Introduction
Cancer metastasis is the process of cancer cells
disseminating from the primary tumor to a distal site through
lymphatic tissue and blood vessels. Cancer metastasis is
responsible for approximately 90% of cancer deaths, indicating that
it is the primary cause of morbidity and mortality (1). Even though most solid tumors are now
manageable or curable by advances in early cancer detection and
treatment, cancers spreading beyond the initial primary site are
usually highly incurable (2). Lack
of understanding of the mechanism underlying the metastatic process
has meant that the predominant cancer treatments focus on
inhibition of cancer growth with little emphasis on metastasis,
meaning that the overall survival of metastatic cancer patients has
not been improved significantly.
Transforming growth factor-β (TGFβ) is one of the
master factors of metastasis in that it induces the
epithelial-mesenchymal transition (EMT), which is associated with
cancer. EMT is the reversible orchestrated transcriptional program
in which well-organized, tightly connected epithelial cells
transdifferentiate into disorganized and motile mesenchymal cells.
TGF-β signaling mediated by SMAD or non-SMAD pathways plays a
fundamental role in activating the transcriptional network to
induce the expression of mesenchymal components and to suppress the
expression of epithelial genes (3,4). As a
result, epithelial cancer cells undergo dramatic remodeling of the
cytoskeleton along with dissolution of tight junctions to acquire
mesenchymal features that exhibit a significantly enhanced
metastatic dissemination potential into distal organs. This event
is induced by the activity of master regulators of EMT, which
include SNAIL, SLUG, ZEB1/delta EF1 and ZEB2/SIP1 (5–8).
ELK3 is an ETS domain-containing protein capable of
forming a ternary complex with DNA and serum response factor
(9). ELK3 is reported to be involved
in the migration and invasion of various cancer cells including
aggressive basal-like breast cancer cells and liver cancer stem
cells (10,11). Previously, we reported that ELK3
suppression impairs the ability of TGFβ signaling to activate the
expression of mesenchymal markers such as Vimentin, Slug and SNAIL
in the triple negative breast cancer cells, which suggests that
ELK3 is implicated in the TGFβ signaling pathway to regulate the
metastatic process of aggressive cancer cells (12,13).
In the present study, to extend our understanding of
the molecular implication of ELK3 to the TGFβ signaling pathway in
cancer cells, we analyzed the regulatory mechanism of TGFβ
signaling on ELK3 expression. We found that TGFβ stimulates the
transcriptional expression of ELK3 in the representative triple
negative breast cancer cell line, MDA-MB231. Furthermore, based on
the biochemical and molecular biology study, we demonstrated that
TGFβ-mediated phosphorylation of SMAD3 functions as a
transcriptional activator of ELK3. Taken together, our data reveal
that ELK3 is a direct downstream target of TGFβ-SMAD3 signaling
pathway in MDA-MB231 cells.
Materials and methods
Plasmids, siRNA and primers
Information on the plasmids and siRNAs is summarized
in the supplementary Tables SI and
SII.
Cell culture and transfection
The triple negative breast cancer cell line
MDA-MB231 and the human breast adenocarcinoma cell line MCF7 and
the human embryonic kidney 293T cells were purchased from American
Type Culture Collection (Manassas, VA, USA). These cells were
maintained in DMEM (Gibco BRL Life Technologies, Rockville, MD,
USA) containing 10% (v/v) heat-inactivated fetal bovine serum
(Gibco BRL). 293T cells were used for the luciferase assay with the
pGL3-ELK3 plasmid. Transient transfection of plasmid DNA or siRNA
was performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA,
USA) according to the manufacturer's protocols.
RNA extraction and reverse
transcription-quantitative (RT-q)PCR
Total RNA was extracted by manual methods using
TRIZol (Invitrogen), and 1 µg of cDNA was synthesized using the
LeGene Express 1st Strand cDNA Synthesis System (LeGene Biosciences
Inc., San Diego, CA, USA) according to the manufacturer's
instructions. RT-qPCR was performed using synthetic cDNAs and
TOPreal™ qPCR 2X PreMIX (Enzynomics, Daejeon, Korea). The
expression of the target genes was normalized to that of
glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The PCR primers
are listed in Table SIII.
Immunoblot analysis
Cells were lysed with RIPA buffer (Cell Signaling
Technology, Beverly, MA, USA) and total cell lysates were separated
by sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and then transferred to polyvinylidene difluoride
membranes (Bio-Rad, Hercules, CA, USA). The membranes were blotted
with the indicated primary antibodies at 4°C overnight. After
washing with TBST, the membranes were incubated for 1 h at room
temperature with secondary antibodies. Immunoreactivity was
detected with an ECL kit (Thermo Scientific, Rochester, NY, USA).
The antibodies used in this study are summarized in Table SIV.
Luciferase assay
The 293T cells were transfected with the indicated
plasmids using Lipofectamine 2000 (Invitrogen) according to the
manufacturer's protocols. Cells were harvested 48 h after
transfection, and luciferase activity was measured using the
Dual-Luciferase Reporter Assay System (Promega) according to the
manufacturer's protocols. The values of firefly luciferase were
normalized to the respective values of Renilla
luciferase.
Chromatin immunoprecipitation
In brief, 37% formaldehyde was added to the cell
culture medium to a final concentration of 1% and incubated for 15
min at RT. Glycine was added to a final concentration of 125 mM for
5 min at RT, and the cells were washed three times with cold PBS.
The cells were lysed in 400 µl of 1X cell lysis buffer (Cell
Signaling) containing protease/phosphatase inhibitor cocktail
(Pierce Biotechnology). After eight rounds of sonication, the
lysates were cleared by centrifugation at 13,000 rpm for 15 min at
4°C. The supernatants were mixed with 40 µl of Dynabead protein G
and 2 µg of primary antibodies for 2 h at RT or overnight at 4°C.
The complexes were washed sequentially with 1X RIPA buffer, 1X RIPA
buffer (500 mM NaCl), LiCl buffer and TE buffer twice for 10 min
each. Then, 3 µl of 10% SDS and 5 µl of 20 mg/ml proteinase K were
added to separate the DNA-protein complex. The DNA was purified by
the phenol/chloroform extraction method, and then it was used in
PCR with primers targeting the ELK3 promoter.
Statistical analysis
Samples were analyzed with Student's t-test or ANOVA
with Duncan's multiple range procedure for multiple comparisons.
All statistical analyses were performed using GraphPad Prism 5
(GraphPad Prism, USA) or the SigmaPlot 11.2 program (Systat
Software, USA). All statistical analyses were performed using
GraphPad Prism 5 (GraphPad Prism, USA). The error bars represent
the standard errors from three independent experiments, which were
each performed using triplicate samples. P-values less than 0.05
were considered statistically significant.
Results
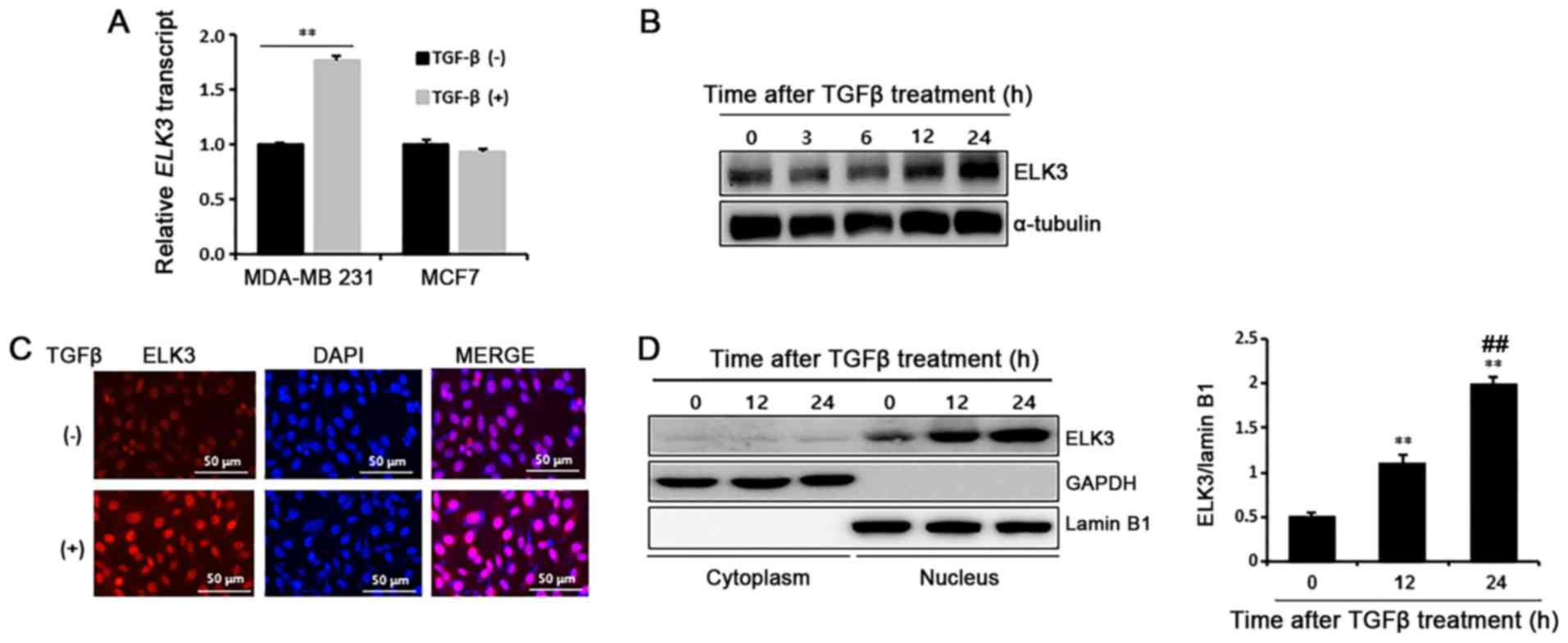
TGFβ induces accumulation of ELK3 in
the nucleus of MDA-MB231 cells, but not in MCF7 cells
Cancer cells treated with TGFβ undergo the EMT
process by developing a fibroblast-like morphological appearance
and changing epithelial and mesenchymal phenotype marker
expression. Unlike MDA-MB231 cells, TGFβ-treated MCF7 cells that
display morphological changes of EMT do not show suppression of
E-cadherin, a typical epithelial phenotype marker (14). Recently, we reported that ELK3
is highly expressed in TNBC-like MDA-MB231 cells, where it
functions as a transcriptional repressor of E-cadherin by
collaborating with ZEB1 (15).
Therefore, we hypothesized that ELK3 is the missing link that
explains the different molecular responses of MDA-MB231 and MCF7
cells when they are treated with TGFβ. We first compared the
expression of ELK3 between MDA-MB231 and MCF7 cells
following TGFβ treatment. As expected, TGFβ stimulated ELK3
expression in MDA-MB231 cells but not in MCF7 cells (Fig. 1A). Consistently, ELK3 protein was
also accumulate in the TGFβ-treated MDA-MB231 cells (Fig. 1B). Immunocytochemical analysis and
subcellular fractionation assays of the cytosol and nucleus
confirmed that ELK3 accumulates in the TGFβ-treated MDA-MB231 cells
(Fig. 1C and D). Overall, these data
indicate that TGFβ induces transcriptional activation of
ELK3 in MDA-MB231 cells but not in MCF7 cells.
TGFβ activates ELK3 expression via
SMAD3
To understand the underlying mechanism of ELK3
activation by TGFβ treatment, we examined the effect of SB431542,
an inhibitor of TGFβ type I receptor, on the TGFβ-mediated
ELK3 expression. As shown in Fig.
2A, pretreatment with SB431542 inhibited mRNA and protein
accumulation of ELK3 in the TGFβ-treated MDA-MB231 cells.
Consistent with the result of chemical inhibition, transfection of
siRNA targeting TGFβ type I receptor abolished the effect of TGFβ
on the transcriptional activation of ELK3 (Fig. 2B). We next questioned whether
ELK3 expression is regulated by SMAD2 or SMAD3. Like
SNAIL, which is a downstream target of SMAD3, transfection
of an siRNA targeting SMAD3 (siSMAD3) hindered the
TGFβ-mediated expression of ELK3, whereas an siRNA targeting
SMAD2 (siSMAD2) did not interfere with the TGFβ effect on
ELK3 or SNAIL expression (Fig.
2C). The increase in expression of ELK3 over time in
TGFβ-treated MDA-MB231 cells was similar between the control and
siSMAD2 transfected MDA-MB231 cells, whereas siSMAD3
transfection abolished the effect of TGFβ on ELK3 expression
(Fig. 2D). Taken together, these
results suggest that TGFβ-mediated transcriptional activation of
ELK3 is mediated by SMAD3.
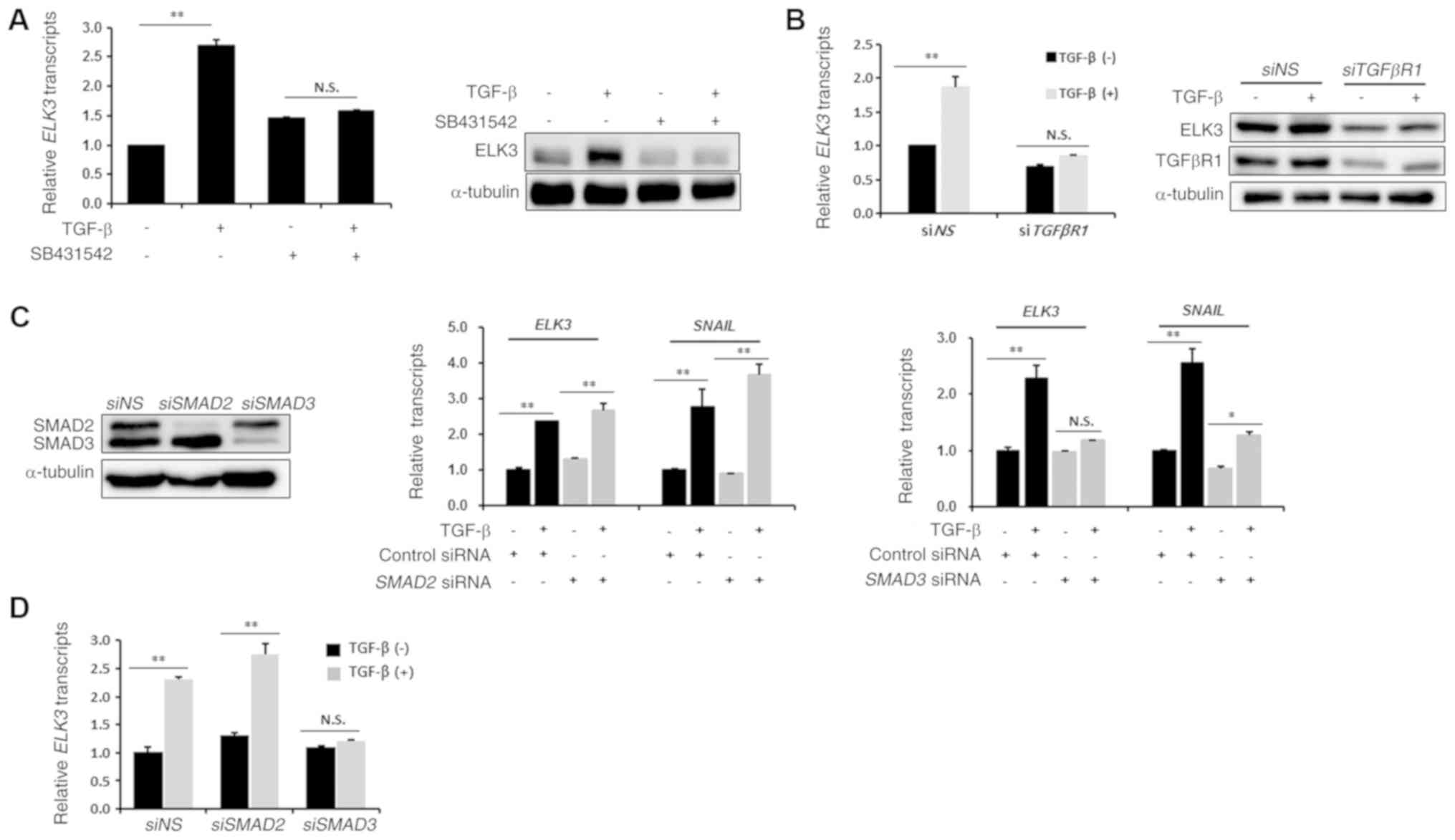 | Figure 2.Effect of TGFβ on ELK3
expression is mediated by SMAD3 but not by SMAD2. (A) Effect of
chemical inhibition of the TGFβ receptor on the TGFβ-mediated
activation of ELK3. Cells were pre-incubated with 10 µM
SB431542 for 1 h and then treated with 10 ng/ml TGFβ for 24 h.
Relative transcripts and protein levels of ELK3 and TGFβ receptor-I
were analyzed by RT-qPCR (left panel) and immunoblot analysis
(right panel), respectively. (B) Effect of molecular inhibition of
TGFβ receptor-I on the TGFβ-mediated activation of ELK3
expression. Non-specific or TGFβ receptor-I targeting siRNAs were
transfected into MDA-MB231 cells for 24 h followed by treatment
with 5 ng/ml of TGFβ for 24 h. Relative transcripts and protein
levels of ELK3 and TGFβ receptor-I were analyzed by RT-qPCR (left
panel) and immunoblot analysis (right panel), respectively. (C)
Effect of molecular inhibition of SMAD2 or SMAD3 on
the TGFβ-mediated activation of ELK3 expression.
Non-specific, SMAD2 or SMAD3 targeting siRNAs were
transfected into MDA-MB231 cells for 24 h, which was followed by
treatment with 5 ng/ml of TGFβ for 24 h. Knockdown effect of
SMAD2 and SMAD3 was analyzed by immunoblot analysis
(left panel). Relative transcripts and protein levels of ELK3 and
SNAIL, which is a target of SMAD3, were analyzed by RT-qPCR (middle
and right panel). (D) Effect of molecular inhibition of
SMAD2 or SMAD3 on the TGFβ-treated, time-dependent
activation of ELK3 expression. Nonspecific, SMAD2 or
SMAD3 targeting siRNAs were transfected into MDA-MB231 cells
for 24 h, which was followed by treatment with 5 ng/ml of TGFβ for
additional 24 h. The expression of ELK3 was quantified by
RT-qPCR. The error bars represent the standard errors from three
independent experiments, which were each performed using triplicate
samples. *P<0.05, **P<0.01. N.S, not significant; TGFβ,
transforming growth factor-β; RT-qPCR, reverse
transcription-quantitative PCR; si, small interfering; NS,
non-specific. |
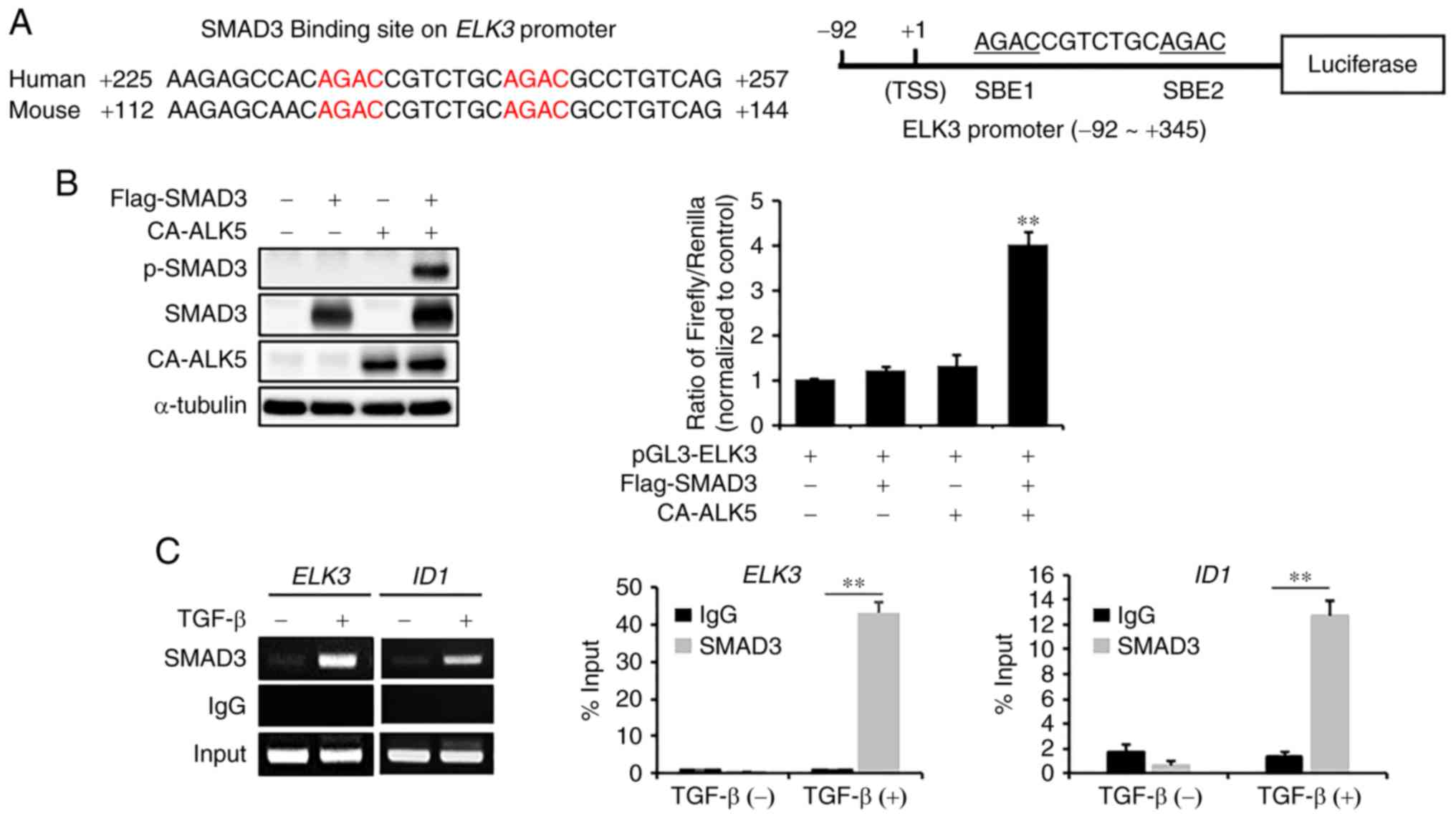
SMAD3 binds to the ELK3 promoter to
activate the transcription of ELK3 upon TGFβ treatment
To analyze whether SMAD3 functions as a direct
transcriptional activator of ELK3, we examined the sequences
of mouse and human ELK3 promoters from −2,000 bp to the
first exon region. We found that two SMAD3 binding sites (SBE) are
conserved at the first exon of the ELK3 promoter in the
human and mouse genomes. Therefore, we constructed a luciferase
reporter construct containing the promoter region of ELK3
from −92 bp to +345 bp (Fig. 3A). To
assess whether SMAD3 functions as a direct transcriptional
activator of the ELK3 promoter, the reporter plasmid
containing the ELK3 promoter was cotransfected into 293T
cells with plasmids encoding a constitutively active form of the
TGFβ type I receptor (CA-ALK5) or SMAD3. As shown in Fig. 3B, the ELK3 reporter plasmid is
activated only when ectopically expressed SMAD3 is phosphorylated
by cotransfection of the CA-ALK5-expressing plasmid in 293T cells.
To confirm that SMAD3 directly binds to the SBE of the ELK3
promoter, we performed chromatin immunoprecipitation (ChIP)
analysis with anti-SMAD3 antibody against genomic DNA of MDA-MB231
with or without TGFβ treatment. Since ID1, an inhibitor of
differentiation, is a direct downstream target of SMAD3 (16), it was used as a positive control in
the ChIP analysis. Like ID1, the SBE region of the ELK3
promoter was significantly enriched by immunoprecipitation with the
anti-SMAD3 antibody upon TGFβ stimulation (Fig. 3C). Taken together, we concluded that
SMAD3 activates transcription of ELK3 by directly binding to
the SBE region of the ELK3 promoter following TGFβ
treatment.
Discussion
During cancer development and progression in
malignancy, the TGFβ signaling pathway acts as a tumor promotor by
driving EMT, which induces tumor cell migration, invasion and
ultimately metastasis to distant organs. ELK3 is constitutively
activated in basal triple negative breast cancer cells (TNBCs) and
functions as a master regulator of cancer metastasis (10,12).
Previously, we suggested that the TGFβ signaling pathway is
interconnected with ELK3 activity, based on the fact that ELK3
knockdown in TNBCs induces collapse of TGFβ signaling (12). In this study, we demonstrated that
ELK3 is transcriptionally activated by TGFβ treatment in TNBCs.
Pharmacological and molecular analysis revealed that ELK3 is a
direct downstream target of SMAD3. In addition, TGFβ induced
migration was decreased in ELK3 knockdowned MDA-MB231 cells (data
not shown).
There are numerous reports that the TGFβ signaling
pathway is strictly regulated by a finely tuned system of negative
and positive feedback loops. The expression of SMAD7, a
representative inhibitory SMAD, is stimulated by TGFβ treatment and
forms a complex with E3 ubiquitin ligase to degrade the TGFβ
receptor, which results in the SMAD pathway inhibiting hyper
activation of TGFβ signaling (17).
During late stages of colorectal cancer, TGFβ activates miR-1269a
expression targeting SMAD7, hence forming a positive feedback loop
to promote metastasis (18). Since
TGFβ signaling is impaired by ELK3 suppression and ELK3 expression
is increased by TGFβ treatment, we suggest that TGFβ and ELK3 might
form a positive autofeedback loop to promote the EMT process.
Numerous studies have shown that inhibition of EMT
is considered an appropriate approach towards the prevention of
metastasis of cancer. Since TGFβ functions as an inducer of EMT,
blocking the TGFβ pathway is considered a promising strategy to
inhibit EMT in cancer cells; cytotoxic drugs such as paclitaxel,
which targets TGFβ receptor kinase, have been used to target the
metastatic potential of breast cancer cells to colonize the lung
(19). In line with this concept,
ELK3 can be a prominent therapeutic target to prevent TGFβ-mediated
metastasis of cancer cells. The potential value of ELK3 as a target
of anticancer drug development is supported by the fact that TNBCs
with reduced ELK3 activity completely lost their metastatic
characteristics (12). It was shown
that small molecule based inhibition of Ras/ERK-mediated ELK3
activity results in the inhibition of prostate cancer progression
and metastasis in mice (20). It
would be interesting to investigate whether simultaneous inhibition
of the TGFβ pathway and ELK3 activity produces clinically effective
therapeutic outcomes.
In summary, we suggest that ELK3 is a novel
downstream target of the TGFβ-SMAD3 signaling pathway and that it
performs a major role in directing the metastasis of cancer. TGF-β1
is preferentially expressed at the advancing tumor edges, where it
promotes malignant progression and metastasis (21–23). To
strengthen our findings, follow-up immunohistochemical studies are
needed to demonstrate the accumulation of ELK3 at the site of
excessive TGFβ expression on invasive tumors.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Seong-Jin Kim
(Seoul National University) for providing the expression plasmids
containing FLAG-SMAD3 and constitutively active ALK5 (CA-ALK5).
Funding
The present study was supported by The Ministry of
Education, Science, and Technology (NRF-2019R1A2C1003581) and by
Basic Science Research Program through The National Research
Foundation of Korea (NRF) funded by The Ministry of Education
(grant no. 2019R1A6A1A03032888).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JHP designed the experiment and performed all
experiments. KSP made substantial contributions to the analysis and
interpretation of data. KSP has also been involved in drafting the
manuscript and revising it critically for important intellectual
content. JHP agreed to the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TGFβ
|
transforming growth factor-β
|
|
SBE
|
SMAD binding element
|
|
TNBCs
|
triple negative breast cancer
cells
|
|
EMT
|
epithelial-mesenchymal transition
|
|
siTGFR1
|
small interfering RNA targeting to
TGFβ receptor R1
|
|
CA-ALK5
|
constitutively active form of TGFβ
receptor-I
|
References
|
1
|
Seyfried TN and Huysentruyt LC: On the
origin of cancer metastasis. Crit Rev Oncog. 18:43–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wells A, Grahovac J, Wheeler S, Ma B and
Lauffenburger D: Targeting tumor cell motility as a strategy
against invasion and metastasis. Trends Pharmacol Sci. 34:283–289.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Savagner P, Yamada KM and Thiery JP: The
zinc-finger protein slug causes desmosome dissociation, an initial
and necessary step for growth factor-induced epithelial-mesenchymal
transition. J Cell Biol. 137:1403–1419. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eger A, Aigner K, Sonderegger S, Dampier
B, Oehler S, Schreiber M, Berx G, Cano A, Beug H and Foisner R:
DeltaEF1 is a transcriptional repressor of E-cadherin and regulates
epithelial plasticity in breast cancer cells. Oncogene.
24:2375–2385. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Comijn J, Berx G, Vermassen P, Verschueren
K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D and van Roy
F: The two-handed E box binding zinc finger protein SIP1
downregulates E-cadherin and induces invasion. Mol Cell.
7:1267–1278. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Buchwalter G, Gross C and Wasylyk B: Ets
ternary complex transcription factors. Gene. 324:1–14. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Heo SH, Lee JY, Yang KM and Park KS: ELK3
expression correlates with cell migration, invasion, and membrane
type 1-matrix metalloproteinase expression in MDA-MB-231 breast
cancer cells. Gene Expr. 16:197–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee JH, Hur W, Hong SW, Kim JH, Kim SM,
Lee EB and Yoon SK: ELK3 promotes the migration and invasion of
liver cancer stem cells by targeting HIF-1α. Oncol Rep. 37:813–822.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kong SY, Kim KS, Kim J, Kim MK, Lee KH,
Lee JY, Oh N, Park JI, Park JH, Heo SH, et al: The ELK3-GATA3 axis
orchestrates invasion and metastasis of breast cancer cells in
vitro and in vivo. Oncotarget. 7:65137–65146. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim KS, Kim J, Oh N, Kim MY and Park KS:
ELK3-GATA3 axis modulates MDA-MB-231 metastasis by regulating
cell-cell adhesion-related genes. Biochem Biophys Res Commun.
498:509–515. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lv ZD, Kong B, Li JG, Qu HL, Wang XG, Cao
WH, Liu XY, Wang Y, Yang ZC, Xu HM and Wang HB: Transforming growth
factor-beta 1 enhances the invasiveness of breast cancer cells by
inducing a Smad2-dependent epithelial-to-mesenchymal transition.
Oncol Rep. 29:219–225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cho HJ, Oh N, Park JH, Kim KS, Kim HK, Lee
E, Hwang S, Kim SJ and Park KS: ZEB1 collaborates with ELK3 to
repress E-cadherin expression in triple negative breast cancer
cells. Mol Cancer Res. 17:2257–2266. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang YY, Brunicardi FC and Lin X: Smad3
mediates immediate early induction of Id1 by TGF-beta. Cell Res.
19:140–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kavsak P, Rasmussen RK, Causing CG, Bonni
S, Zhu H, Thomsen GH and Wrana JL: Smad7 binds to Smurf2 to form an
E3 ubiquitin ligase that targets the TGF beta receptor for
degradation. Mol Cell. 6:1365–1375. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bu P, Wang L, Chen KY, Rakhilin N, Sun J,
Closa A, Tung KL, King S, Kristine Varanko A, Xu Y, et al: miR-1269
promotes metastasis and forms a positive feedback loop with TGF-β.
Nat Commun. 6:68792015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park SY, Kim MJ, Park SA, Kim JS, Min KN,
Kim DK, Lim W, Nam JS and Sheen YY: Combinatorial TGF-β attenuation
with paclitaxel inhibits the epithelial-to-mesenchymal transition
and breast cancer stem-like cells. Oncotarget. 6:37526–37543. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Semenchenko K, Wasylyk C, Cheung H,
Tourrette Y, Maas P, Schalken JA, van der Pluijm G and Wasylyk B:
XRP44X, an inhibitor of Ras/Erk activation of the transcription
factor Elk3, inhibits tumour growth and metastasis in mice. PLoS
One. 11:e01595312016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pickup M, Novitskiy S and Moses HL: The
roles of TGFβ in the tumour microenvironment. Nat Rev Cancer.
13:788–799. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dalal BI, Keown PA and Greenberg AH:
Immunocytochemical localization of secreted transforming growth
factor-beta 1 to the advancing edges of primary tumors and to lymph
node metastases of human mammary carcinoma. Am J Pathol.
143:381–389. 1993.PubMed/NCBI
|
|
23
|
Steiner MS, Zhou ZZ, Tonb DC and Barrack
ER: Expression of transforming growth factor-beta 1 in prostate
cancer. Endocrinology. 135:2240–2247. 1994. View Article : Google Scholar : PubMed/NCBI
|