhibit stem cell properties,
including dormancy, self-renewal, infinite proliferation and
multi-lineage differentiation (4,5). Studies
in various types of cancer, in particular BC, have demonstrated
that cancer stem cells (CSCs) subpopulations play a role in cancer
recurrence, therapy resistance, metastasis and aggressive relapse
(6–8). Therefore, CSCs are becoming important
targets for cancer therapy (8,9). In a
number of studies investigating BC, distinct cell subpopulations
with high tumorigenicity and stem cell properties have been
isolated (10,11). A tumorigenic subpopulation of BC
cells that express CD44 with low or negative CD24 expression
(CD44+/CD24−/low) has been identified as a
major breast CSC population (12).
Dendritic cell (DC)-based treatment has been widely
studied over the past two decades (13–15).
Most of the clinical trials of DC-based treatment in melanoma,
prostate cancer, malignant glioma and renal cell carcinoma
demonstrated an increase in median overall survival with minimal
toxicity (16). In a previous study,
human breast CSCs were injected into NOD/SCID mouse mammary fat
pads and used to prime human DCs; after mature DCs were re-injected
into mice, they exhibited longer survival times and lower tumor
masses compared with mice that had not received DC treatment
(17). These studies suggested that
tumor antigen-activated DCs may be a promising therapy for BC and
that CSC antigens may be used for the production of efficacious
DC-based treatment in treating refractory human cancers in
general.
To the best of our knowledge, no study has evaluated
DC-based therapy using human breast CSC antigens to activate
effector T cells for cancer cell killing to date. Thus, the aim of
this study was to explore whether CSC antigens may evoke an
effective immune response against both CSC and non-CSC cells, which
may cause tumor regression and prevent tumor resistance.
Materials and methods
Isolation and characterization of
primary BC cell culture
The study protocol was evaluated and approved by the
Siriraj Institutional Review Board (approval nos. Si520/2010 and
Si321/2016; Bangkok, Thailand). Written informed consent was
obtained from all individual participants prior to enrollment in
the study between January 2010 and December 2018. A fresh luminal
BC tissue sample (0.5×0.5×1.0 cm3) isolated from BC
tissues surgically resected from a 48-year old Thai female patient
with T2N1aM0 (stage IIB) BC was incubated in 10X antibiotic mixture
(1 U/ml penicillin G sodium and 1 mg/ml streptomycin; Thermo Fisher
Scientific, Inc.) diluted in DMEM/F12. Following incubation, the
cells were washed three times with PBS to remove red blood cells
(RBC) and debris. The obtained tissue was chopped into 0.1×0.1×0.1
cm3 sections and plated in a culture dish. Cell
trypsinization was performed using 0.25% trypsin and 0.9 mM EDTA
(Thermo Fisher Scientific, Inc.) to activate cell proliferation.
The cancer cell line obtained using the aforementioned protocol was
termed BCA55-121, and karyotype analysis was performed by Giemsa
staining (Division of Medical Genetics, Office for Research and
Development, Faculty of Medicine Siriraj Hospital). Chromosomes
were observed and counted under microscope. Growth curves of
BCA55-121 were generated using a Live-Cell imager
(IncuCyte® Zoom; Sartorius AG) according to the
manufacturer's instructions. HLA typing was performed at the
Department of Transfusion Medicine, Faculty of Medicine Siriraj
Hospital. BCA55-121 cells were cultured in DMEM/F12 (Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS;
Thermo Fisher Scientific, Inc.) and placed in a 5% CO2
incubator at 37°C. The presence of protein markers including
pan-cytokeratin (pan-CK), programmed death ligand 1 (PD-L1),
fibroblast-activation protein (FAP) was confirmed in BCA55-121
cells by immunofluorescence following incubation with anti-panCK
(1:100; cat. no. sc-8018; Santa Cruz Biotechnology, Inc.),
anti-PD-L1 (1:100; cat. no. ab205921; Abcam), and anti-FAP (1:100;
cat. no. ab53066; Abcam) for 2–3 h at room temperature. Cells were
then incubated with appropriate secondary antibodies, including
anti-mouse horseradish peroxidase (HRP) conjugate Cy3 (1:2,000;
cat. no. 115-166-071) and anti-rabbit HRP conjugated FITC (1:400;
cat. no. ab6717; Abcam) at room temperature for 1 h in the dark.
Nucleus was stained using Hoechst 33342 (1:1,000; Invitrogen;
Thermo Fisher Scientific, Inc.) for 1 h at room temperature. The
proliferation of BCA55-121 cells was analyzed by
Incucyte® and analyzed in Incucyte® Zoom
System, and was compared with the proliferation of the commercial
cell lines MCF-7 and MDA-MB-231.
Breast CSC isolation and analysis
BCA55-121 cells were incubated with Allophycocyanin
(APC)-labeled anti-CD44 (1:5; cat. no. 21270446; ImmunoTools GmbH)
and FITC-labeled anti-CD24 (1:5; cat. no. 21270443; ImmunoTools
GmbH) antibodies for 30 min at 4°C. A CytoFLEX flow cytometer
(Beckman Coulter, Inc.) was used for flow cytometric analysis using
CytExpert software version 2.1 (Beckman Coulter, Inc.). For CSC
isolation, 1×107 BCA55-121 cells labeled with the fluorescent
antibodies were suspended in 2 ml 2% FBS/PBS, and
CD44+CD24- cells were collected using a FACSAria II cell
sorter (BD Biosciences). The purity of the obtained cells was
assessed using the CytoFLEX flow cytometer.
Soft agar colony formation assay
A 0.5% base agar layer was prepared by mixing 1%
agarose with 2X DMEM (or DMEM/F12) supplemented with 20% FBS in a
1:1 ratio. An over-layer of 0.35% agar cell suspension was prepared
by mixing 0.7% agarose with 2X DMEM (or DMEM/F12) in a ratio of
1:1. BCA55-121 cells and their CSCs were suspended (4×104
cells/ml), in this mixture, which was incubated at 37°C in 5%
CO2 for 14–21 days. Subsequently, cells were fixed with
5% glutaraldehyde and stained with 0.5% crystal violet at room
temperature for 15 and 30 min, respectively. Colonies with a
diameter >500 µm were counted and surface areas of the formed
colonies were measured using an inverted microscope IX71 (original
magnification, ×100) at 1 h (baseline) and at the end of the
experiment. The data obtained from CSCs are presented compared with
the whole population of cultured BC cells.
Western blotting of CSC-related
proteins and programmed death ligand-1 (PD-L1)
Proteins (20 µg) extracted from BCA55-121 cells
using extraction buffer [0.25 M Tris-HCl pH 6.8, 5% glycerol, 4%
SDS, 10% β-mercaptoethanol, 0.001% (w/v) bromophenol blue] were
separated using 12% polyacrylamide gels (Sigma-Aldrich; Merck
KGaA). Proteins were then transferred to 0.45-µm nitrocellulose
membranes. The membranes were blocked for 1 h with 5% BSA in TBST
(25 mM Tris-HCl, pH 7.5; 125 mM NaCl; 0.05% Tween 20) at room
temperature and incubated overnight at 4°C with anti-human SOX2
rabbit monoclonal antibody (1:1,000; cat. no. 3579; Cell Signaling
Technology, Inc.), anti-human OCT4A rabbit monoclonal antibody
(1:1,000; cat. no. 2840; Cell Signaling Technology, Inc.),
anti-human homeobox protein Nanog (NANOG) rabbit monoclonal
antibody (1:1,000; cat. no. 4903, Cell Signaling Technology, Inc.)
or anti-human PD-L1 rabbit monoclonal antibody (1:500; cat. no.
ab205921; Abcam). The membranes were then washed three times with
TBST and incubated with a HRP-conjugated goat anti-rabbit IgG
antibody (1:1,000; cat. no. ab6721; Abcam) for 2 h at room
temperature. The proteins were visualized using SuperSignal West
Pico Chemi-luminescent Substrate (Thermo Fisher Scientific, Inc.)
under a Gel Documentation System (G:Box; version EF; Syngene).
Anti-β-actin antibody (1:5,000; Santa Cruz Biotechnology, Inc.;
cat. no. sc-47778) was incubated for 1 h at room temperature to
detect β-actin as a loading control.
Dendritic cell activation and
immunophenotyping assay
Venous blood (50 ml) from three healthy donors with
aged between 25 and 30 years with no underlying diseases was
collected in 0.1% heparin solution and peripheral blood mononuclear
cells (PBMCS) were isolated using Lymphosep® lymphocyte
separation media (Biowest) according to the manufacturer's
instructions. Briefly, heparinized blood was overlaid on
Lymphosep® media and centrifuged at 400 × g at 20°C for
30 min. PBMCs were collected and washed three times at room
temperature using 200 × g centrifugations for 10 min with PBS or
RBC lysis buffer (150 mM NH4Cl, 10 mM NaHCO3
and 1.26 mM EDTA), and then resuspended in AIM-V® medium
(Thermo Fisher Scientific, Inc.) and plated onto 6-well plates at
5×106 cells/well. After 2 h of incubation at 37°C and pH 6.8–7.3,
non-adherenT cells were gently removed and suspended in 900 µl
human type AB serum (Merck KGaA) with 100 µl DMSO and cryopreserved
at −80°C as a source of lymphocytes. The adherent monocyte-enriched
cells were cultured at 37°C in 5% CO2 incubator in
AIM-V® medium containing granulocyte-macrophage colony
stimulating factor (50 ng/ml) and interleukin (IL)-4 (25 ng/ml)
(ImmunoTools GmbH) for 6 days to produce immature dendritic cells
(iDCs). The iDCs were then matured to mature dendritic cells (mDCs)
after incubation at 37°C in 5% CO2 with tumor necrosis
factor-α (50 ng/ml) and interferon (IFN)-γ (50 ng/ml) (ImmunoTools
GmbH) in AIM-V® medium for 2 days. Subsequently, wells
containing ~5×105 mDCs were pulsed with 10 µg cancer cell-derived
RNA. For phenotypic analysis of DCs, 5×105 DCs were detached with 5
mM EDTA, washed in 2% FBS/PBS and incubated with
fluorescence-conjugated monoclonal antibodies (1:50) at 4°C for 30
min. Following washing in 2% FBS/PBS twice, cells were analyzed
using a CytoFLEX flow cytometer. To determine the phenotype of
monocytes and DCs, anti-CD11c APC (1:50; cat. no. 21487116;
ImmunoTools GmbH), anti-CD14 APC (1:50; 21620146; ImmunoTools
GmbH), anti-CD14 FITC (1:50; cat. no. 21620143; ImmunoTools GmbH),
anti-CD40 FITC (1:50, cat. no. 21270403; ImmunoTools GmbH),
anti-CD83 phycoerythrin (PE; 1:20; cat. no. 12-0839-42; Thermo
Fisher Scientific, Inc.), anti-CD86 PE (1:20; cat. no. 12-0869-42;
Thermo Fisher Scientific, Inc.) and anti-HLA-DR FITC (1:50; cat.
no. 21278993; ImmunoTools GmbH) monoclonal antibodies were used. To
determine human leukocyte antigen-A2 (HLA-A2) expression, PBMCs
from healthy donors were collected using the method described above
and analyzed using anti-HLA-A2 APC antibody (1:50; cat. no.
17-9876-42; ImmunoTools GmbH) and the CytoFLEX Flow cytometer.
Total effector lymphocytes and
CD8+ T cell preparation
Cryopreserved lymphocytes were thawed and
resuspended in RPMI medium Thermo Fisher Scientific, Inc.)
supplemented with 5% human AB serum (Merck KGaA). The lymphocytes
were activated by co-culture with RNA-pulsed mDCs at a ratio of
10:1 for 1 day. Subsequently, lymphocytes were cultured at 37°C in
RPMI medium supplemented with 5% human AB serum, IL-2 (20 ng/ml),
IL-7 (10 ng/ml) and IL-15 (20 ng/ml) (ImmunoTools GmbH) for 9 days.
The culture medium was replaced every other day.
Cytotoxic T cells were isolated using a
CD8+ T cell Isolation kit (Miltenyi Biotec, Inc.)
according to the manufacturer's protocol. In brief,
1×107 lymphocytes were centrifuged at 300 × g for 10 min
at room temperature and resuspended in 40 µl buffer containing PBS,
0.5% bovine serum albumin (Capricorn Scientific) and 2 mM EDTA. The
cell suspension was mixed and incubated with 10 µl of a
CD8+ T cell biotin-antibody cocktail (Miltenyi Biotec,
Inc.) for 5 min, followed by 20 µl of a CD8+ T cell
micro-bead cocktail (Miltenyi Biotec, Inc.) for 10 min at room
temperature. The cell mixture was then magnetically separated using
a magnetic-activated cell sorting (MACS) column and MACS separator
(Miltenyi Biotec, Inc.), and the flow-through enriched in
CD8+ T cells were collected. To determine the phenotype
of effector immune cells, anti-CD3 FITC (cat. no. 21850033;
ImmunoTools GmbH), anti-CD4 APC (cat. no. 21850046; ImmunoTools
GmbH), anti-CD8 APC (cat. no. 21620086; ImmunoTools GmbH),
anti-CD16 APC (cat. no. 21278166; ImmunoTools GmbH) and anti-CD56
PE (BioLegend, Inc.) monoclonal antibodies were used. Phenotype
markers of lymphocytes, including CD3, CD4, CD8, CD16 and CD56 were
analyzed using a FACSCalibur flow cytometer (BD Biosciences) using
Flow Jo (Treestar) software version X.
IFN-γ measurement by ELISA
Amounts of IFN-γ secreted by CSC RNA-pulsed
DC-activated T cells in comparison with naïve T cells was measured
using an IFN-γ ELISA kit (cat. no. DIF50, R&D Systems, Ltd.)
according to the manufacturer's protocol. Cytokine concentration
was determined by measuring optical density using a microplate
reader at 450/570 nm.
Tumor cell killing by effector T
cells
BC target cells (T), either the whole culture
population (BCA55-121-WP) or the enriched BCA55-121-CSCs, were
plated into separate wells of 96-well plates (~3,000 cells/well)
for 24 h for the assessment of immune cell killing activity.
Subsequently, 50 µl Caspase-3/7 green reagent (Sartorius AG) at a
20 µM concentration was added to detect apoptosis. Total
lymphocytes (E) from healthy donors with HLA matched to that of
BCA55-121 cells (HLA-A2) were added as effectors and co-cultured
with the tumor target cells (T) at effector to target (E:T) ratios
of 5:1, 10:1 and 20:1 for 24 h. Cancer cells with apoptotic
activity were detected using an IncuCyte® live-cell
analysis system.
Statistical analysis
All data are presented as the mean ± standard
deviation (SD). The data were tested for normal distribution by the
Shapiro-Wilk test. The data from two groups were analyzed by paired
Student's t-test and from multiple groups by one-way
repeated-measure analysis of variance (ANOVA) followed by Tukey's
post-hoc test using GraphPad Prism software version 7.04 (GraphPad
Software, Inc.) or SigmaPlot 16.0 (Systat Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Characterization of the primary BC
cell line BCA55-121
The tumor size measured 4.5×4×2 cm3, and
was diagnosed as an invasive ductal carcinoma
(moderately-differentiated) positive for estrogen receptor
(>75%) and progesterone receptor (<10%) and negative for
HER2/neu. Angiolymphatic invasion was absent, however, one lymph
node was positive for macro-metastatic cancer cells. The cancer
cells were visualized as cobblestone shaped under phase-contrast
microscopy (data not shown). The primary cell line was positive for
pan-cytokeratin, confirming that the cells were of epithelial
origin (18) and without stromal
fibroblast contamination in the culture (Fig. 1A). The cell line also tested negative
for fibroblast activation protein (FAP), and PD-L1 was expressed at
a very low level (Fig. 1A).
Chromosomal analysis revealed an aneuploid aberration of chromosome
copies. BCA55-121 cells exhibited a higher proliferation rate
compared with the MCF-7 cell line, but lower than that of
MDA-MB-231 cells (Fig. 1A). Breast
CSCs, defined as CD44+/CD24− cells, were
separated from the BCA55-121-WP cells by fluorescence-activated
cell sorting. These CSC markers were expressed in ~75.2±0.45% of
the BCA55-121-WP population (Fig.
1B). CSCs were subsequently sorted to enrich the cells with
this phenotype to 92.9±5.83% purity (Fig. 1B). The tumorigenic ability of the
BCA55-121-CSCs were tested, and the results were compared with
BCA55-121-WP cells. Soft agar colony formation assay, a 3D spheroid
cell culture assay that measures cell proliferation in semi-solid
matrices (11) was conducted. The
assay selectively enriches cells that exhibit a malignant
transformation, anchorage-independent proliferation and progenitor
cell-like tumorigenic properties. Significantly greater
proliferative and tumorigenic abilities (2–3 folds) of the
BCA55-121-CSC population were indicated by colony count and colony
size compared with unsorted BCA55-121-WP cells (Fig. 1C). The level of OCT4 was increased in
BCA55-121-CSCs compared with the unsorted cells (Fig. 1D). NANOG expression did not change in
BCA55-121-CSC compared with BCA55-121 WP, and the level of SOX2 was
very low. These results confirmed the greater tumorigenic and
stemness properties of the CSC-enriched population. In addition,
the expression of PD-L1 was significantly increased in the CSC
population of the CSC culture compared with the WP culture
(Fig. 1E).
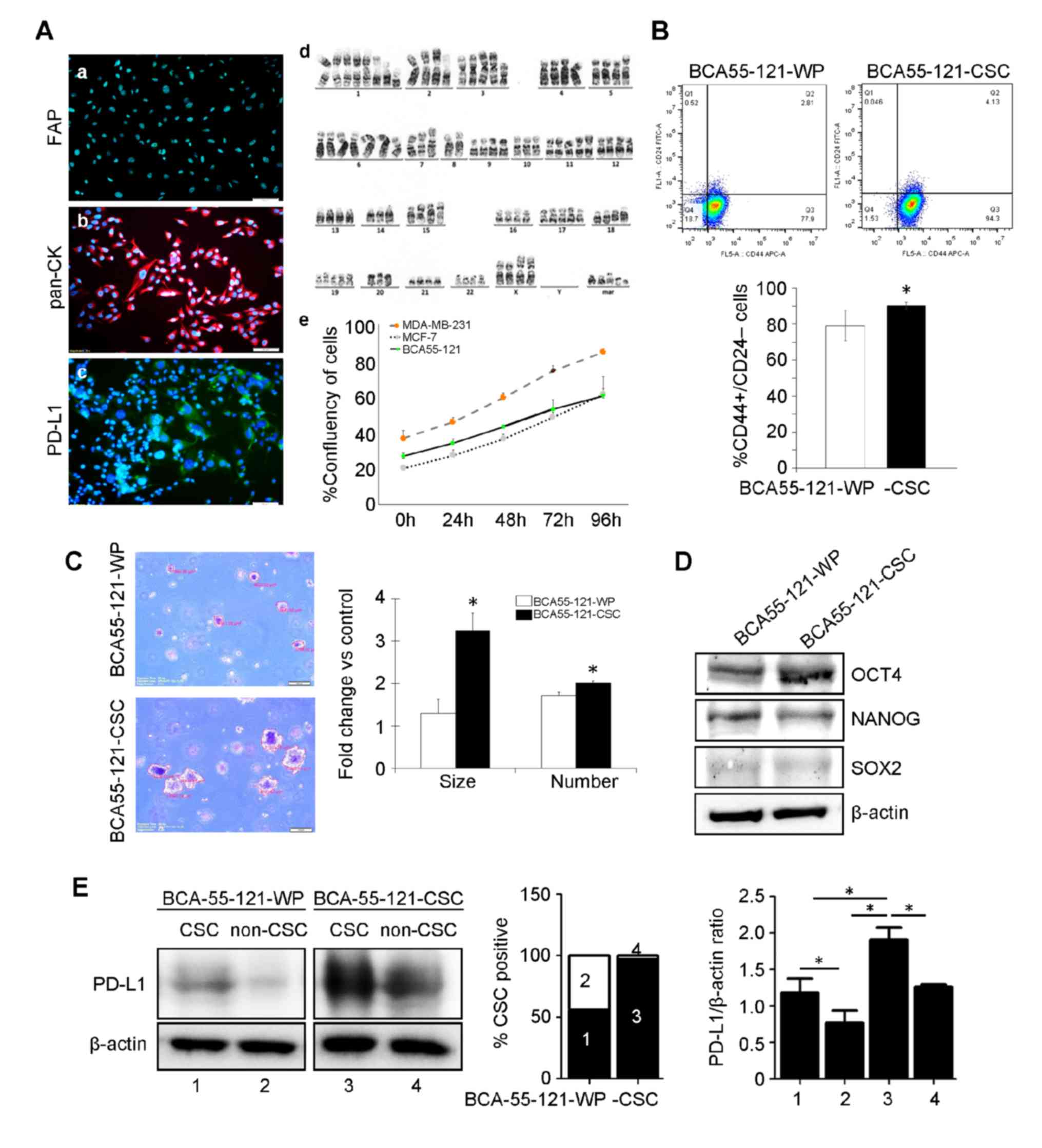 | Figure 1.Characterization of the BCA55-121-WP
and BCA55-121-CSC BC cell lines. (A) Immunofluorescent staining of
(a) pan-CK, (b) PD-L1 and (c) FAP. (d) Chromosomal analysis of
BCA55-121 cells displaying aneuploid chromosomes. (e) Growth curve
of BCA55-121-WP cells compared with reference BC cell lines,
MDA-MB-231 and MCF-7. (B) Flow cytometric analysis of
CD44+/CD24− cells in BCA55-121 before and
after stem-cell enrichment. (C) Representative fields of cancer
cell colonies from the soft agar colony formation assay. Colony
size and colony count are presented as fold-changes between
BCA55-121-CSC and BCA55-121-WP cells. (D) Protein expression levels
of stem cell-associated proteins OCT4, NANOG and SOX2. (E) PD-L1
expression in the CSC and non-CSC populations in BCA55-121-WP and
BCA55-121-CSC culture demonstrated using western blot analysis with
β-actin as a loading control. Bars represent mean ± SD of two
independent experiments. *P<0.05. WP, whole population; BC,
breast cancer; CD, cluster of differentiation; CSC, cancer stem
cell; pan-CK, pan-cytokeratin; PD-L1, programmed death ligand 1;
FAP, fibroblast-activation protein. |
Activation of PBMC-derived DCs and
lymphocytes
DC phenotype was analyzed on days 1 (monocytes), 6
(iDCs) and 9 (mDCs). Monocytes are the only cell type that
expresses CD14 (19), and this was
diminished during subsequent maturation and replaced by a DC marker
(CD11c), a DC maturation marker (CD83), an antigen-presenting
molecule (HLA-DR) and co-stimulatory molecules (CD40 and CD86;
Fig. 2A) demonstrating DC maturation
in vitro. The subsets of cells in the activated lymphocyte
population following co-culture with DCs were analyzed by flow
cytometry. The highest proportion of cytotoxic T lymphocytes in the
total lymphocytes was observed after co-culture with RNA-pulsed DCs
followed by CD4+ T cells (Fig. 2B).
NK cell and NKT cell populations were also identified among the
activated lymphocytes (Fig. 2B). In
addition, a significantly increased number of IFN-γ-positive CD8+ T
cells were observed on day 18 of the lymphocyte activation protocol
(day 9 after co-culture with RNA-pulsed DC; Fig. 2C). For each of the three healthy
donors, higher levels of secreted IFN-γ were present in the
supernatants of activated T cells compared with the supernatants of
naïve T cells (Fig. 2D).
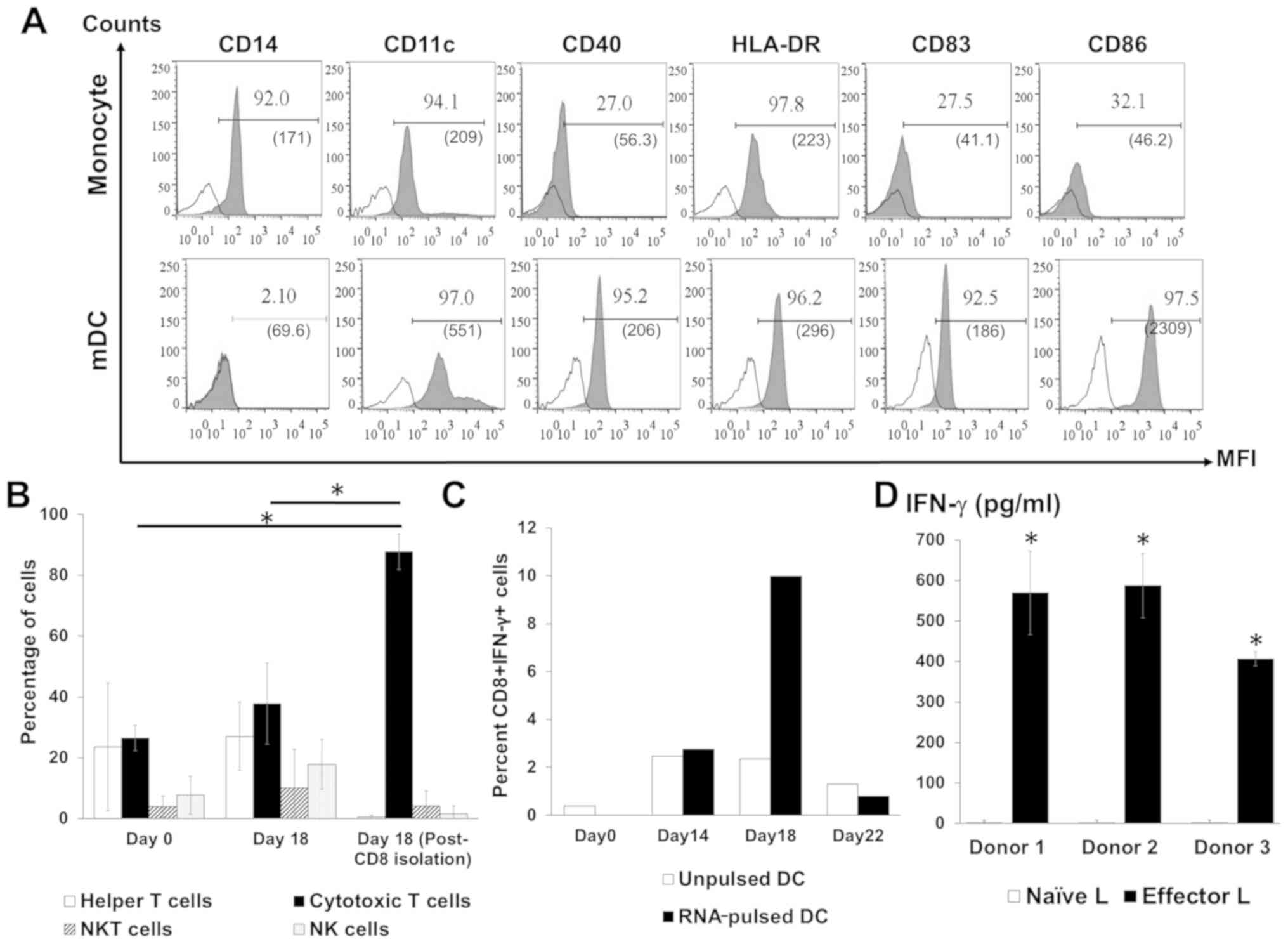 | Figure 2.Analysis of activated DCs and
lymphocyte phenotypes. (A) Profiles of the specific markers CD14,
CD11c, CD40, HLA-DR, CD83 and CD86 on monocytes and mDCs. (B)
Percentage distribution of lymphocyte subpopulations before and
after activation and following the isolation of CD8+ T
cells. (C) The percentage of IFN-γ-positive CD8+ T
lymphocytes after co-culture with unpulsed or RNA-pulsed DCs from
one donor. (D) Level of IFN-γ in the culture media of effector
lymphocytes from three donors. Bars represent mean ± SD of three
independent experiments. *P<0.05. CD, cluster of
differentiation; DCs, dendritic cells; mDC, mature dendritic cell;
HLA-DR, human leukocyte antigen DR; IFN, interferon; L, lymphocyte;
MFI, mean fluorescence intensity; NK, natural killer. |
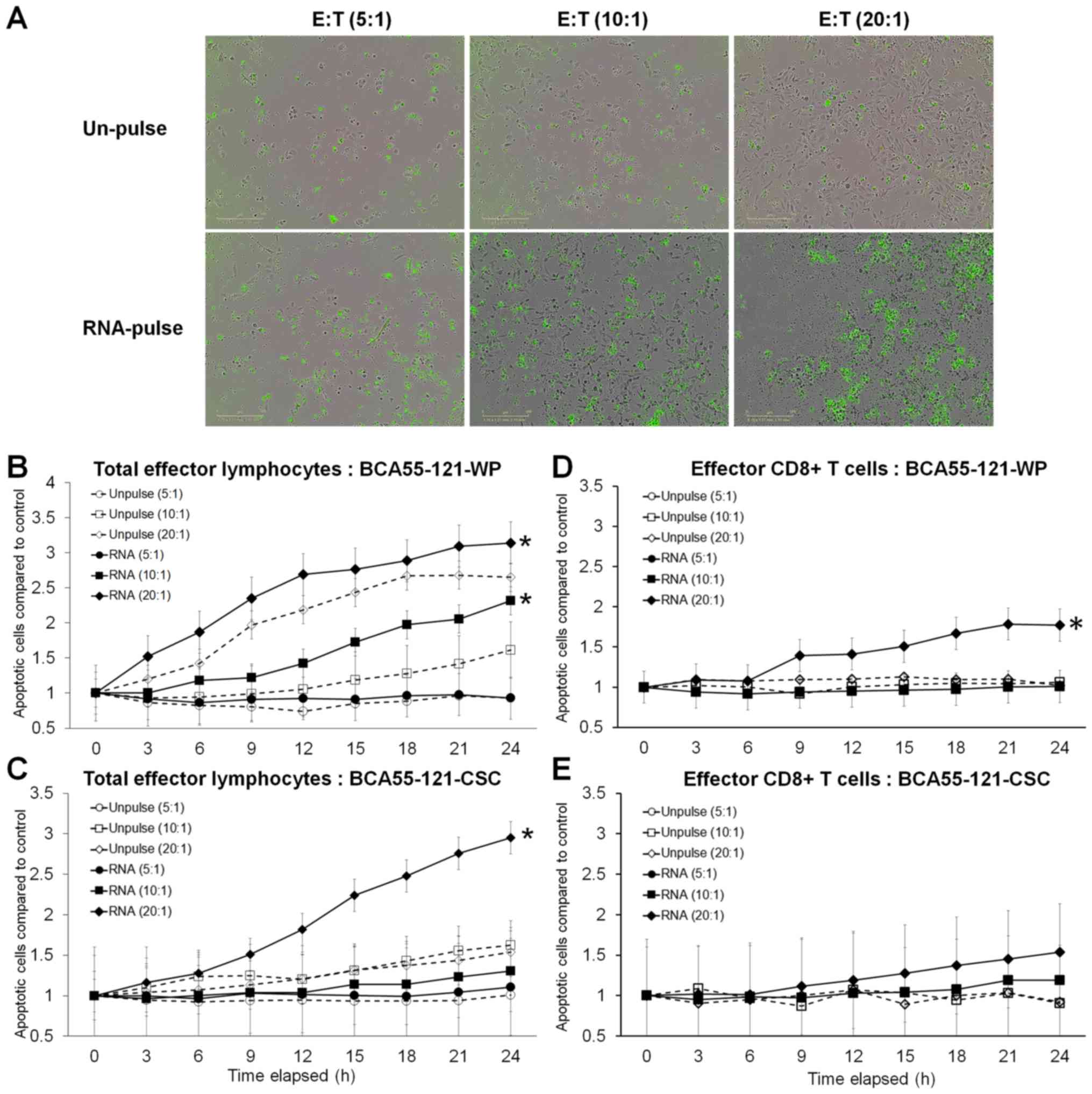
Cancer cell killing assay
Total RNA from BCA55-121-CSC was loaded onto DCs,
and the efficacy in activating effector lymphocytes to kill cancer
cells was measured using Caspase-3/7 reagents for apoptosis with
the Live-cell imager. To prevent the killing of cancer cells by
other immune cells, not effector CD8+ T cells, HLA-A2
matched lymphocytes and BCA55-121 cells were used. The efficacy of
tumor cell killing between lymphocytes activated by BCA55-121-WP
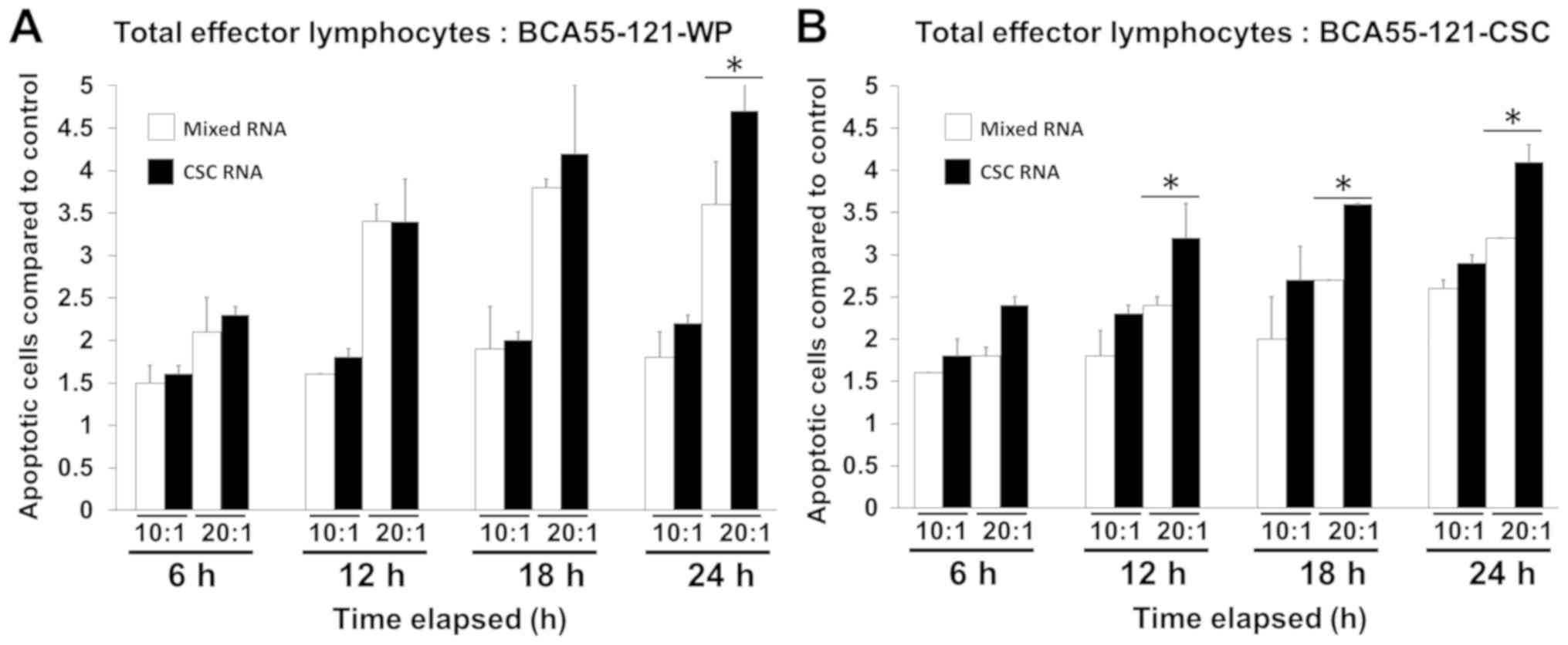
and CSC RNA-pulsed DCs was compared (Fig. 3). The killing activity in the CSC
RNA-activated lymphocytes was greater compared with that of
BCA55-121-WP or CSCs, and this was at either 10:1 or 20:1 E:T tumor
cells with significance reached at 20:1 E:T ratio (Fig. 3). Green fluorescence, representing
cells with activated Caspase-3/7 at different E:T ratios, is
displayed in Fig. 4A. Although
increasing apoptotic activity over time was observed in both
experiments, a 10:1 ratio of total activated lymphocytes did not
result in enhanced killing of CSC targets, although BCA55-121-WP
cells were killed (Fig. 4B and C).
Additionally, at a 20:1 ratio, more BCA55-121-WP cells were killed
compared with CSCs at all time points (Fig. 4B and C). These results demonstrated
that apoptotic resistance may be a property of CSCs. In addition,
CD8+ effector T cells isolated from the CSC
RNA-activated lymphocyte population were less effective compared
with the whole lymphocyte population against either BCA55-121-WP or
BCA55-121-CSC targets (Fig. 4D and
E). Limited killing activity was observed by CD8+ T
cells that were activated by DCs without antigen exposure, which
suggested that cancer cell killing was antigen specific.
Discussion
The present study explored the potential of
DC-primed T cells to alleviate treatment-refractory BC. CSCs were
selected as a major target to alleviate cancer recurrence and
aggressiveness. CD44+/CD24− was used as a CSC
marker, and subsequent analyses of this subpopulation revealed its
superior proliferative and tumorigenic capability in an
anchorage-independent 3D culture assay compared with BCA55-121-WP.
This ability was associated with high expression of stem
cell-associated proteins, confirming the stem cell properties of a
new in-house BC cell line, BCA55-121-CSC. In the present study,
higher expression of PD-L1 was observed in the CSC population of
the CSC culture compared with the CSC population of the WP culture.
These results were supported by previous studies, in which PD-L1
expression was positively associated with the expression of
stemness markers (20,21), and supported the notion that
BCA55-121-CSC cultures possessed CSC properties. In the present
study, CSC RNA could be presented as tumor antigens by DCs and
activated T cells in the whole T cell population and enriched
CD8+ T cells, to stimulate an enhanced ability to kill
cancer cells superior to that obtained using RNA from the total
BCA55-121 cell population. BCA55-121-CSCs were more resistant to
activated effector T cells compared with the whole BCA55-121 cell
population. In the current study, total lymphocytes yielded an
improved killing effect on both BCA55-121-WP and BCA55-121-CSC
populations compared with effector CD8+ T cells as the
total lymphocyte population contains CD4+, NK and NKT
cells, which may have an effect on cancer cell killing.
Tumor-associated antigens (TAAs), in the form of
peptides, protein lysates or total RNA, were capable of priming DCs
for effector lymphocyte activation (22,23).
Peptide antigens can be loaded directly onto MHC molecules;
however, it is mandatory to match the immunogenic epitopes relevant
to a particular cancer cell with the complementary specific HLA
haplotype (15). The single best
immunogenic TAA epitope in BC is yet to be defined, cancer-derived
RNA has been used to present a broad range of TAA epitopes on
various MHC haplotypes present on DCs (24,25). The
RNA-based antigen approach has been most commonly used in clinical
studies, offering HLA class I and II cross-presentation and in turn
inducing humoral and cellular immune responses in CD4+
and CD8+ T cells (26–28). The
present study compared the cancer cell killing ability of T cells
activated by DCs pulsed with total RNA with those activated by
protein lysate-pulsed DCs and demonstrated that the ability to
induce cancer cell apoptosis was greater in the former (data not
shown). Moreover, the use of RNA avoids the effects of the
immunosuppressive factors present in the tumor protein lysate;
therefore, pulsing DCs with RNA may result in improved function
(24,25). Taken together, this evidence supports
the selection of total RNA as the antigen source in the present
study. The present study demonstrated that CSC-RNA-pulsed DCs
induced effector lymphocytes to kill both BCA55-121-WP and
BCA55-121-CSCs, and the killing activity was superior compared with
that of DCs that received RNA from whole cancer cells, which
contained a mixture of both non-CSCs and CSCs. These results
emphasized the potential of DC-based protocols using CSC RNA to
prime DCs for eradication of tumor cells via tumor antigen-specific
effector T cells.
In the present study, the T cell population
activated by co-culture with RNA-primed DCs consisted of mainly
cytotoxic and helper T cells together with some NKT and NK cells.
These activated T cells produced high levels of IFN-γ, which is
characteristic of effector T cells (29). These findings were consistent with
another report that confirmed the potential of ex vivo
activation of effector T cells with an enhanced specificity for
cancer antigens appropriate for use in adoptive transfer (30). In the present study, cancer cell
killing by the effector T cells generated with CSC RNA-pulsed DCs
was significantly greater compared with T cells activated with
unpulsed DCs, and the effect was dose-dependent. Substantial
killing was observed in cultures of unpulsed DC-activated T cells,
which may be due to two main factors. The first is the presence of
non-antigen specific cancer killing cells in the total lymphocyte
population, comprising NK and NKT cells, which are also activated
by DCs. In the present study, non-specific killing by other immune
cells was addressed by isolating CD8+ T cells from the
activated total lymphocyte population. The tumor-killing effect of
the activated CD8+ T cells demonstrated a greater
specificity of killing, potentially by antigen-dependent cytotoxic
T cells, since little or no killing was observed among
unpulsed-DC-activated CTLs. The efficacy of tumor killing between
lymphocytes activated by whole culture RNA-pulsed DCs and CSC
RNA-pulsed DCs was further compared. The results of the present
study revealed superior tumor killing activity with the CSC
RNA-activated cells, especially at 20:1 and 10:1 E:T ratios.
Inferior apoptotic activity was observed towards BCA55-121-CSC
target cells. The second potential mechanism of killing in unpulsed
cultures maybe the mismatch of HLA molecules between effector cells
derived from healthy donors and the target cancer cells. HLA-A2
matched donors were used in the present study as the BCA55-121
cancer cell line is HLA-A2 positive; however, residual HLA
haplotype mismatch may have stimulated a small amount of
non-specific killing and prevented optimal antigen presentation and
effector cell activation leading to lower specific cytotoxicity in
RNA-pulsed cultures. However, in the clinical application of
DC-based vaccines, autologous DCs are activated by autologous tumor
antigens and thus eliminate this confounder (14,16). The
limitation of this study was the lack of investigation of HLA class
I expression in DC. In addition, using only one donor was a
limitation of the experiment presented in this study.
The present study revealed that CSCs were more
resistant compared with the whole cancer cell population to
effector T cells. This may be explained by the finding that the CSC
population in CSC cultures expressed high levels of PD-L1 (20), which may induce apoptosis of effector
T cells (31). These in vitro
findings need to be investigated in an ex vivo system and in
clinical trials for the development of DC-based activation of T
cells against breast CSCs. A combination of drugs targeting CSCs
and activated effector T cells may be a potential approach for
resolving the issue of the resistant CSC population. This treatment
needs to be developed alone or in conjuction with other treatments
to optimize antitumor immune responses and overcome the relatively
immunosuppressive stage, during which DCs in the tumor and the
surrounding microenvironment develop tolerance (32,33).
Personalized cancer therapy may be achieved by the activation of
patient-derived DCs by using autologous tumor antigens from
surgical tissue-derived primary cell culture. Several studies
performed using the personalized DC-based vaccine have confirmed
potent anti-tumor immunity activity mediated by a broad range of
TAAs relevant to each patient (34–36).
This approach has the potential to improve the efficacy of
treatment and quality of life for patients with BC.
Acknowledgments
Not applicable.
Funding
This research project was supported by the Faculty
of Medicine, Siriraj Hospital, Mahidol University (grant no.
R015933006) and by the Thailand Science Research and Innovation
(grant no. RSA6280091), Ministry of Higher Education, Science,
Research and Innovation, Thailand awarded to CT.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NS, NJ, PJ and ST performed the experiments and
acquired the data. CT conceived and designed the study, acquired,
analyzed and interpreted the data. TC, PY and PT analyzed the data.
MW and PO collected the breast cancer tissue samples and verified
clinicopathological data. NS, NJ and CT drafted the article. CT
revised the manuscript for important intellectual content. All
authors have read and approved the manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
individual participants prior to enrollment in the study. The
present study was approved by the Siriraj Institutional Review
Board (approval nos. Si520/2010 and Si321/2016).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Global Burden of Disease Cancer
Collaboration, ; Fitzmaurice C, Allen C, Barber RM, Barregard L,
Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, et al:
Global, regional, and national cancer incidence, mortality, years
of life lost, years lived with disability, and disability-adjusted
life-years for 32 cancer groups, 1990 to 2015: A systematic
analysis for the global burden of disease study. JAMA Oncol.
3:524–548. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Colleoni M, Sun Z, Price KN, Karlsson P,
Forbes JF, Thürlimann B, Gianni L, Castiglione M, Gelber RD, Coates
AS and Goldhirsch A: Annual hazard rates of recurrence for breast
cancer during 24 years of follow-up: Results from the international
breast cancer study group trials I to V. J Clin Oncol. 34:927–935.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gerber B, Freund M and Reimer T: Recurrent
breast cancer: Treatment strategies for maintaining and prolonging
good quality of life. Dtsch Arztebl Int. 107:85–91. 2010.PubMed/NCBI
|
|
4
|
Dawood S, Austin L and Cristofanilli M:
Cancer stem cells: Implications for cancer therapy. Oncology
(Williston Park). 28:1101–1107, 1110. 2014.PubMed/NCBI
|
|
5
|
Palucka K and Banchereau J:
Dendritic-cell-based therapeutic cancer vaccines. Immunity.
39:38–48. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abdullah LN and Chow EK: Mechanisms of
chemoresistance in cancer stem cells. Clin Transl Med. 2:32013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carrasco E, Alvarez PJ, Prados J, Melguizo
C, Rama AR, Aránega A and Rodríguez-Serrano F: Cancer stem cells
and their implication in breast cancer. Eur J Clin Invest.
44:678–687. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dey P, Rathod M and De A: Targeting stem
cells in the realm of drug-resistant breast cancer. Breast Cancer
(Dove Med Press). 11:115–135. 2019.PubMed/NCBI
|
|
9
|
Islam F, Gopalan V, Smith RA and Lam AK:
Translational potential of cancer stem cells: A review of the
detection of cancer stem cells and their roles in cancer recurrence
and cancer treatment. Exp Cell Res. 335:135–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Britton KM, Kirby JA, Lennard TW and
Meeson AP: Cancer stem cells and side population cells in breast
cancer and metastasis. Cancers (Basel). 3:2106–2130. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Akrap N, Andersson D, Bom E, Gregersson P,
Ståhlberg A and Landberg G: Identification of distinct breast
cancer stem cell populations based on single-cell analyses of
functionally enriched stem and progenitor pools. Stem Cell Reports.
6:121–136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shao J, Fan W, Ma B and Wu Y: Breast
cancer stem cells expressing different stem cell markers exhibit
distinct biological characteristics. Mol Med Rep. 14:4991–4998.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gelao L, Criscitiello C, Esposito A, De
Laurentiis M, Fumagalli L, Locatelli MA, Minchella I, Santangelo M,
De Placido S, Goldhirsch A and Curigliano G: Dendritic cell-based
vaccines: Clinical applications in breast cancer. Immunotherapy.
6:349–360. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Constantino J, Gomes C, Falcão A, Cruz MT
and Neves BM: Antitumor dendritic cell-based vaccines: Lessons from
20 years of clinical trials and future perspectives. Transl Res.
168:74–95. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sabado RL, Balan S and Bhardwaj N:
Dendritic cell-based immunotherapy. Cell Res. 27:74–95. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Anguille S, Smits EL, Lion E, van Tendeloo
VF and Berneman ZN: Clinical use of dendritic cells for cancer
therapy. Lancet Oncol. 15:e257–e267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pham PV, Le HT, Vu BT, Pham VQ, Le PM,
Phan NL, Trinh NV, Nguyen HT, Nguyen ST, Nguyen TL and Phan NK:
Targeting breast cancer stem cells by dendritic cell vaccination in
humanized mice with breast tumor: Preliminary results. Onco Targets
Ther. 9:4441–4451. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Andergassen U, Vogl A, Mumm JN, Kölbl AC,
Hutter S, Rack B, Friese K and Jeschke U: Immunocytochemical
characterization of disseminated tumour cells from bone marrow of
breast cancer patients. Anticancer Res. 36:3217–3222.
2016.PubMed/NCBI
|
|
19
|
Zarif JC, Hernandez JR, Verdone JE,
Campbell SP, Drake CG and Pienta KJ: A phased strategy to
differentiate human CD14+monocytes into classically and
alternatively activated macrophages and dendritic cells.
Biotechniques. 61:33–41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Darvin P, Sasidharan Nair V and Elkord E:
PD-L1 Expression in human breast cancer stem cells is
epigenetically regulated through posttranslational histone
modifications. J Oncol. 2019:39589082019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao L, Guo Q, Li X, Yang X, Ni H, Wang T,
Zhao Q, Liu H, Xing Y, Xi T and Zheng L: MiR-873/PD-L1 axis
regulates the stemness of breast cancer cells. EBioMedicine.
41:395–407. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin T, Shi P, Gou S, Shen Q and Wang C:
Dendritic cells loaded with pancreatic cancer stem cells (CSCs)
lysates induce antitumor immune killing effect in vitro. PLoS One.
9:e1145812014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xie BH, Yang JY, Li HP, Zhang B, Chen W,
Zhou B, Peng BG, Liang LJ and He Q: Dendritic cells transfected
with hepatocellular carcinoma (HCC) total RNA induce specific
immune responses against HCC in vitro and in vivo. Clin Transl
Oncol. 16:753–760. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheever MA, Allison JP, Ferris AS, Finn
OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL,
Weiner LM and Matrisian LM: The prioritization of cancer antigens:
A national cancer institute pilot project for the acceleration of
translational research. Clin Cancer Res. 15:5323–5337. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Criscitiello C: Tumor-associated antigens
in breast cancer. Breast Care (Basel). 7:262–266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Garg NK, Dwivedi P, Prabha P and Tyagi RK:
RNA pulsed dendritic cells: An approach for cancer immunotherapy.
Vaccine. 31:1141–1156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McNamara MA, Nair SK and Holl EK:
RNA-based vaccines in cancer immunotherapy. J Immunol Res.
2015:7945282015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen DS and Mellman I: Oncology meets
immunology: The cancer-immunity cycle. Immunity. 39:1–10. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhat P, Leggatt G, Waterhouse N and Frazer
IH: Interferon-γ derived from cytotoxic lymphocytes directly
enhances their motility and cytotoxicity. Cell Death Dis.
8:e28362017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nguyen ST, Nguyen HL, Pham VQ, Nguyen GT,
Tran CD, Phan NK and Pham PV: Targeting specificity of dendritic
cells on breast cancer stem cells: In vitro and in vivo
evaluations. Onco Targets Ther. 8:323–334. 2015.PubMed/NCBI
|
|
31
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Duraiswamy J, Kaluza KM, Freeman GJ and
Coukos G: Dual blockade of PD-1 and CTLA-4 combined with tumor
vaccine effectively restores T-cell rejection function in tumors.
Cancer Res. 73:3591–3603. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zong J, Keskinov AA, Shurin GV and Shurin
MR: Tumor-derived factors modulating dendritic cell function.
Cancer Immunol Immunother. 65:821–833. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shimodaira S, Sano K, Hirabayashi K, Koya
T, Higuchi Y, Mizuno Y, Yamaoka N, Yuzawa M, Kobayashi T, Ito K and
Koizumi T: Dendritic cell-based adjuvant vaccination targeting
Wilms' tumor 1 in patients with advanced colorectal cancer.
Vaccines (Basel). 3:1004–1018. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen Y, Yao K, Wang B, Qing J and Liu G:
Potent dendritic cell vaccine loaded with latent membrane protein
2A (LMP2A). Cell Mol Immunol. 5:365–372. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hobo W, Strobbe L, Maas F, Fredrix H,
Greupink-Draaisma A, Esendam B, de Witte T, Preijers F, Levenga H,
van Rees B, et al: Immunogenicity of dendritic cells pulsed with
MAGE3, survivin and B-cell maturation antigen mRNA for vaccination
of multiple myeloma patients. Cancer Immunol Immunother.
62:1381–1392. 2013. View Article : Google Scholar : PubMed/NCBI
|