Introduction
Glioblastoma is the most common malignant intrinsic
brain tumor in adults (1). These
tumors are believed to originate from molecular genetic lesions in
neuroglial stem or progenitor cells and can arise at any age, but
the risk of developing glioblastoma increases with age (2). The WHO classification characterizes
glioblastomas as glial tumors exhibiting the histopathological
features of microvascular proliferation and necrosis which are
characteristics of WHO grade IV tumors (3). Typical glioblastomas do not show
mutations in the isocitrate dehydrogenase genes, but genetic
alterations in three major pathways: The phosphoinositide 3-kinase
(PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR),
the p53 and the retinoblastoma (RB) pathways; furthermore, tyrosine
kinases such as epidermal growth factor receptor and c-MET mediate
and maintain the malignant phenotype of glioblastoma (4).
There has been essentially no progress in the
pharmacological therapy of this tumor since the introduction of
temozolomide in 2005 (5,6). Notably targeted therapies have failed
at least in unselected patient populations that were not
pre-enriched for certain molecular markers (7). Type I interferons (IFN) have already
been tested for potential anti-glioma activity in the early periods
of cancer immunotherapy. IFN are endogenous cytokines that have a
major role in combatting viral infections. Moreover, there is
increasing interest in a role of type I IFN in suppressing tumor
growth, notably a role in negatively regulating the cancer stem
cell phenotype (8). We and others
have previously characterized profound anti-glioblastoma stem cell
properties of type I IFN in vitro that involved suppression
of sphere formation but relatively little induction of cell death.
The failure of IFN to induced cell death was surprising since the
IFN-induced transcriptional changes in glioma cells were predicted
to favour activation of the intrinsic caspase-dependent cell death
pathway (9,10). These promising cell culture data
indicating efficacy of IFN against glioblastoma appear to be in
conflict with the failure to confirm activity of type I IFN in
clinical trials with human glioblastoma patients (11). Yet, interest in IFN signaling in
glioblastoma persists and current efforts focus on integrating IFN
into combined modality treatments.
TG02 is a novel orally available inhibitor of
multiple cyclin-dependent kinases (CDK) that exhibits strong in
vitro activity in glioma models while single agent activity in
rodent glioma models remains moderate (12,13).
TG02 is currently explored in clinical trials in recurrent and in
newly diagnosed glioblastoma (NCT02942264, NCT03224104) (14). The present study sought to explore
whether the combination of type I IFN with TG02 might exhibit
synergistic activity and might be suitable to activate
caspase-dependent cell death pathways in human glioma models in
vitro.
Materials and methods
Reagents
Human IFN-β1a was provided by Biogen Inc. Stock
solutions for in vitro experiments were prepared in 20 mM
sodium acetate, pH 8.4, containing 150 mM arginine hydrochloride.
TG02 was provided by Adastra. Staurosporine was purchased from
AppliChem, acetyl-Asp-Glu-Val-Asp-7-amino-4-methyl coumarin
(ac-DEVD-amc) and benzyloxycarbonyl-Val-Ala-Asp
(OMe)-fluoromethylketone (zVAD-fmk) were obtained from Bachem.
ON-TARGET plus human siRNA SMART pool targeting human X-linked
inhibitor of apoptosis protein (XIAP)-associated factor (XAF)-1 or
caspase 3 was purchased from Dharmacon.
Cell culture
The human long-term cell line, LN-229, and the human
glioma-initiating cell lines (GIC), T-325, ZH-161, S-24, and
ZH-305, and their sensitivity to TG02 have been described in detail
(12). LN-229 cells were cultured in
Dulbecco's modified Eagle's medium supplemented with 10% fetal calf
serum (Invitrogen) and 1% glutamine (Invitrogen). After approval of
the local ethics committees of the procedure and after obtaining
informed consent from patients, GIC were isolated from freshly
resected tumors using the Papain Dissociation System (Worthington
Biochemical Corporation). GIC were maintained in Neurobasal Medium
(NB) with B-27 supplement (20 µl/ml) (Thermo Fisher Scientific,
Inc.), L-glutamine (10 µl/ml), fibroblast growth factor-2 and
epidermal growth factor (20 ng/ml each; Peprotech) and
penicillin/streptomycin (pen-strep, Sigma-Aldrich/Merck). The GIC
were studied within a range of maximum passages of 40–50. All cells
were sent for short tandem repeat analysis (DSMZ) and are regularly
tested for mycoplasma contamination, last in November 2018.
Acute growth inhibition assay
After seeding cells and after a recovery time of 24
h in complete medium, the cells were exposed to TG02 for acute
growth inhibition assays for 72 h (high seeding density,
1.5×104 cells for LN-229 or 104 cells for GIC
per well) or for the determination of clonogenic survival (low
seeding density, 50 cells for LN-229 or 150 cells per well for GIC)
for more than ten days in 96 well plates. Human IFN-β1a was added
24 h prior to TG02 exposure and maintained throughout the
experiment as indicated. Metabolic activity was assessed by MTT
assay (Sigma Aldrich).
RT-qPCR
For gene expression analyses, total mRNA was
extracted after 24 h incubation in serum-free medium. Gene
expression was determined by the Real-Time PCR System QuantStudio 6
using PowerUp SYBR™ Green Master Mix (Thermo Fisher Scientific,
Inc.) using primers at optimized concentrations. Relative
quantification was calculated using the ΔCT-method (15) and specific target gene expression was
normalized to the housekeeping genes hypoxanthine
phosphoribosyltransferase 1 (HPRT1) or ADP-ribosylation factor 1
(ARF1). The following human-specific primers were used: IFNAR2
(Bio-Rad; qHsaCED0045841), XAF-1 (Bio-Rad; qHsaCED0045841), ARF1
(forward 5′-GACCACGATCCTCTACAAGC-3′, reverse
5′-TCCCACACAGTGAAGCTG-3′), caspase 3 (forward
5′-TGGAGATCAGCTCCCGAGATG-3′, reverse 5′-ATTGCCCACAGCCACTCTG-3′),
HPRT1 (forward 5′-TGAGGATTTGGAAAGGGTGT-3′, reverse
5′-AGCACACAGAGGGCTACAA-3′), IFNAR1 (forward
5′-TATGCTGCGAAAGTCTTCTTGAG-3′, reverse
5′-TCTTGGCTAGTTTGGGAACTGTA-3′) and myxovirus resistance protein A
(MxA) (forward 5′-TGGAGATCAGCTCCCGAGATG-3′, reverse
5′-ATTGCCCACAGCCACTCTG-3′) (all Microsynth).
Immunoblotting
Immunoblot analysis was performed as described
(12). Whole cell lysates were
prepared using radioimmunoprecipitation assay (RIPA) buffer [10 mM
Tris pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS]
supplemented with 1% complete protease inhibitor mix (Roche
Diagnostics) and phosphatase inhibitor cocktails 1 and 2
(Sigma-Aldrich), and protein levels were determined by Bradford
assay. After loading equal protein amounts, SDS-PAGE (10%
acrylamide gels) was performed under reducing conditions followed
by protein transfer to nitrocellulose membranes. To avoid
unspecific antibody binding, membranes were blocked in
Tris-buffered saline containing 0.1% Tween 20 and 5% skim milk or
5% bovine serum albumin. For primary antibody incubation the
following antibodies were used: Rabbit anti-IFNAR1 (LifeSpan
BioSciences Inc., Seattle, WI, LS-C185508), mouse anti-MxA
hybridoma supernatant (provided by J. Pavlovic, Zurich,
Switzerland, clone 143), rabbit anti-XAF-1 (Santa Cruz
Biotechnology; sc-19194), rabbit anti-caspase 3 (Santa Cruz
Biotechnology; sc-9662), rabbit anti-phosphorylated-Rpb1 (RNAPII)
(Cell Signaling Technology, 13499), rabbit anti-MCL-1 Cell
Signaling Technology; 5453), rabbit anti-c-MYC (Cell Signaling
Technology; 5605), rabbit anti-CDK9 (Santa Cruz Biotechnology;
sc-13130) or rabbit anti-β-actin (Santa Cruz Biotechnology;
sc1616). As secondary antibodies Amersham ECL sheep anti-mouse
IgG-HRP (GE Healthcare Lifesciences; NA931V) or goat anti-rabbit
IgG-HRP (Santa Cruz Biotechnology; sc-2004) were used.
Flow cytometry
Flow cytometry was performed according to standard
protocols using phycoerythrin (PE)-conjugated monoclonal mouse
anti-human IFNAR2 (PBL Assay Science; clone MMHAR-2, 10 µg/ml) or
PE-conjugated mouse IgG1κ isotype control (BD Biosciences; clone
MOPC-21, 10 µg/ml). Induction of cell death was analyzed using
annexin V (AnxV) (BioLegend) and propidium iodide (PI)
(Sigma-Aldrich) staining. Cells were washed with annexin buffer (10
mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) and stained
with 5 µl AnxV and 5 µl PI [50 µg/ml] for 15 min at room
temperature in the dark. Staurosporine was used as positive control
for apoptosis. Fluorescence intensity was recorded using a BD FACS
Verse flow cytometer (BD Biosciences). Data (10,000 events per
condition) were analyzed and median fluorescent intensities were
calculated using FlowJo Version 10.0.7 (Tree Star).
Caspase activity assay
After exposure to TG02 or IFN-β1a alone or in
combination in the absence or presence of zVAD-fmk, the cells were
lysed with lysis buffer followed by incubation with the fluorescent
substrate Ac-DEVD-amc [6.25 µM]. Measurement of DEVD-amc cleaving
activity was performed using a Tecan Infinite M200 Pro plate reader
(Tecan Trading AG) (480 nm emission; 360 nm excitation (12).
Transfection
For transient gene silencing, cells were transfected
at 20 µM of specific or scramble control small interfering (si) RNA
by electroporation. Control or ON-TARGET plus siRNA SMARTpool were
purchased from Dharmacon (caspase 3, L-004307-00-0005;
xaf, L-004357-00-0005). At 72 h post transfection, samples
were treated with IFN-β1a or TG02 or both as indicated and
processed for further treatment. For stable gene overexpression,
cells were transfected with the sequence-verified human
bcl-2/pcDNA3 plasmid (Addgene no. 19279) using the X-tremeGENE™ HP
DNA Transfection Reagent. G418 sulfate was used for selection of
clones with transgene expression (12).
Statistical analysis
Data are representative of experiments performed two
to three times with similar results. Statistical analysis was
performed using GraphPad Prism 5 or 7 software. Statistical
significance was assessed using either two-sided unpaired and
paired Student's t-test or one-way ANOVA with Tukey's post hoc test
for multiple comparison analysis. Quantitative data are represented
as mean ± standard deviation or standard error of the mean.
P<0.05 was considered to indicate a statistically significant
difference. Synergy was assessed using the fractional product
method (16) and differences
exceeding 10% of observed versus predicted (additive) effect were
considered to indicate synergy.
Results
IFN-β promotes TG02-induced cell
death
The expression of IFN-α/β receptors 1 and 2 in the
human glioma cell line models used here and their uniform
responsiveness to IFN-β in terms of MxA gene induction have been
partially published (10,17) and are summarized in Fig. S1. Growth inhibitory effects of TG02
alone in the cell line models studied here have also been
characterized (12). We first
explored whether pre-exposure to IFN-β sensitized glioma cells to
TG02 in short-term growth inhibition or clonogenicity respectively
spherogenicity assays. GIC were exposed to lower concentrations of
IFN-β than the LN-229 long-term cell line because of the higher
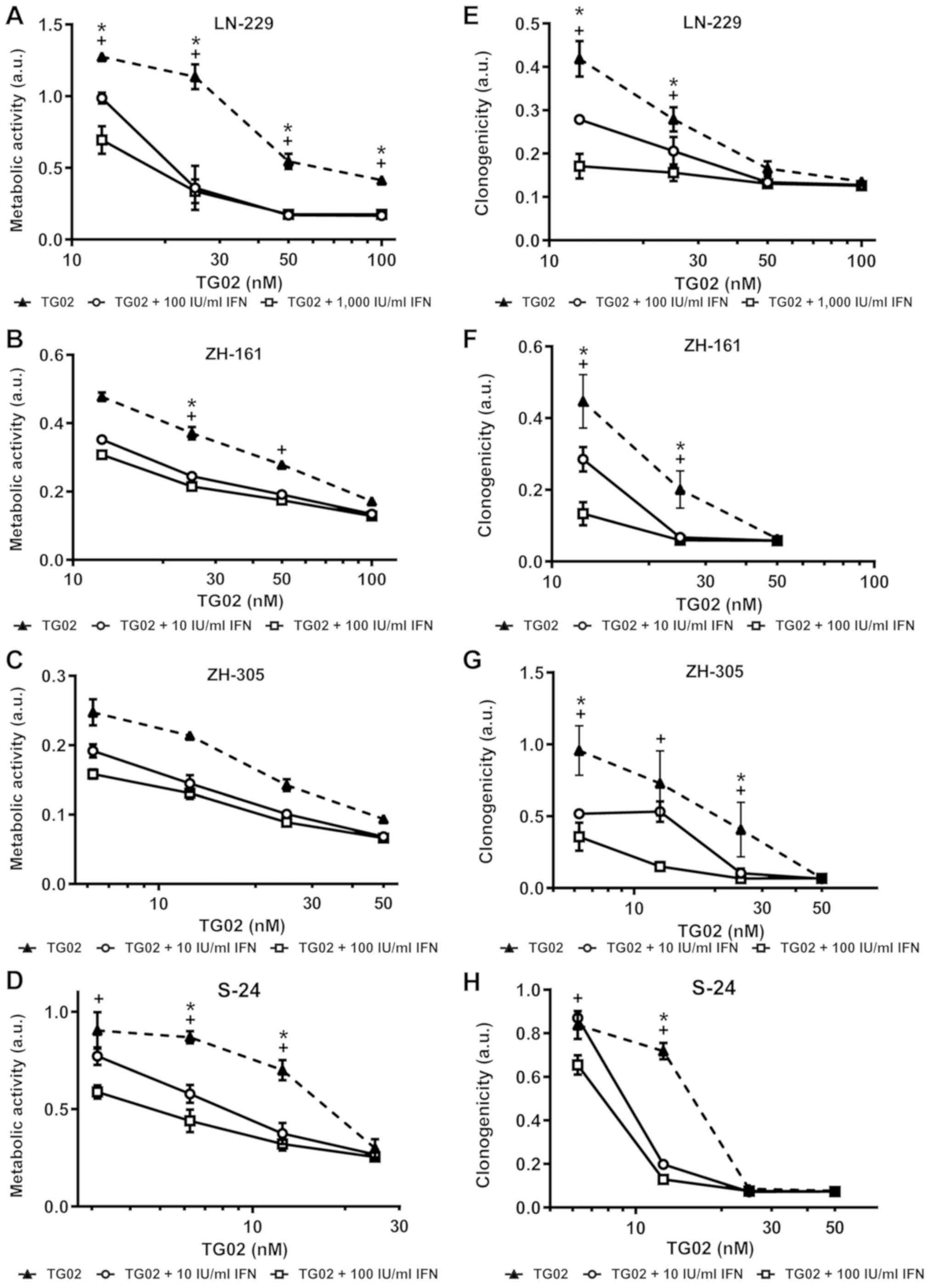
intrinisic sensitivity to IFN-β of GIC (10). We noted strong sensitization by IFN-β
to TG02 in LN-229 and S-24 cells in acute growth inhibition assays
and in S-24 cells in spherogenicity assays and largely additive
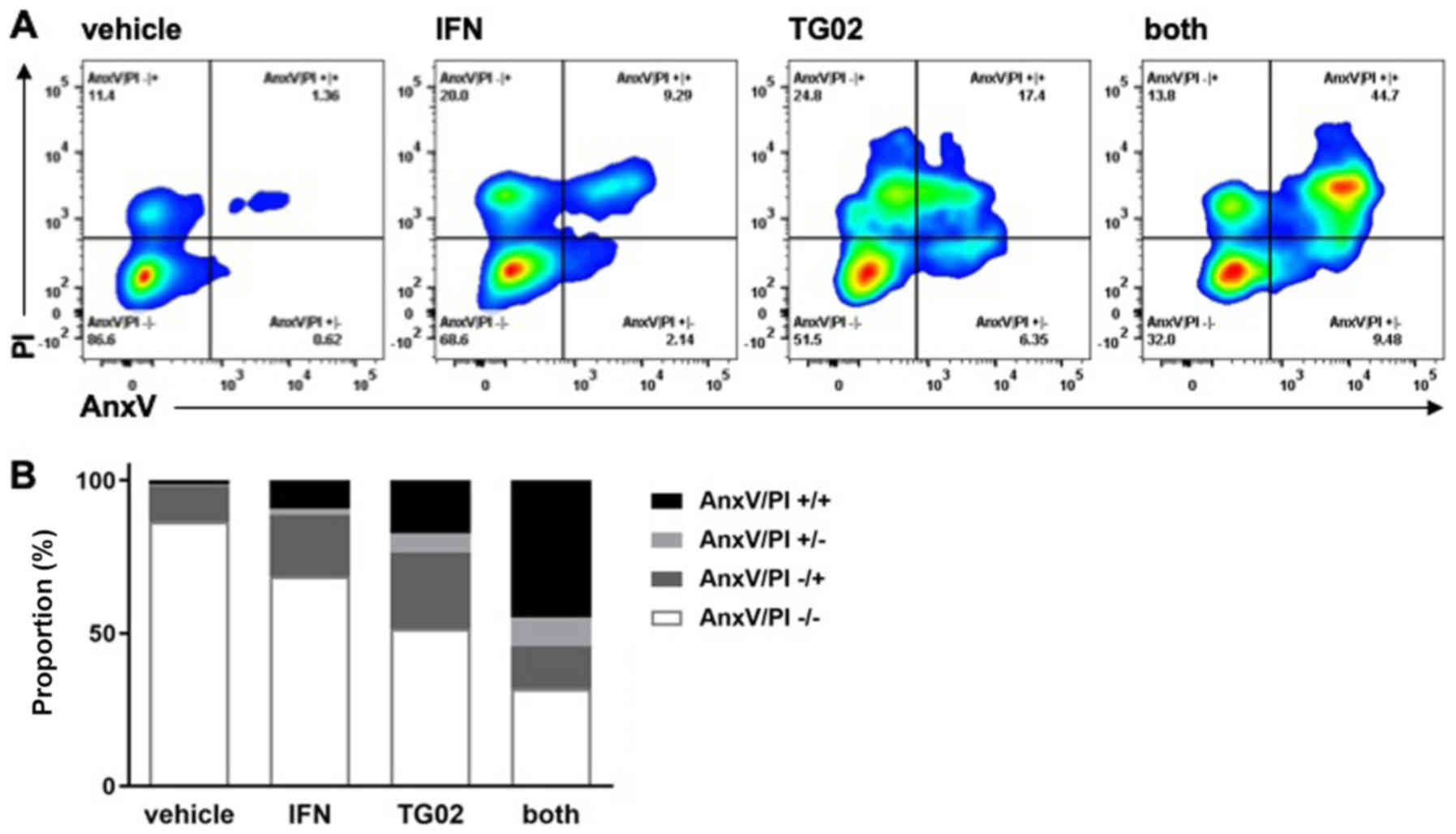
effects in both assays in the other model systems (Fig. 1A-H). Synergistic induction of cell
death by IFN-β and TG02 in LN-229 cells was confirmed by AnxV/PI
flow cytometry (Fig. 2).
Synergistic induction of caspase
activation does not mediate synergistic growth inhibition by IFN-β
and TG02
We had previously observed that exposure to IFN-β
induced changes in gene expression, notably induction of xaf
expression, predicting enhanced vulnerability to caspase-dependent
apoptosis (10) and that TG02 alone
induces largely caspase-independent apoptosis (12). Indeed, pre-exposure of LN-229 cells
to IFN-β facilitated processing of caspase 3 in response to TG02 as
confirmed by immunoblot, albeit only to a minor degree compared
with exposure to staurosporine which was used as a positive control
(18) (Fig. 3A). Facilitation of induction of
DEVD-amc-cleaving caspase activity by pre-exposure to IFN-β was
confirmed by a fluorimetric assay. Specificity was confirmed by
demonstrating the abrogation of this caspase activity by the broad
spectrum caspase inhibitor, zVAD-fmk, under all conditions
(Fig. 3B). Similarly, BCL-2 gene
transfer into LN-229 cells abrogated the promotion of caspase
activation induced by the combination of IFN-β and TG02, again
using staurosporine as a positive control (Fig. 3C). We have previously reported that
BCL-2 moderately attenuates TG02-induced cell death, likely in a
caspase-independent manner since zVAD-fmk had no such effect except
for high TG02 concentrations in selected models (12). Here we report that neither zVAD-fmk
nor BCL-2 interfered specifically with the sensitizing properties
of IFN-β for TG02-induced cell death (Fig. 3D and E). This challenges the view
that the enhanced caspase activity demonstrated in Fig. 3A-C was responsible for synergistic
growth inhibition.
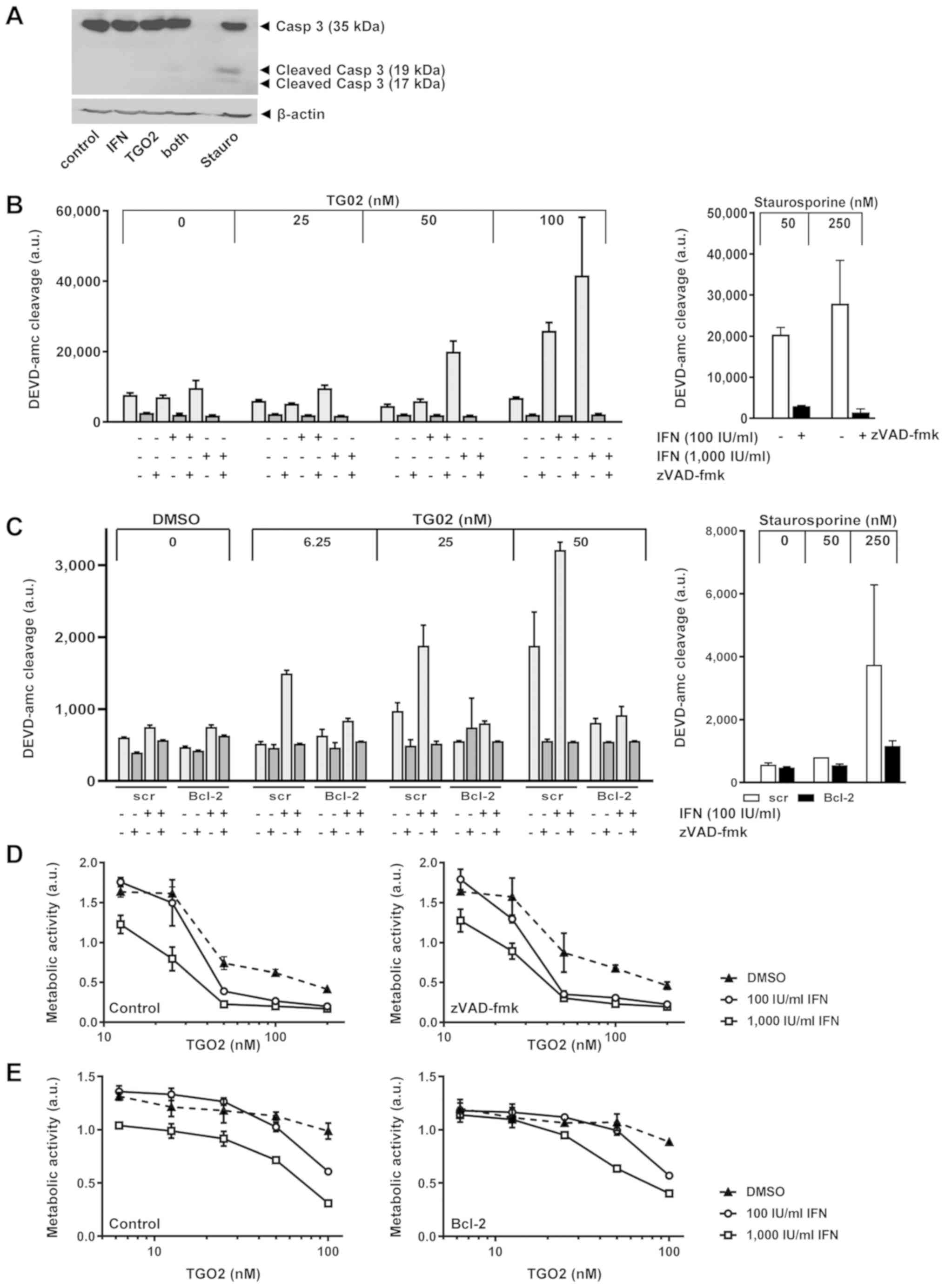 | Figure 3.IFN-β promotes DEVD-amc-cleavage
activity induced by TG02. (A) Caspase 3 processing was monitored in
LN-229 cells pre-exposed or not to IFN-β (1,000 IU/ml) for 24 h and
then treated with TG02 (50 nM) in the absence or presence of IFN-β
for another 24 h. (B) LN-229 cells were pretreated or not with
IFN-β (100 or 1,000 IU/ml) for 24 h and then exposed to TG02 in the
absence or presence of zVAD-fmk (100 µM) for another 24 h prior to
fluorimetric determination of DEVD-amc cleavage. (C) A similar
experiment was conducted to compare the effects of IFN-β and TG02
and their combination in bcl-2-transfected versus control
transfected cells. In (B) and (C), stauro treatment for 24 h was
used as a positive control. (D) LN-229 cells were treated as in
Fig. 1A, but co-exposed to zVAD-fmk
(100 µM) during TG02 exposure. (E) Bcl-2-transfected vs.
control transfected LN-229 cells were pretreated or not with IFN-β
(100 or 1,000 IU/ml) for 24 h and then exposed to TG02. Metabolic
activity was assessed at 72 h by MTT assay. Casp 3, caspase 3; IFN,
interferon; stauro, staurosporine; ac-DEVD-amc,
acetyl-Asp-Glu-Val-Asp-7-amino-4-methyl coumarin; zVAD-fmk,
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone; a.u., arbitrary
units. |
Neither xaf nor caspase 3 gene
silencing block IFN-β induced sensitization to TG02
To determine in more depth whether caspase-dependent
apoptotic signalling mediates the synergy of IFN-β and TG02, we
explored whether xaf induction or expression of caspase 3
were required for IFN-β-mediated sensitization to TG02. XAF is a
previously identified IFN-β response gene in human glioma cells
including GIC (10) (Fig. S2A). We silenced xaf expression
(Fig. S2B) which led to a strong
reduction of XAF protein in response to IFN-β (Fig. 4A). However, depletion of XAF did not
abrogate the sensitization of glioma cells to TG02 mediated by
pre-exposure to IFN-β (Fig. 4B).
Similarly, depletion of caspase 3 failed to affect the activity of
this combination (Fig. 4C and D). We
had previously observed features of senescence in one of two
long-term glioma cell lines chronically exposed to TG02 (12). Accordingly, we determined
senescence-associated β-galactosidase activity in two GIC models
upon exposure to IFN-β or TG02 or both, using irradiation at 20 Gy
as a positive control (19). S-24
cells stimulated with IFN-β or TG02 showed no increase in
β-galactosidase activity whereas co-treatment did, but no such
effect was seen in ZH-161 cells (Fig.
S3).
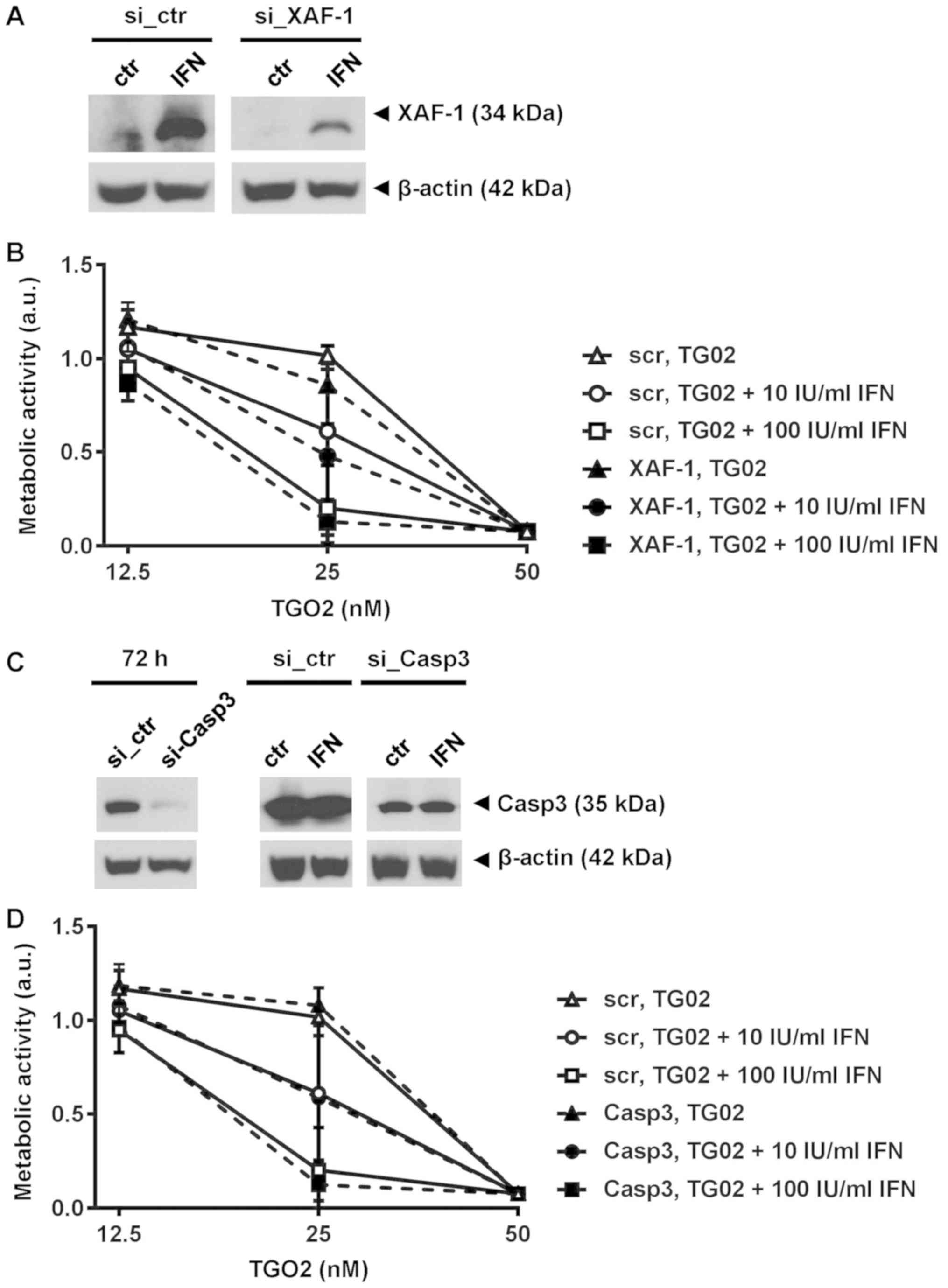 | Figure 4.IFN-β-mediated sensitization does not
require XAF-1 or caspase 3. (A) XAF-1 protein levels were
determined with or without IFN-β1a stimulation (1,000 IU/ml) for 24
h started at 72 h after electroporation to induce XAF-1 silencing
in ZH-161 cells. (B) ZH-161 were pretreated or not with IFN-β at 10
or 100 IU/ml for 24 h and then exposed to increasing concentrations
of TG02. Metabolic activity was assessed after >10 days by MTT
assay. (C) Caspase 3 gene silencing in ZH-161 cells assessed by
immunoblot at 72 h, maintained in the presence of IFN-β [(1,000
IU/ml) for 24 h]. β-actin served as loading control. (D)
Control-transfected or caspase 3-silenced ZH-161 cells treated as
in (B). XAF, XIAP-associated factor; si, small interfering RNA;
ctr, control; IFN, interferon; Casp3, caspase 3; a.u., arbitrary
units; scr, scrambled control. |
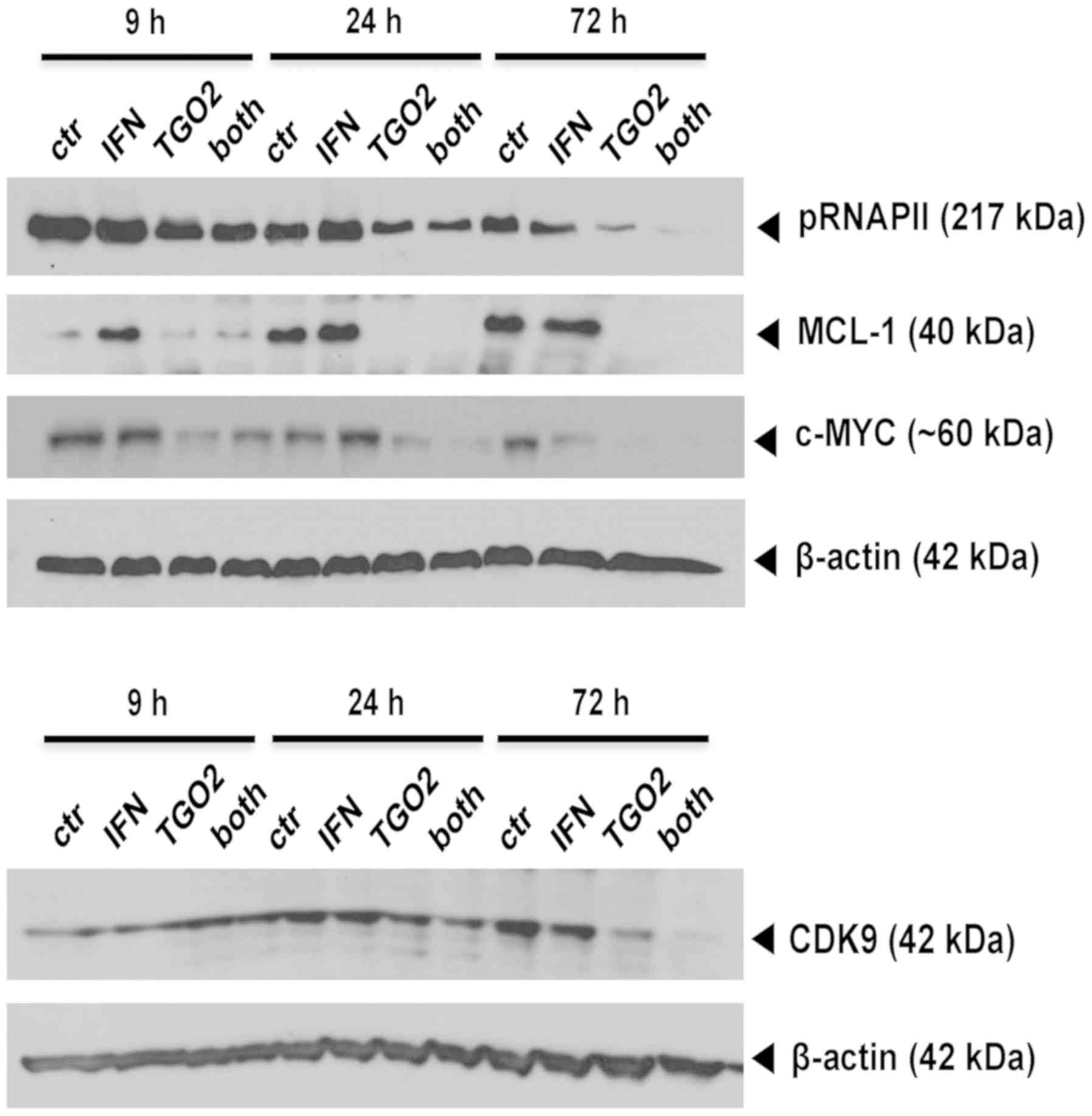
IFN-β facilitates direct TG02 target
inhibition
Altogether these data indicated that the synergistic
growth inhibition mediated by pre-exposure to IFN-β followed by
TG02 treatment might operate further up-stream, prior to the cell
death pathways involving caspase inhibition or senescence. Indeed,
immunoblot analyses allowed to demonstrate a time-dependent
suppression of RNA polymerase II phosphorylation by TG02 that was
facilitated in glioma cells pre-exposed to IFN-β. Yet, the
TG02-induced decreases of two major putative down-stream effectors
of TG02, MCL-1 and c-MYC, were not enforced by IFN-β. In contrast,
the protein levels of CDK9, the bona fide main target of TG02 and
the kinase responsible for RNA polymerase II phosphorylation, were
decreased by TG02 alone and more so by the combination, at 72 h
(Fig. 5).
Discussion
Effective pharmacological treatment options for
glioblastoma have not been developed since the introduction of
temozolomide in 2005 (5,6). Immunotherapy may have failed because
glioblastomas are currently considered as lymphocyte-depleted or
cold tumors (20). Targeted therapy
has not been successful because glioblastoma in most instances, if
not all, is not a single pathway-driven disease (7). This provides space for the development
of innovative, more broadly acting anti-cancer agents in
glioblastoma. A prominent example currently explored in a pivotal
trial is the proteasome inhibitor, marizomib (NCT03345095). Another
example is TG02, an orally available inhibitor of multiple CDK with
good blood brain barrier penetration (NCT03224104). This drug has
shown promising single agent activity at least in cell culture
models (12,13).
We report that pre-exposure to type I interferon
sensitizes human glioma models including GIC cultures to subsequent
exposure to TG02, both in acute growth inhibition and in clonogenic
survival assays (Fig. 1). We
hypothesized that this synergy was related to the promotion of
caspase-dependent apoptosis since IFN-β induces a shift in gene
expression that should facilitate caspase-mediated cell death
(10). Furthermore, TG02 alone is
not a potent activator of the caspase-dependent cell death pathway,
but activates alternative death pathways (12). Consistent with the expectation,
immunoblot and caspase activity assays confirmed synergy in
promoting caspase activation when IFN-β and TG02 were combined.
Consistent with a mitochondrial type of caspase-mediated cell
death, overexpression of BCL2 was protective (Fig. 3). A likely candidate promoting
synergistic cell death was the proapoptotic protein, XAF1, which is
highly inducible by type I interferon (10). Yet, a role of XAF1 in mediating
synergistic cell death induction was excluded by XAF gene
silencing. Further, somewhat unexpectedly, gene silencing of
caspase 3 did not abrogate the synergy of type I interferon and
TG02 either, and, expectedly, also not the activity of either agent
administered alone (Fig. 4).
Finally, we reconsidered that potentially synergy
might operate more up-stream at the level where TG02 proximately
triggers the cell death cascade. Indeed, immunoblot confirmed that
there was a time-dependent decrease in the phosphorylation of
RNAPII which was greatly facilitated by pre-exposure to IFN-β
(Fig. 5). This loss of pRNAPII is
predicted in response to TG02 since RNAPII is phosphorylated by
CDK9, the principal target of TG02. That total CDK9 protein was
also decreased, points to cdk9 being one of the genes with
short-lived expression that is subject to TG02-induced
transcriptional perturbations. Exploring how IFN signaling primes
glioma cells for TG02-mediated direct target inhibition may help to
design novel, more effective pharmacological approaches to
glioblastoma.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by an unrestricted
grant from Adastra to MW and by a Swiss National Science Foundation
grant to MW (grant no. 31003A_166634/1).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ELR and MW conceived and supervised the study. BL,
ELR and MW designed experiments. BL, ELR, MS and ME performed
experiments. BL, ELR, MS and MW analysed data. MW wrote the first
draft of the manuscript. BL and ELR made manuscript revisions. All
authors approved the final version of the manuscript.
Ethics approval and consent to
participate
The generation of human GIC was approved by the
Ethics Committee of the Canton of Zurich, Switzerland (approval no.
KEK 2016-00456). Informed consent was obtained from the patients
from whose tumors the GIC lines were generated.
Patient consent for publication
Not applicable.
Competing interests
MW received an unrestricted research grant from
Adastra (San Diego, CA). The other authors declare that they have
no competing interests.
Glossary
Abbreviations
Abbreviations:
|
AnxV
|
AnnexinV
|
|
ARF1
|
ADP-ribosylation factor 1
|
|
CDK
|
cyclin-dependent kinase
|
|
GIC
|
glioma-initiating cell
|
|
HPRT1
|
hypoxanthine phosphoribosyltransferase
1
|
|
IFN
|
interferon
|
|
IFNAR
|
IFN-α/β receptors
|
|
MxA
|
myxovirus resistance protein A
|
|
PE
|
phycoerythrin
|
|
pRNAPII
|
phosphorylated RNA polymerase II
|
|
PI
|
propidium iodide
|
|
RT-PCR
|
reverse transcriptase polymerase chain
reaction
|
|
si
|
small interfering
|
|
XAF
|
XIAP-associated factor
|
References
|
1
|
Ostrom QT, Gittleman H, Truitt G, Boscia
A, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical report:
Primary brain and other central nervous system tumors diagnosed in
the united states in 2011–2015. Neuro Oncol. 20 (Suppl-4):iv1–iv86.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weller M, Wick W, Aldape K, Brada M,
Berger M, Pfister SM, Nishikawa R, Rosenthal M, Wen PY, Stupp R and
Reifenberger G: Glioma. Nat Rev Dis Primers. 1:150172015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
Classification of Tumors of the Central Nervous System: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brennan CW, Verhaak RGW, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al: The somatic genomic landscape of glioblastoma.
Cell. 155:462–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weller M, van den Bent M, Tonn JC, Stupp
R, Preusser M, Cohen-Jonathan-Moyal E, Henriksson R, Le Rhun E,
Balana C, Chinot O, et al: European association for neuro-oncology
(EANO) guideline on the diagnosis and treatment of adult astrocytic
and oligodendroglial gliomas. Lancet Oncol. 18:e315–e329. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Prados MD, Byron SA, Tran NL, Phillips JJ,
Molinaro AM, Ligon KL, Wen PY, Kuhn JG, Mellinghoff IK, de Groot
JF, et al: Toward precision medicine in glioblastoma: The promise
and the challenges. Neuro Oncol. 17:1051–1063. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yue C, Xu J, Tan Estioko MD, Kotredes KP,
Lopez-Otalora Y, Hilliard BA, Baker DP, Gallucci S and Gamero AM:
Host STAT2/type I interferon axis controls tumor growth. Int J
Cancer. 136:117–126. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Du Z, Cai C, Sims M, Boop FA, Davidoff AM
and Pfeffer LM: The effects of type I interferon on glioblastoma
cancer stem cells. Biochem Biophys Res Commun. 491:343–348. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Happold C, Roth P, Silginer M, Florea AM,
Lamszus K, Frei K, Deenen R, Reifenberger G and Weller M:
Interferon-β induces loss of spherogenicity and overcomes therapy
resistance of glioblastoma stem cells. Mol Cancer Ther. 13:948–961.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wakabayashi T, Natsume A, Mizusawa J,
Katayama H, Fukuda H, Sumi M, Nishikawa R, Narita Y, Muragaki Y,
Maruyama T, et al: JCOG0911 INTEGRA study: A randomized screening
phase II trial of interferonβ plus temozolomide in comparison with
temozolomide alone for newly diagnosed glioblastoma. J Neurooncol.
138:627–636. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Le Rhun E, von Achenbach C, Lohmann B,
Silginer M, Schneider H, Meetze K, Szabo E and Weller M: Profound,
durable and MGMT-independent sensitivity of glioblastoma cells to
cyclin-dependent kinase inhibition. Int J Cancer. 145:242–253.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Su YT, Chen R, Wang H, Song H, Zhang Q,
Chen LY, Lappin H, Vasconcelos G, Lita A, Maric D, et al: Novel
targeting of transcription and metabolism in glioblastoma. Clin
Cancer Res. 24:1124–1137. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu J, Bryla C, McCoy A, Lisa B, Garren N,
Siegel C, Grajkowska E, Theeler B, Park DM, Parrott T, et al:
ACTR-69. Phase I trial of TG02 plus dose-dense or metronomic
temozolomide for adults with recurrent anaplastic astrocytoma and
glioblastoma. Neuro Oncol. 19 (Suppl-6):vi15. 2017. View Article : Google Scholar
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Greco WR, Bravo G and Parsons JC: The
search for synergy: A critical review from a response surface
perspective. Pharmacol Rev. 47:331–385. 1995.PubMed/NCBI
|
|
17
|
Silginer M, Nagy S, Happold C, Schneider
H, Weller M and Roth P: Autocrine activation of the IFN signaling
pathway may promote immune escape in glioblastoma. Neuro Oncol.
19:1338–1349. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weller M, Trepel M, Grimmel C, Schabet M,
Bremen D, Krajewski S and Reed JC: Hypericin-induced apoptosis of
human malignant glioma cells is light-dependent, independent of
bcl-2 expression, and does not require wild-type p53. Neurol Res.
19:459–470. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fumagalli M, Rossiello F, Mondello C and
d'Adda di Fagagna F: Stable cellular senescence is associated with
persistent DDR activation. PLoS One. 9:e1109692014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lim M, Xia Y, Bettegowda C and Weller M:
Current state of immunotherapy for glioblastoma. Nat Rev Clin
Oncol. 15:422–442. 2018. View Article : Google Scholar : PubMed/NCBI
|