Introduction
Lung cancer is the most common type of malignant
tumor and was the leading cause of cancer-associated death in the
USA in 2019 (1). Lung cancer can be
categorized into two main histological subtypes, non-small-cell
lung cancer (NSCLC) and small-cell lung cancer, accounting for ~85%
and ~15% of cases, respectively (2,3). Lung
adenocarcinoma (LUAD) is one of the major subtypes of NSCLC with a
low 5-year survival rate of ~20% in the USA (4,5). It is
currently understood that two of the driver oncogenes involved in
tumorigenesis and progression of LUAD are epidermal growth factor
receptor and anaplastic lymphoma kinase (6–8). These
biomarkers can be used for diagnosis and targeted in the treatment
of LUAD; however, prognosis remains unsatisfactory due to the
underlying molecular heterogeneity and diverse etiology of LUAD.
Therefore, it is important that novel valuable biomarkers, which
have associations with prognosis in LUAD are identified.
Long non-coding RNA (lncRNA) is a type of non-coding
RNA which is >200 nucleotides in length (9). Previous research has demonstrated that
lncRNAs serve diverse functions in cellular processes and are
involved in tumorigenesis, progression and metastasis (10,11).
However, only a few lncRNAs involved in the development of LUAD
have been identified, including histocompatibility leukocyte
antigen complex P5, chromatin-associated RNA 10 and
metastasis-associated lung adenocarcinoma transcript 1 (12–14). The
underlying molecular mechanisms for the functions of numerous
lncRNAs remains unclear, to the best of our knowledge, but it is
hypothesized that lncRNAs may have valuable clinical applications
in the future.
Previous studies have described several novel
lncRNAs with potential use as biomarkers differentially expressed
in human LUAD tissue (15,16) and further investigation into these
lncRNAs may elude their prognostic value. Integrated prognostic
analysis of lncRNAs combined with overall outcomes, pathological
stages and other clinical parameters may improve the prediction of
overall survival time of patients with LUAD.
In the present study, the construction method for a
previous lncRNA gene signature was amended (17,18),
aiming to improve its accuracy. Based on lncRNA expression
profiling, a prognostic model was constructed using a comprehensive
approach involving the least absolute shrinkage and selection
operator (LASSO) algorithm and multivariate Cox regression. Model
robustness and accuracy were assessed using area under the
calculated curve (AUC) and Kaplan-Meier (K-M) survival analyses in
high- and low-risk groups. Finally, potential biomarkers associated
with survival status were identified using K-M survival analysis
and possible functions of these biomarkers were predicted using
enrichment analysis of co-expressed mRNAs.
Materials and methods
Datasets
RNA sequencing data and corresponding clinical
information were obtained from The Cancer Genome Atlas (TCGA)
database (cancer.gov/tcga), including data for 535
LUAD tissue samples and 59 control adjacent normal tissues. The
LUAD group consisted of 249 males and 286 females with an age range
of 33–88 years and a median age of 66 years, whereas the control
group consisted of 25 males and 34 females with an age range of
42–86 years and a median age of 66 years. LUAD patients with
incomplete survival data were excluded, therefore 500 samples were
used for analysis.
Identification of differentially
expressed lncRNAs
All analyses were performed using R software
(version 3.5.3; r-project.org/) and R packages by R
studio version 1.1.463 (19,20). Raw lncRNA expression profiles were
constructed using an expression matrix and normalized using the
edgeR package (version 3.22.5) (21)
and the lncRNAs differentially expressed between LUAD and control
samples were identified. lncRNAs which met the threshold of
|log2[fold change (FC)]|≥2 and false discovery rate (FDR) <0.05
were screened out for subsequent analysis.
Definition of the gene-related
prognostic model
Univariate Cox, LASSO and multivariate Cox
regression analyses were performed to identify the association
between survival time and lncRNA expression using the glmnet
(version 2.0.18) (22) and survival
(version 2.44.1.1) (23) packages. A
univariate Cox model was established to assess the correlation
between overall survival (OS) and lncRNA expression levels.
P<0.05 was considered to indicate a statistically significant
difference. lncRNAs meeting this criterion were included in LASSO
regression analysis, which was to further select prognostic genes
and to avoid the over-fitting of the signature model. After
selection using the minimum λ value, which represented the optimal
number of variables in this model, a further refined group of
lncRNAs was included in a multivariate Cox analysis to determine
the independent contribution of each lncRNA to prognosis and hazard
ratios (HR) and 95% confidence intervals (CI) were calculated. The
Cox regression coefficient (β) and expression levels of lncRNAs
were used to calculate prognosis-related risk scores according to
the formula: Σ (explncRNAn × βlncRNAn). Samples were further
divided into high- and low-risk groups using the median risk
score.
Evaluation of the risk model and
identification of potential biomarkers
Time-dependent receiver operator characteristic
(ROC) curves were plotted to evaluate performance of the prognostic
model. K-M survival analyses were performed to predict survival
times for the high and low risk groups and to calculate AUCs value
for the prognostic model. Multivariate Cox analysis results were
used to identify potential biomarkers. A threshold of P<0.05 was
used to identify eligible lncRNAs for survival and K-M curve
analyses and to predict the OS associated with each lncRNA.
Subsequently, potential biomarkers were identified if this
P<0.05 criterion from the survival curves was met.
Functional enrichment analysis
The biological function of the lncRNAs with
significant P-values for OS in the comparison between the high and
low risk groups was predicted using their co-expressed
protein-coding mRNAs. The mRNAs that correlated with prognostic
lncRNAs were identified using Pearson correlation analysis. An
absolute correlation coefficient value >0.4 was considered
significant. The Database for Annotation, Visualization and
Integrated Discovery (DAVID) Bioinformatics Tool (version 6.8)
(24,25) was used to perform the Gene Ontology
(GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional
enrichment analysis.
Results
lncRNAs are differentially expressed
between LUAD and control samples
A total of 1,684 differentially expressed lncRNAs
(Table SI) were identified, meeting
the conditions of |log2 (FC)|≥2 and FDR <0.05. Of these, 1,499
lncRNAs were upregulated and 185 lncRNAs were downregulated
(Fig. 1). The lncRNAs which had
significant differential expression levels between the LUAD and
control tissue groups were used for subsequent prognostic
analysis.
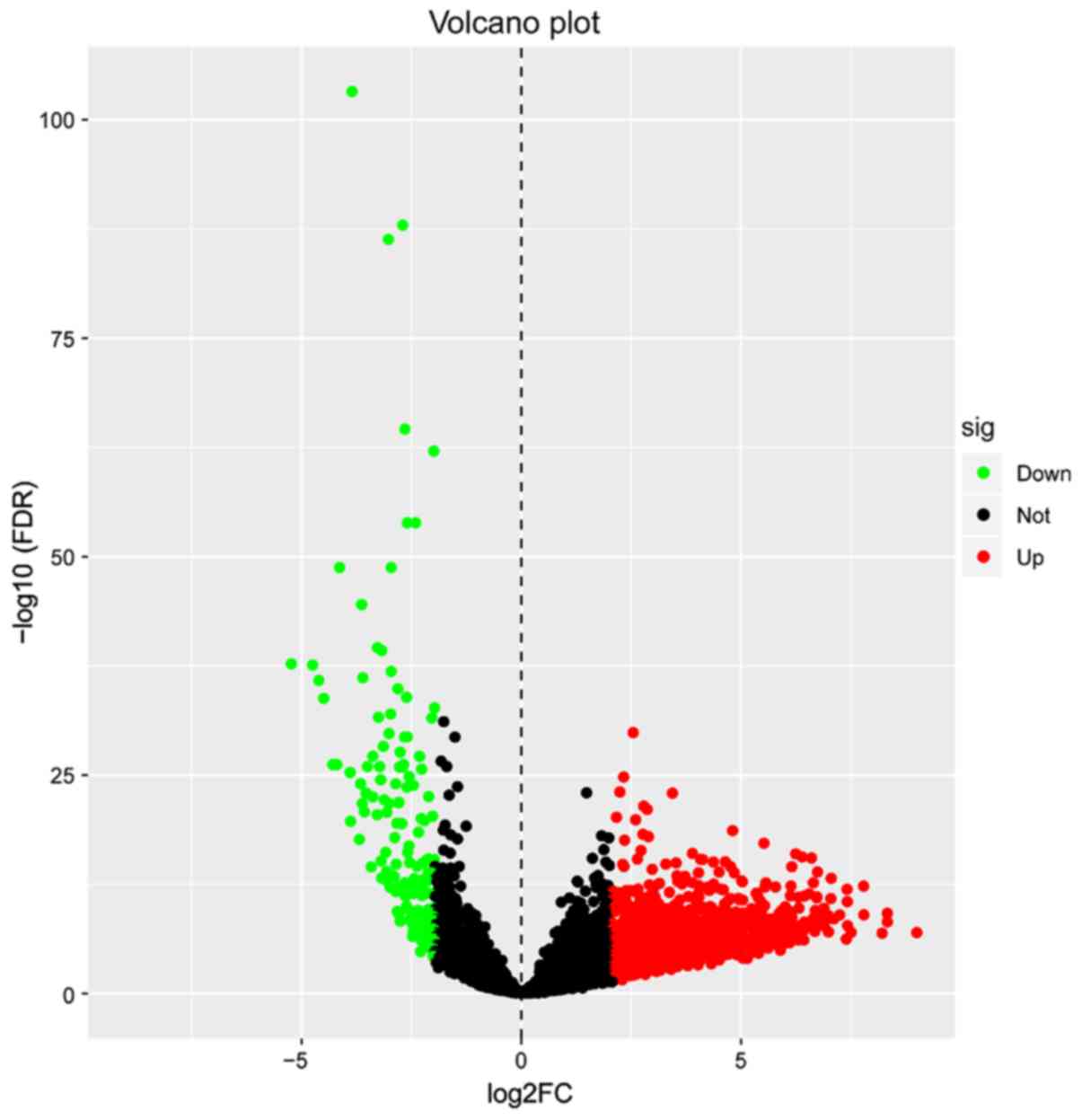 | Figure 1.Identification of 1,684
differentially expressed lncRNAs. Red dots represent 1,499
upregulated lncRNAs, green dots represent 185 downregulated lncRNAs
and black dots represent 9,736 non-differentially expressed
lncRNAs. lncRNA, long non-coding RNA; FC, fold change; sig,
significance; down, downregulated; not, not significant; up,
upregulated; FDR, false discovery rate. |
Construction of the prognostic
model
Univariate Cox regression was used to analyze the
correlations between differentially expressed lncRNA profiles and
the OS of patients with LUAD. A total of 107 lncRNAs were
significantly correlated with patient OS (P<0.05) and these
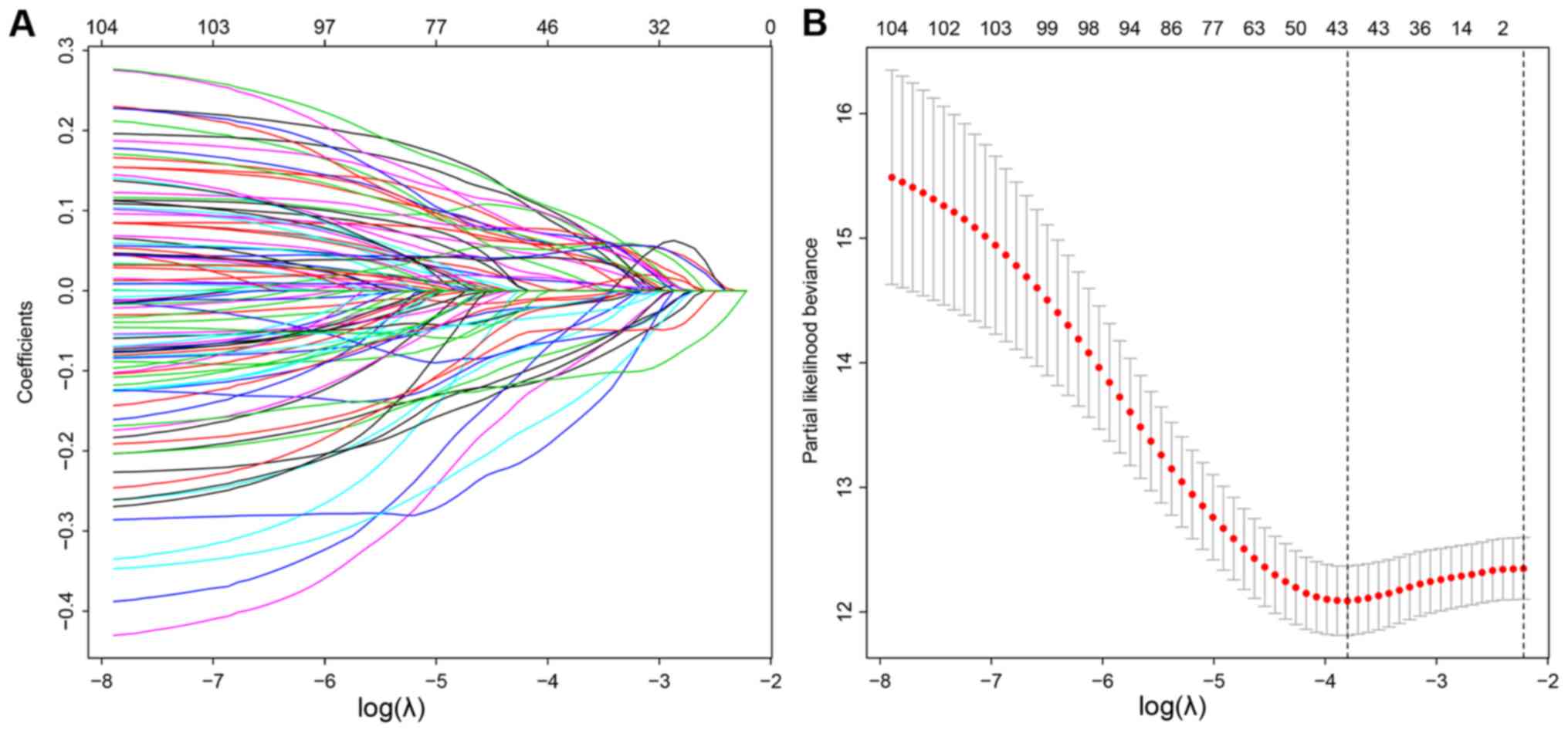
lncRNAs were used in a LASSO regression analysis (Table SII). Overall, 44 key lncRNAs were
selected when the λ value was at a minimum (Fig. 2A and B). Finally, a multivariate Cox
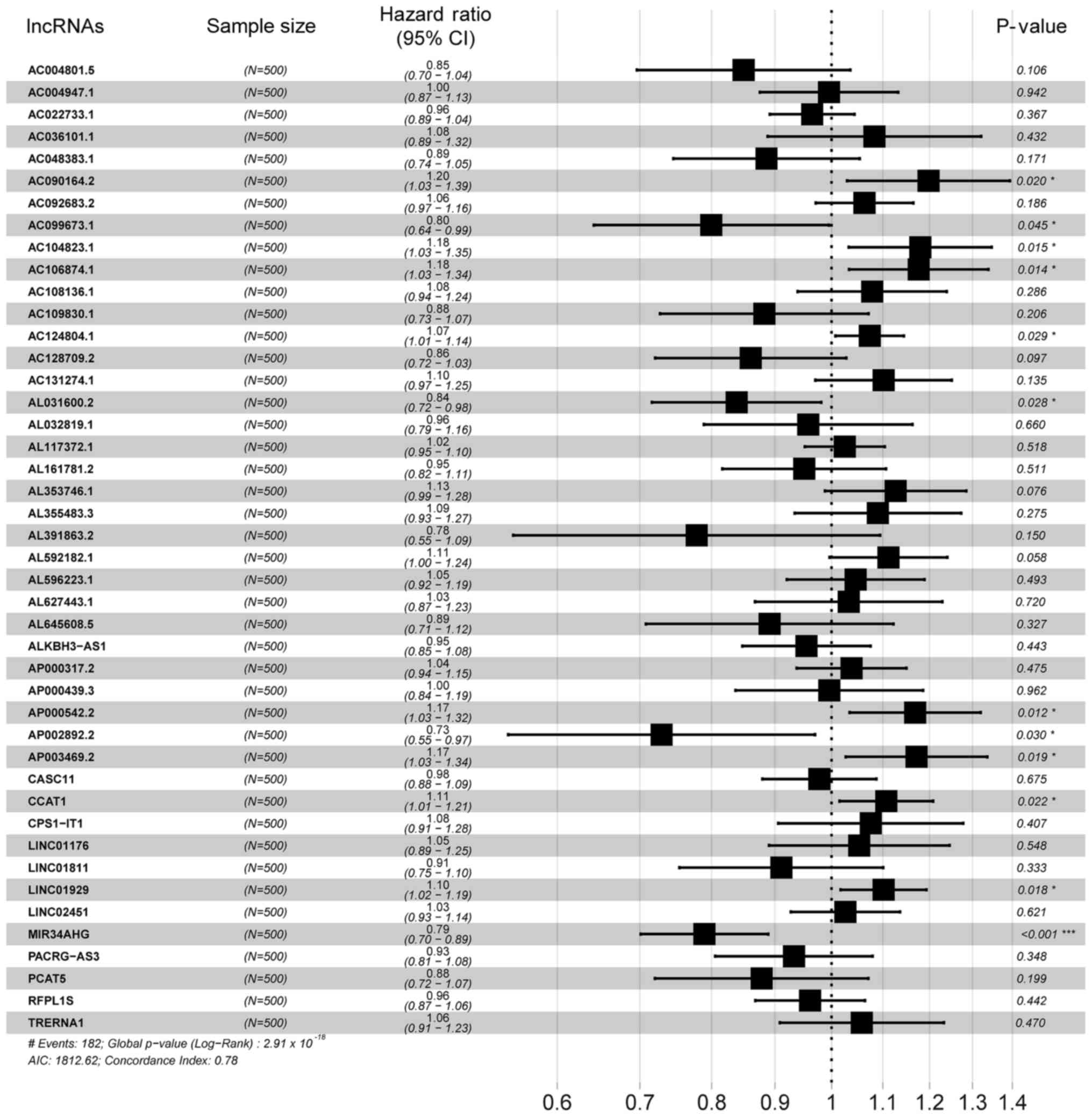
regression analysis was performed to obtain HR values and 95% CIs
for each key lncRNA (Fig. 3).
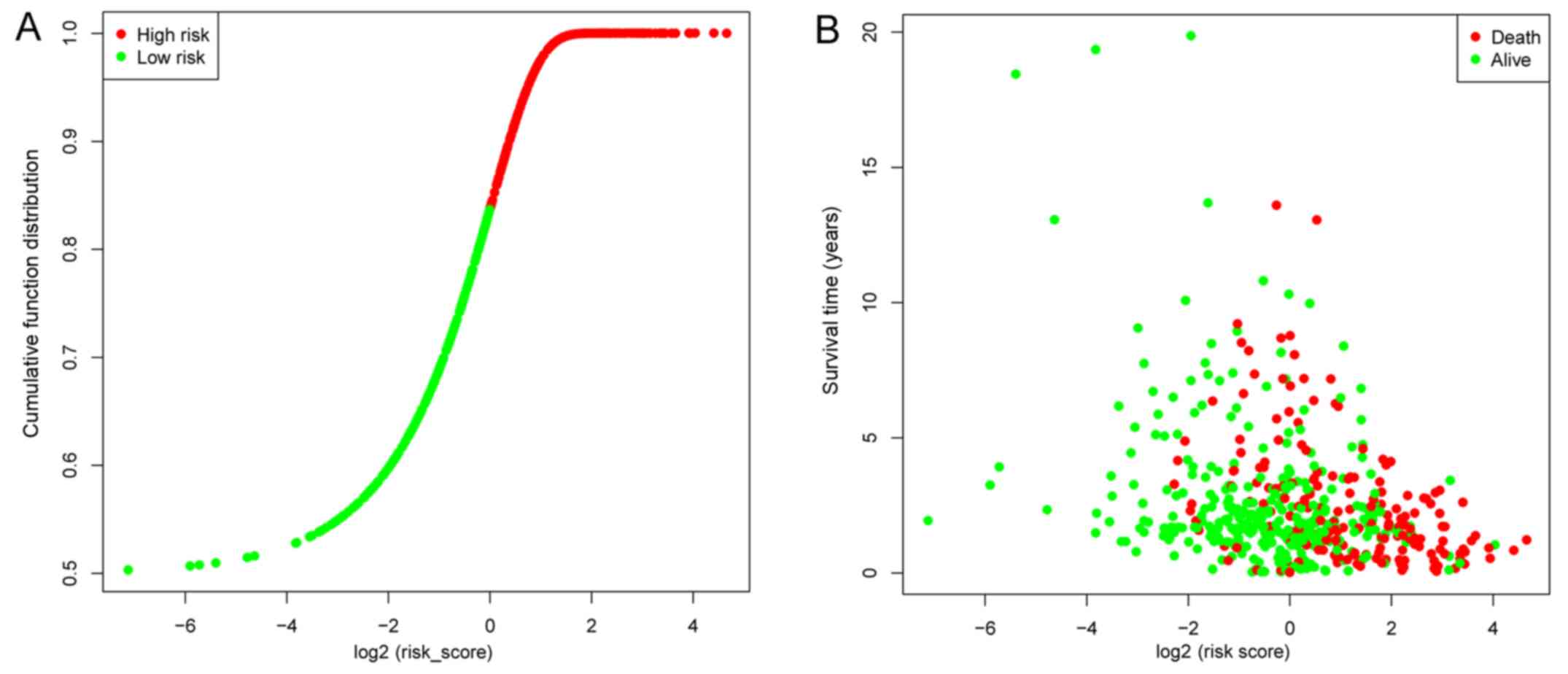
Samples were classified into high- and low-risk groups according to
respective median risk scores (Fig. 4A
and B).
Evaluation of the prognostic
model
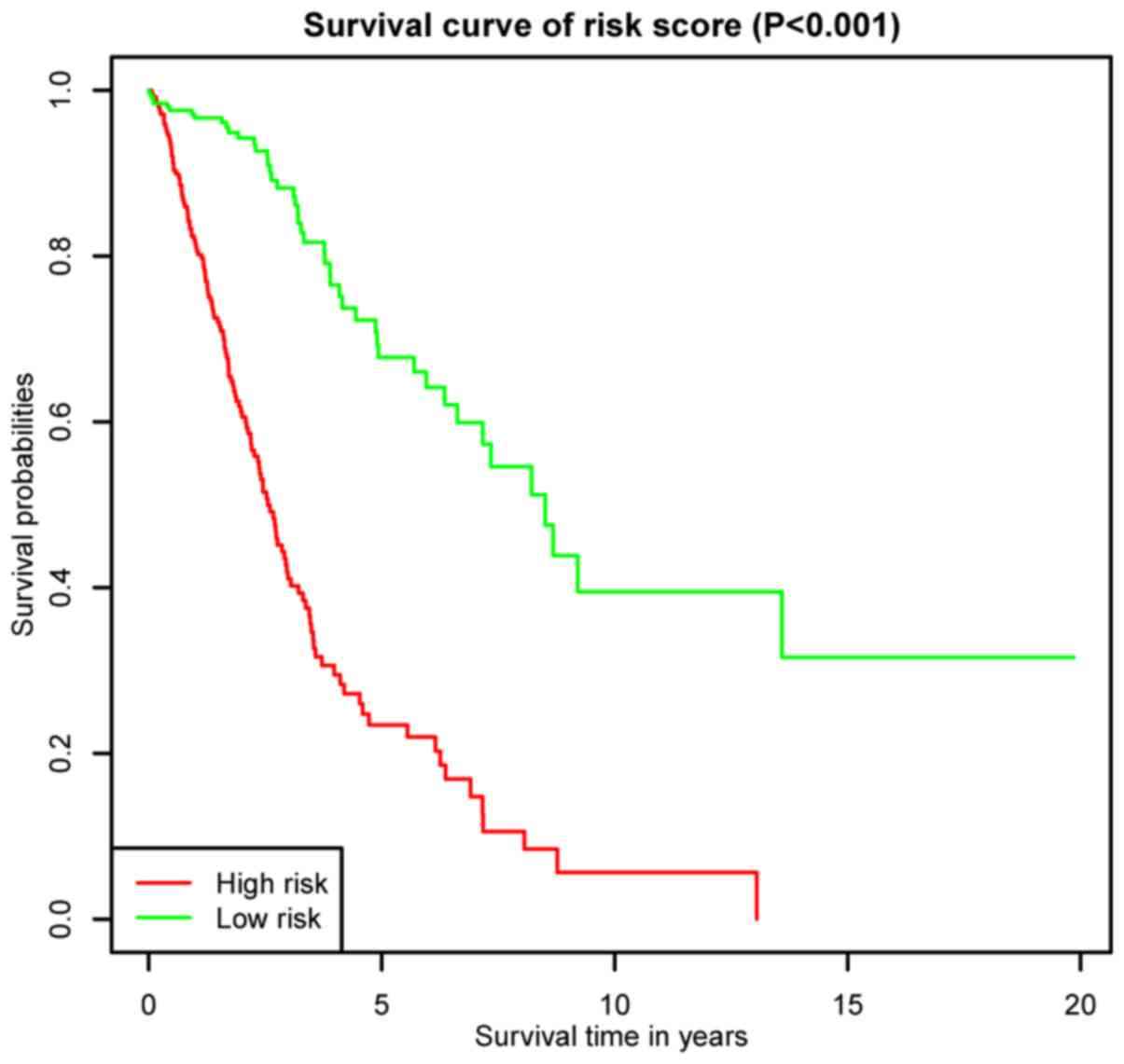
K-M survival curves for OS outcomes were performed
according to median risk score values, demonstrating that the
predicted survival time of the high-risk group was significantly
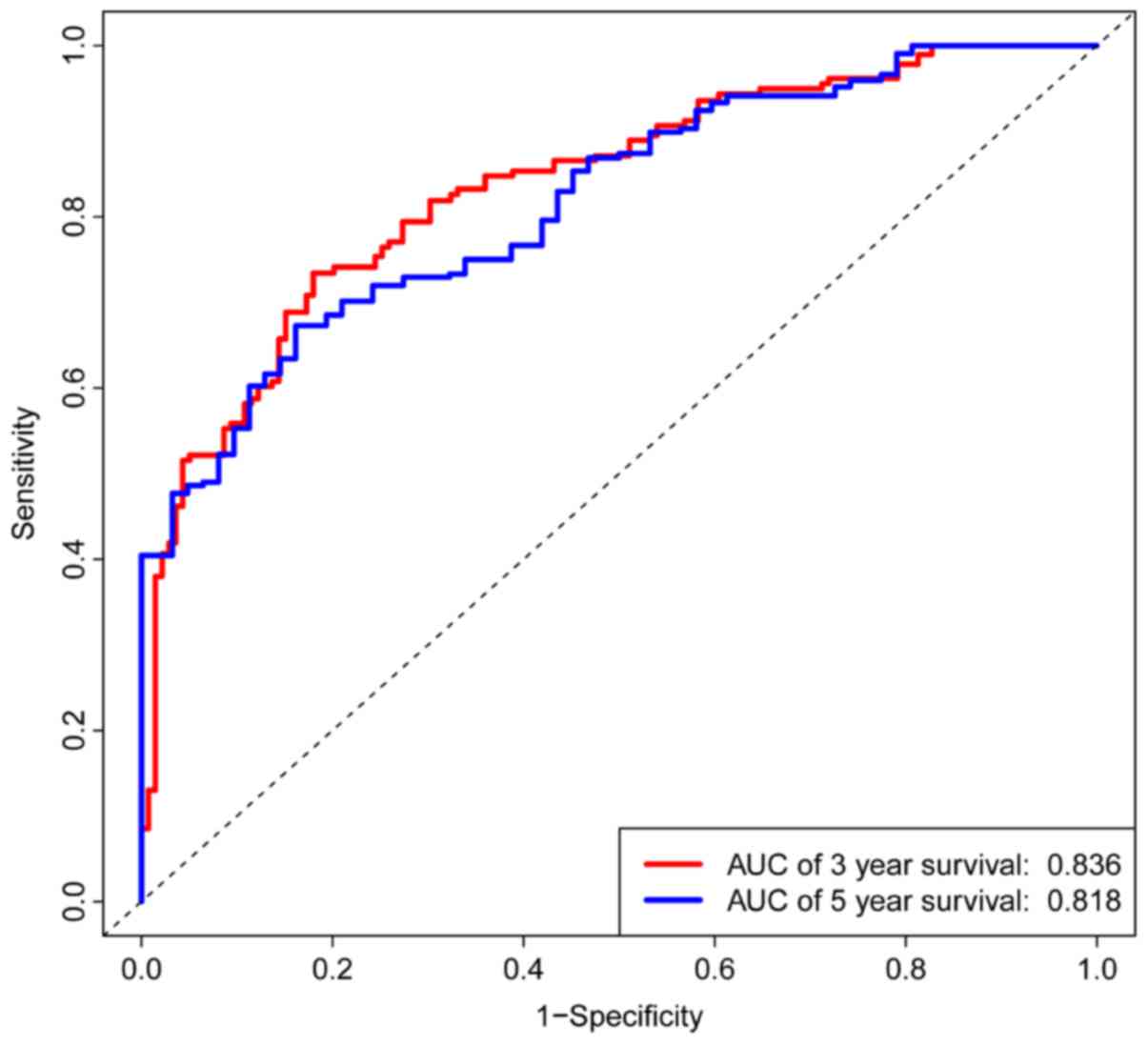
shorter compared with the low risk group (P< 0.001; Fig. 5). The AUCs for 3- and 5-year
survivals were 0.836 and 0.818, respectively, of a time-dependent
ROC curve demonstrating that the risk score model had stable
performance (Fig. 6).
Identification of potential
biomarkers
Multivariate Cox regression showed that lncRNAs with
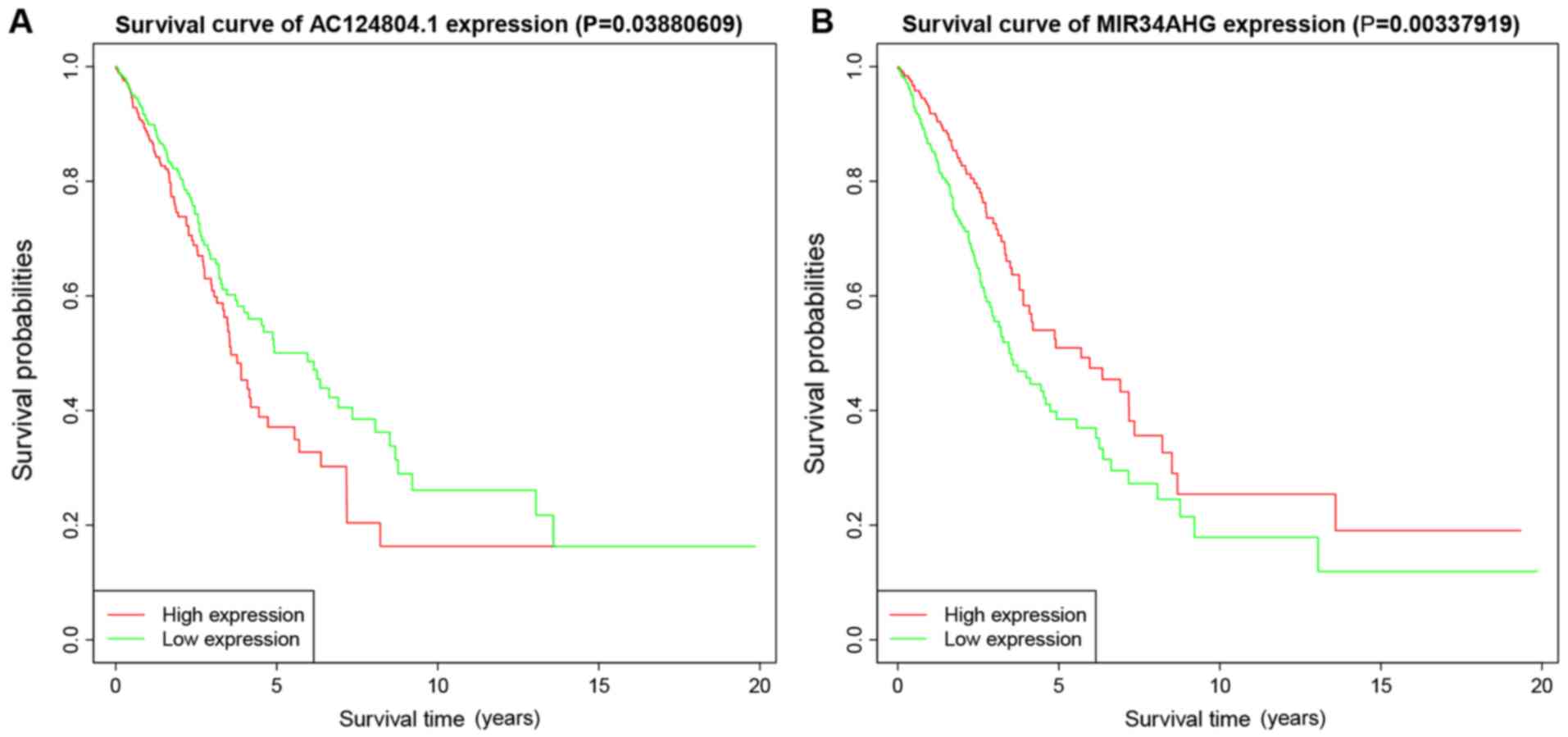
P<0.05 were correlated with survival status (Fig. 3). K-M survival curves enabled
screening out of two lncRNAs, AC124804.1 and MIR34AHG, with
significantly different predicted survival times between the high-
and low-risk score groups (Fig. 7A and
B). These two lncRNAs had the greatest correlation with OS time
and may have potential as prognostic biomarkers for patients with
LUAD.
Functional enrichment
To investigate the underlying molecular mechanisms
of the functions of AC124804.1 and MIR34AHG, the co-expressed mRNAs
of these lncRNAs were identified using Pearson's correlation
analysis. For MIR34AHG, no mRNAs met the screening condition of a
correlation coefficient value of >0.4. For AC124804.1, a total
of 818 protein coding mRNAs were identified for further functional
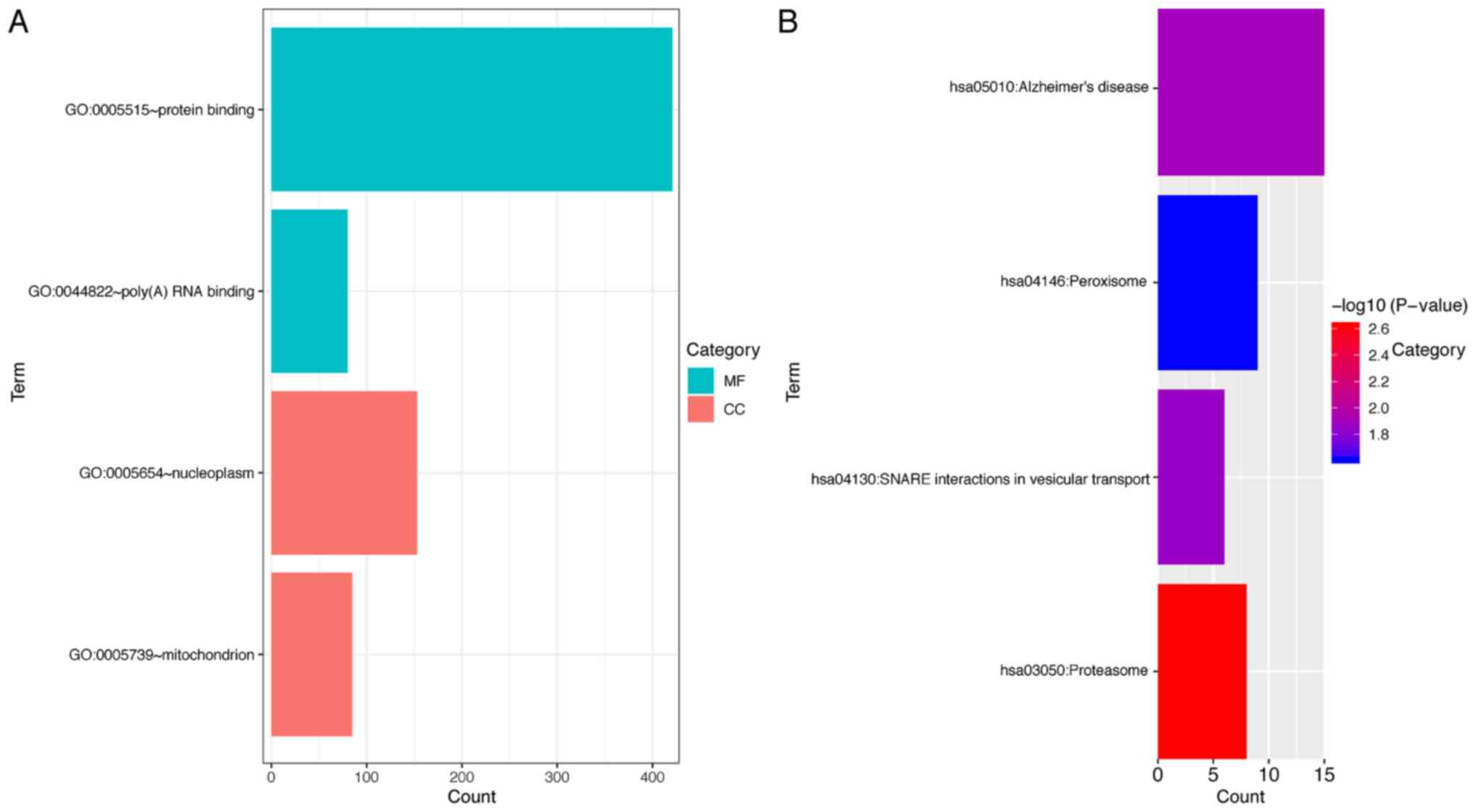
enrichment. Overall, four GO terms and four KEGG pathways had
confirmed association with AC124804.1. The 818 co-expressed mRNAs
were primarily clustered in the poly(A) RNA binding and protein
binding of molecular functions, and to the mitochondrion and
nucleoplasm cellular components (P<0.05; Fig. 8A). KEGG pathway enrichment analysis
demonstrated that these mRNAs were primarily associated with
Proteasome, Alzheimer's disease, SNARE interactions in vesicular
transport and Peroxisome pathways (Fig.
8B).
Discussion
LUAD is a large global health challenge (1,26) and
our knowledge of its underlying mechanisms may be improved by
preclinical next-generation sequencing bioinformatics technology
(27,28). However, despite advances in the
diagnosis and treatment of LUAD, the OS time of patients with LUAD
remains poor (29). Recent studies
have identified molecular biomarkers throughout the various
clinical stages and pathological tissue types (30,31).
However, patients with the same clinical and pathological features
at diagnosis often have a completely different final prognoses
(32). Molecular gene signatures
associated with prognosis may aid the identification of novel
molecular biomarkers for use in diagnosis and treatment of LUAD in
combination with patients' genetic profiles and clinical parameters
(17,18). Recent studies pertaining to LUAD have
investigated gene signatures based on lncRNA data and the selected
lncRNAs may be important risk factors and have potential predictive
power for patient survival (17,18).
However, due to complex underlying mechanisms, the biological
functions of the majority of lncRNAs have not been resolved
(33). The prognostic lncRNA
signature constructed in the present study, demonstrating
correlation with clinical features of LUAD, may be aid the
screening out of potential lncRNAs for further investigation.
To improve the accuracy and robustness of the
present model compared with previous studies (15,17,18), a
different approach was taken to data preprocessing and construction
of the model. First, the present study utilized larger sample sizes
compared with previous studies (15,17,18)
resulting from differences in inclusion criteria, including a total
of 535 LUAD tissues and 59 adjacent normal tissues (controls).
Second, the present study used different methods of data
preprocessing and normalization. For example, previous studies
using some online tools for analysis, whereas the present study
performed analysis using edgeR package in R software, which could
analyze the latest raw counts of RNA-seq data of TCGA. Third, the
present group selected variables using univariate Cox analysis and
LASSO regression, which were not employed in previous studies. In
addition, the eligible lncRNAs in the present study were confirmed
using P-values of multivariate Cox regression and survival
analyses.
In the present study, lncRNA profiles were used to
construct the prognostic model, using data from tissue samples and
clinical information of 500 patients held in the TCGA database. A
total of 1,684 differentially expressed lncRNAs were investigated
as potential biomarkers using univariate Cox regression to screen
out lncRNAs that were significantly associated with the clinical
characteristics. The results of univariate Cox regression were
further filtered using the LASSO algorithm, which weakens
collinearity of the risk model and enables the most influential
variables to be selected (34,35).
This approach is suitable for dealing with datasets containing a
large volume of variables, as occurs in genomics (36). The final 44 lncRNAs identified using
LASSO regression were then analyzed using multivariate Cox
regression. The correlation of each lncRNA with survival of
patients with LUAD was presented as an HR value. Patients were
divided into high and low risk groups according to the median risk
scores which demonstrated that 3 and 5-year survival times were
significantly different between the high and low risk score groups.
These data combined with the AUC score results confirmed the
accuracy and robustness of this model for predicting prognosis of
patient with LUAD.
A multivariate Cox regression forest plot identified
lncRNAs with significant P-values as potential prognostic
biomarkers for LUAD. The respective survival times correlated with
these lncRNAs were calculated prospectively using K-M survival
analysis, identifying candidate prognostic factors. According to
the prognostic signature constructed in the present study, the
expression levels of AC124804.1 and MIR34AHG were significantly
correlated with differential clinical outcomes and predicted OS
times of the two risk groups. These two lncRNAs may have value as
novel biomarkers for estimating prognosis of patients with LUAD. To
the best of our knowledge, there is no published work describing
the relationships between these lncRNAs and LUAD to date. To
explore the potential functions of AC124804.1 and MIR34AHG, the
co-expressed protein-coding mRNAs of these lncRNAs were
investigated using a Pearson correlation coefficient test followed
by GO and KEGG functional enrichment analyses. These analyses
demonstrated that the potential underlying molecular mechanisms of
AC124804.1 function were pathways involving proteasome, peroxisome
and SNARE interactions in vesicular transport, which have confirmed
roles in tumorigenesis and progression in previous studies
(37–39). MIR34AHG is the host gene of miR-34a
and consequently may have biological functions associated with the
miR-34 family, which may have suppressive functions in LUAD as
previously demonstrated (40,41). The
relationship between these two lncRNAs and LUAD should be studied
further.
There are several limitations to the present study.
First, the data for the lncRNA signatures were from the TCGA
database and these samples primarily originated from Caucasians and
African Americans, limiting extrapolation of the results of the
present study to other ethnic groups. Second, the present study
used a database mining design without validation in fresh samples
and prospective experimental studies. Third, the clinical
parameters included only OS time, with histological subtypes and
other risk factors for LUAD were excluded from the analysis. The
primary purpose of the present study was to identify novel
biomarkers for the diagnosis and treatment of LUAD. The lncRNA
prognosis signature should be further investigated incorporating
more specific clinical characteristics to fully understand the
associations involved.
In conclusion, a robust lncRNA signature that could
stratify and predict survival time of patients with LUAD was
constructed. Furthermore, Cox regression was used to identify two
novel survival associated lncRNA biomarkers and provided potential
targets for diagnostic and therapeutic application. The biological
functions of the two lncRNAs identified in the present study need
confirmation in further experimental studies.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by The National Natural
Science Foundation of China (grant no. 81603577).
Availability of data and materials
The datasets used and/or analyzed during the present
study available from the corresponding author on reasonable
request.
Authors' contributions
YZ conceived and designed the study. XY conducted
the analysis and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AUC
|
area under the curve
|
|
CI
|
confidence interval
|
|
FDR
|
false discovery rate
|
|
GO
|
Gene Ontology
|
|
HR
|
hazard ratio
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
K-M analysis
|
Kaplan-Meier analysis
|
|
LASSO
|
least absolute shrinkage and selection
operator
|
|
lncRNA
|
long non-coding RNA
|
|
LUAD
|
lung adenocarcinoma
|
|
NSCLC
|
non-small-cell lung cancer
|
|
OS
|
overall survival
|
|
ROC
|
receive operator characteristic
|
|
TCGA
|
The Cancer Genome Atlas.
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Simó M, Vaquero L, Ripollés P,
Gurtubay-Antolin A, Jové J, Navarro A, Cardenal F, Bruna J and
Rodríguez-Fornells A: Longitudinal brain changes associated with
prophylactic cranial irradiation in lung cancer. J Thorac Oncol.
11:475–486. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shahabi S, Kumaran V, Castillo J, Cong Z,
Nandagopal G, Mullen DJ, Alvarado A, Correa MR, Saizan A, Goel R,
et al: LINC00261 is an epigenetically regulated tumor suppressor
essential for activation of the DNA damage response. Cancer Res.
79:3050–3062. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu GS, Li M, Xu CX and Wang D: APE1
stimulates EGFR-TKI resistance by activating Akt signaling through
a redox-dependent mechanism in lung adenocarcinoma. Cell Death Dis.
9:11112018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Macheleidt IF, Dalvi PS, Lim SY, Meemboor
S, Meder L, Käsgen O, Müller M, Kleemann K, Wang L, Nürnberg P, et
al: Preclinical studies reveal that LSD1 inhibition results in
tumor growth arrest in lung adenocarcinoma independently of driver
mutations. Mol Oncol. 12:1965–1979. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hung MS, Lung JH, Lin YC, Fang YH, Huang
SY, Jiang YY, Hsieh MJ and Tsai YH: Comparative analysis of two
methods for the detection of EGFR mutations in plasma circulating
tumor DNA from lung adenocarcinoma patients. Cancers (Basel).
11:8032019. View Article : Google Scholar
|
|
7
|
Li S, Choi YL, Gong Z, Liu X, Lira M, Kan
Z, Oh E, Wang J, Ting JC, Ye X, et al: Comprehensive
characterization of oncogenic drivers in Asian lung adenocarcinoma.
J Thorac Oncol. 11:2129–2140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang X, Min S, Liu H, Wu N, Liu X, Wang T,
Li W, Shen Y, Wang H, Qian Z, et al: Nf1 loss promotes Kras-driven
lung adenocarcinoma and results in Psat1-mediated glutamate
dependence. EMBO Mol Med. 11:e98562019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang L, Meng X, Zhu XW, Yang DC, Chen R,
Jiang Y and Xu T: Long non-coding RNAs in Oral squamous cell
carcinoma: Biologic function, mechanisms and clinical implications.
Mol Cancer. 18:1022019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan Y, Xu Z, Li Z, Sun L and Gong Z: An
insight into the increasing role of lncRNAs in the pathogenesis of
gliomas. Front Mol Neurosci. 10:532017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu Z, Yan Y, Qian L and Gong Z: Long
non-coding RNAs act as regulators of cell autophagy in diseases
(Review). Oncol Rep. 37:1359–1366. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang L, Wang R, Fang L, Ge X, Chen L,
Zhou M, Zhou Y, Xiong W, Hu Y, Tang X, et al: HCP5 is a
SMAD3-responsive long non-coding RNA that promotes lung
adenocarcinoma metastasis via miR-203/SNAI axis. Theranostics.
9:2460–2474. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ge X, Li GY, Jiang L, Jia L, Zhang Z, Li
X, Wang R, Zhou M, Zhou Y, Zeng Z, et al: Long noncoding RNA CAR10
promotes lung adenocarcinoma metastasis via miR-203/30/SNAI axis.
Oncogene. 38:3061–3076. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen J, Liu X, Xu Y, Zhang K, Huang J, Pan
B, Chen D, Cui S, Song H, Wang R, et al: TFAP2C-activated MALAT1
modulates the chemoresistance of docetaxel-resistant lung
adenocarcinoma cells. Mol Ther Nucleic Acids. 14:567–582. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Salavaty A, Rezvani Z and Najafi A:
Survival analysis and functional annotation of long non-coding RNAs
in lung adenocarcinoma. J Cell Mol Med. 23:5600–5617. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Serghiou S, Kyriakopoulou A and Ioannidis
JP: Long noncoding RNAs as novel predictors of survival in human
cancer: A systematic review and meta-analysis. Mol Cancer.
15:502016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi X, Tan H, Le X, Xian H, Li X, Huang K,
Luo VY, Liu Y, Wu Z, Mo H, et al: An expression signature model to
predict lung adenocarcinoma-specific survival. Cancer Manag Res.
10:3717–3732. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sui J, Yang S, Liu T, Wu W, Xu S, Yin L,
Pu Y, Zhang X, Zhang Y, Shen B, et al: Molecular characterization
of lung adenocarcinoma: A potential four-long noncoding RNA
prognostic signature. J Cell Biochem. 120:705–714. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
R Core Team (2012), . R: A language and
environment for statistical computing. R Foundation for Statistical
Computing; Vienna: http://www.R-project.org/
|
|
20
|
RStudio Team (2015), . RStudio: Integrated
Development for R. RStudio, Inc.; Boston, MA: http://www.rstudio.com/
|
|
21
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Friedman J, Hastie T and Tibshirani R:
Regularization paths for generalized linear models via coordinate
descent. J Stat Softw. 33:1–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Therneau TM and Grambsch PM: Modeling
Survival Data: Extending the Cox Model. Statistics for Biology and
Health. Springer Science and Business Media; New York, NY: pp.
39–76. 2000, View Article : Google Scholar
|
|
24
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:32003.
View Article : Google Scholar
|
|
25
|
Yan Y, Xu Z, Qian L, Zeng S, Zhou Y, Chen
X, Wei J and Gong Z: Identification of CAV1 and DCN as potential
predictive biomarkers for lung adenocarcinoma. Am J Physiol Lung
Cell Mol Physiol. 316:L630–L643. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sivakumar S, San Lucas FA, Jakubek YA,
McDowell TL, Lang W, Kallsen N, Peyton S, Davies GE, Fukuoka J,
Yatabe Y, et al: Genomic landscape of allelic imbalance in
premalignant atypical adenomatous hyperplasias of the lung.
EBioMedicine. 42:296–303. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu C, Tian F, Liu J, Su M, Wu M, Zhu X and
Qian W: Circular RNA cMras inhibits lung adenocarcinoma progression
via modulating miR-567/PTPRG regulatory pathway. Cell Prolif.
52:e126102019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kerdidani D, Chouvardas P, Arjo AR,
Giopanou I, Ntaliarda G, Guo YA, Tsikitis M, Kazamias G, Potaris K,
Stathopoulos GT, et al: Wnt1 silences chemokine genes in dendritic
cells and induces adaptive immune resistance in lung
adenocarcinoma. Nat Commun. 10:14052019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Misono S, Seki N, Mizuno K, Yamada Y,
Uchida A, Sanada H, Moriya S, Kikkawa N, Kumamoto T, Suetsugu T, et
al: Molecular pathogenesis of gene regulation by the miR-150
duplex: miR-150-3p regulates TNS4 in lung adenocarcinoma. Cancers
(Basel). 11:6012019. View Article : Google Scholar
|
|
30
|
Abdel-Aziz A, Ahmed RA and Ibrahiem AT:
Expression of pRb, Ki67 and HER 2/neu in gastric carcinomas:
Relation to different histopathological grades and stages. Ann
Diagn Pathol. 30:1–7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ren Y, Zhao S, Jiang D, Feng X, Zhang Y,
Wei Z, Wang Z, Zhang W, Zhou QF, Li Y, et al: Proteomic biomarkers
for lung cancer progression. Biomarkers Med. 12:205–215. 2018.
View Article : Google Scholar
|
|
32
|
Wang P, Jin M, Sun CH, Yang L, Li YS, Wang
X, Sun YN, Tian LL and Liu M: A three-lncRNA expression signature
predicts survival in head and neck squamous cell carcinoma (HNSCC).
Biosci Rep. 38:BSR201815282018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Peng W, He D, Shan B, Wang J, Shi W, Zhao
W, Peng Z, Luo Q, Duan M, Li B, et al: LINC81507 act as a competing
endogenous RNA of miR-199b-5p to facilitate NSCLC proliferation and
metastasis via regulating the CAV1/STAT3 pathway. Cell Death Dis.
10:5332019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Machado AD, Gómez LM, Marchioni DM, Dos
Anjos FS, Molina MD, Lotufo PA, Benseñor IJ and Titan SM:
Association between dietary intake and coronary artery
calcification in non-dialysis chronic kidney disease: The PROGREDIR
Study. Nutrients. 10:3722018. View Article : Google Scholar
|
|
35
|
Kwon Y, Lee T, Lang A and Burnette D:
Assessment on latitudinal tree species richness using environmental
factors in the southeastern United States. PeerJ. 7:e67812019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Waldmann P, Ferenčaković M, Mészáros G,
Khayatzadeh N, Curik I and Sölkner J: AUTALASSO: An automatic
adaptive LASSO for genome-wide prediction. BMC Bioinformatics.
20:1672019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Follo C, Vidoni C, Morani F, Ferraresi A,
Seca C and Isidoro C: Amino acid response by Halofuginone in cancer
cells triggers autophagy through proteasome degradation of mTOR.
Cell Commun Signal. 17:392019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kaur S, Nag A, Gangenahalli G and Sharma
K: Peroxisome proliferator activated receptor gamma sensitizes
non-small cell lung carcinoma to gamma irradiation induced
apoptosis. Front Genet. 10:5542019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang YS, Tzeng HT, Tsai CH, Cheng HC, Lai
WW, Liu HS and Wang YC: VAMP8, a vesicle-SNARE required for
RAB37-mediated exocytosis, possesses a tumor metastasis suppressor
function. Cancer Lett. 437:79–88. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yun MR, Lim SM, Kim SK, Choi HM, Pyo KH,
Kim SK, Lee JM, Lee YW, Choi JW, Kim HR, et al: Enhancer remodeling
and MicroRNA alterations are associated with acquired resistance to
ALK inhibitors. Cancer Res. 78:3350–3362. 2018.PubMed/NCBI
|
|
41
|
Zhao K, Cheng J, Chen B, Liu Q, Xu D and
Zhang Y: Circulating microRNA-34 family low expression correlates
with poor prognosis in patients with non-small cell lung cancer. J
Thorac Dis. 9:3735–3746. 2017. View Article : Google Scholar : PubMed/NCBI
|