Introduction
Soft tissue sarcoma is a solid tumor that
constitutes less than 1% of all malignancies. On an average, 3 out
of every 100,000 people suffer from soft tissue sarcoma, making it
a rare disease (1). Molecular
classification based on the genetic alteration divides sarcomas
into two main categories: i) sarcomas with specific genetic
alterations; and ii) sarcomas displaying multiple, complex
karyotypic abnormalities with no specific pattern (2). The main anticancer drugs used to treat
their sarcoma are doxorubicin and ifosfamide (3), but the success rate of therapy using
these drugs is approximately 25% (4,5). The
incidence of fibrosarcomas among soft tissue sarcomas is
approximately 2–3% (6), and it is
considered to be a tumor that does not respond easily to anticancer
drugs. Therefore, there is an urgent need of development of novel
drugs that are effective against fibrosarcoma.
Anticancer drugs derived from natural organic
compounds extracted from plants and marine products have gained
widespread attention in the recent years. Eribulin and Trabectedin
have been used in the clinic since 2010, and have been reported to
be effective on soft tissue sarcomas (7). Pristimerin (PM) is a terpenoid
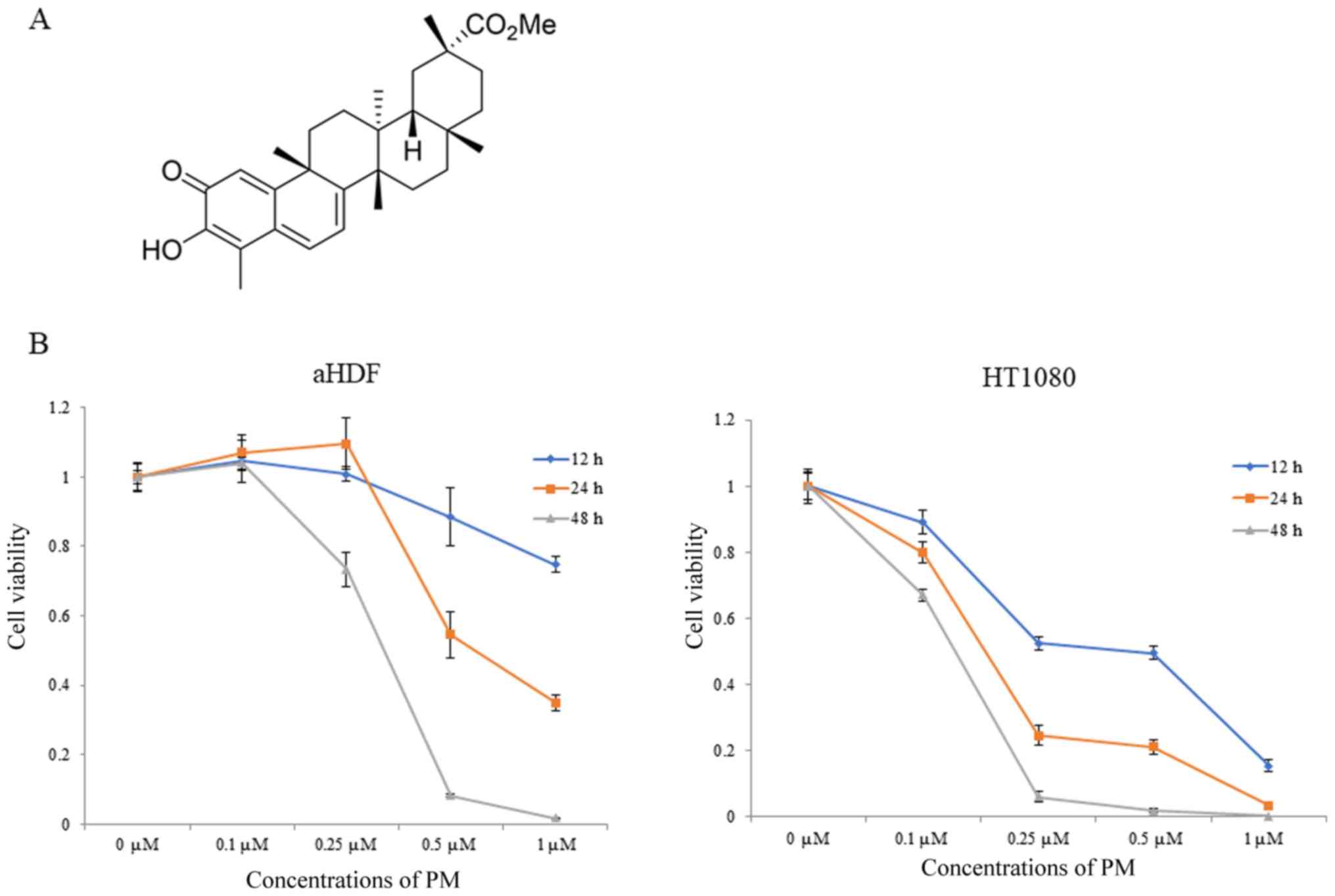
extracted from the Celastraceae family of plants (Fig. 1A). Terpenoids have anti-inflammatory
and anti-tumor effects (8–11), and PM has been reported to show
anti-inflammatory and antioxidant effects (12,13). In
addition, PM has been reported to have anti-tumor effects on lung
cancer, breast cancer, prostate cancer, uterine cancer, gliomas,
and leukemias (14–18). Docetaxel, another commonly used
terpenoid, has demonstrated efficacy against soft tissue sarcomas
and is used clinically (19).
However, there are still no reports on the use of PM. PM is a
terpenoid like docetaxel, but its molecular weight (465 kDa) is
lower than that of docetaxel (808 kDa), and it is believed to have
higher cell penetration ability (20). The purpose of this study was to
clarify if PM is an effective drug against fibrosarcoma.
Materials and methods
Cell lines and cell conditions
The human fibrosarcoma cell line, HT1080 (cat. no.
300216-SF), and aHDF cells (normal human dermal fibroblasts) were
purchased from Cosmo Bio Co., Ltd. Dulbecco's modified Eagle's
medium (DMEM) was purchased from Nacalai Tesque. The cells were
incubated in complete DMEM containing 10% fetal bovine serum (FBS;
Equitech-Bio), 100 units/ml penicillin, and 100 mg/ml streptomycin
(complete DMEM; Nacalai Tesque) at 37°C in a humidified atmosphere
containing 5% CO2. Cells were trypsinized with
trypsin/ethylenediaminetetraacetic acid (Nacalai Tesque) and
subcultured.
Reagents
PM was purchased from Sigma-Aldrich; Merck KGaA,
dissolved in dimethyl sulfoxide (Nacalai Tesque) to yield a 10 mM
stock solution, and stored at −20°C. PM was diluted to the required
concentrations in cell culture medium.
Antibodies
Primary antibodies against AKT (cat. no. 4691S),
p-AKT (cat. no. 9271T), BAX (cat. no. 2772T), BCL-2 (cat. no.
2872T), mTOR (cat. no. 2983S), p-mTOR (cat. no. 5536S), NF-κB p65
(cat. no. 8242S), p-NF-κB p65 (cat. no. 3033S), JNK (cat. no.
9252S), p-JNK (cat. no. 9251S), ERK (cat. no. 9102S), p-ERK (cat.
no. 9101S), and β-actin (cat. no. A2228), and anti-mouse IgG, and
anti-rabbit IgG secondary antibodies were purchased from Cell
Signaling Technology.
Cell viability assay
Cell viability was determined using the RealTime-Glo
MT Cell Viability Assay kit (Promega Corporation). Cells were
seeded in a Poly-l-Lysine (PLL)-coated 96-well plate at a density
of 1×103 cells/well in 100 µl complete DMEM. After 24 h,
MT cell viability substrate and Nanoluc® enzyme were
added to the medium, and the cells were treated with various
concentrations of PM. The CentroXS3 LB 960 System (Berthold
Technologies) was used to measure light emission 12, 24 and 48 h
after PM treatment. The IC50 was calculated as the concentration of
the drug that reduced cell viability by 50% under experimental
conditions.
Apoptosis analysis
Cells were seeded in a 12-well plate at a density of
1×104 cells/well in 1 ml complete DMEM. Cells were
harvested following treatment with PM for 24 h, and stained with 1
µl Annexin V-FITC and 0.5 µl PI solution (Nacalai Tesque) for 20
min at room temperature in dark. Following incubation, binding
buffer was added and the cells were analyzed by flow cytometry
using a FACS Vantage Flow Cytometer (BD Biosciences). For analysis
of each sample, 10,000 events were recorded.
Western blot analysis
Cells were seeded in a 6-well plate at a density of
1×105 cells/well in 2 ml complete DMEM. Cells were
harvested by scraping using 150 µl of radio-immunoprecipitation
assay buffer supplemented with protease and phosphatase inhibitor
cocktails (Sigma-Aldrich; Merck KGaA) and solubilization was
achieved by 10 min. The samples were centrifuged at 15,000 × g for
10 min and the supernatant was aspirated. Protein concentration was
estimated using bicinchoninic acid (BCA) assay, and equal amounts
of total proteins were loaded onto polyacrylamide gels for sodium
dodecyl sulfate polyacrylamide gel electrophoresis (PAGE). Samples
containing 10 µg of protein were separated on 10% Bis-Tris Gel
NuPAGE® electrophoresis using 5% MOPS SDS Running Buffer
(Thermo Fisher Scientific, Inc.). Separated proteins were
dry-blotted onto nitrocellulose iBlot®gel transfer
stacks in iBlot Gel transfer devices (Thermo Fisher Scientific,
Inc.) for 7 min. Nitrocellulose membrane was blocked by shaking in
Blocking One solution (Nacalai Tesque) for 60 min at room
temperature. The blots were subsequently incubated overnight at 4°C
in the same solution containing the following primary antibodies
(1:4,000 dilutions each). β-actin was used as the loading control.
Blots probed for β-actin and other primary antibodies were washed
thrice with TBST and incubated for 60 min at room temperature with
peroxidase-conjugated secondary antibodies in 1:4,000 dilutions.
The blots were again washed thrice with TBST, and chemiluminescent
signal was visualized using Chemi Lumi One imager (Nacalai Tesque).
Protein band intensities were assessed using the ECL Select LAS500
reagent (GE Healthcare).
Animals
Animal experiments were approved by the Experimental
Animals Committee, Kyoto Prefectural University of Medicine.
BALB/C-nu/nu mice (age 4 weeks, females) were purchased from
Shimizu Laboratory Supplies. All procedures were undertaken in
accordance with the NIH Guide for the Care and Use of Laboratory
Animals.
In vivo tumor growth assay
After acclimatization of mice to laboratory
conditions for 1 week, cells (1.0×107) resuspended in
100 µl phosphate-buffered saline were injected into the hypodermis
of the back of each mouse under 5% isoflurane anesthesia. After
tumors grew to approximately 100 mm3, the mice were
injected intraperitoneally (i.p.) with PM at the dose of 1 mg/kg,
every three days for 15 days. Untreated mice (10% DMSO and 90% PBS
i.p) were used as controls (each group contained 4 mice). Tumor
size was measured with a vernier caliper (calculated
volume=shortest diameter2 × longest diameter/2) after
every 3 days. All the mice were sacrificed by cervical dislocation
under 5% isoflurane inhalation anesthesia without causing pain, and
the tumors were weighed on day 15. Liver and renal toxicity were
examined by blood sampling and liver and renal enzyme assays on day
15.
Statistical analysis
All experimental data are represented as means ± SD.
Parametric one-way analysis of variance (ANOVA) was used to examine
statistical differences among the groups. If the result was
significant, the Tukey-Kramer test was used to determine specific
differences between the groups. In all analyses, P<0.05 was
considered to indicate a statistically significant difference.
Results
PM treatment inhibits the
proliferation of HT1080 cells
In order to study the anti-tumor effect of PM, cell
viability was measured at 12, 24 and 48 h after PM administration
(0, 0.1, 0.25, 0.5 and 1 µM). As shown in Fig. 1B, higher concentrations of PM (0.5
and 1 µM) significantly suppressed the viability of aHDF cells at
12 and 24 h. Moreover, the viability of aHDF cells was observed to
significantly lower after 48 h of PM administration. Meanwhile, PM
treatment reduced the viability of HT1080 cells in a
concentration-dependent manner at all the measured time points. In
addition, the IC50 values of PM in aHDF cells after 12, 24 and 48 h
of PM administration was 1 µM or higher, 0.59±0.04 and 0.32±0.02
µM, respectively. Meanwhile, the IC50 values of PM in HT1080 cells
were 0.43±0.10, 0.16±0.01 and 0.13±0.01 µM, respectively.
PM induces apoptosis in HT1080
cells
In order examine the effect of PM on induction of
apoptosis in HT1080 cells; the number of apoptotic cells was
analyzed by flow cytometry after 24 and 48 h of PM administration.
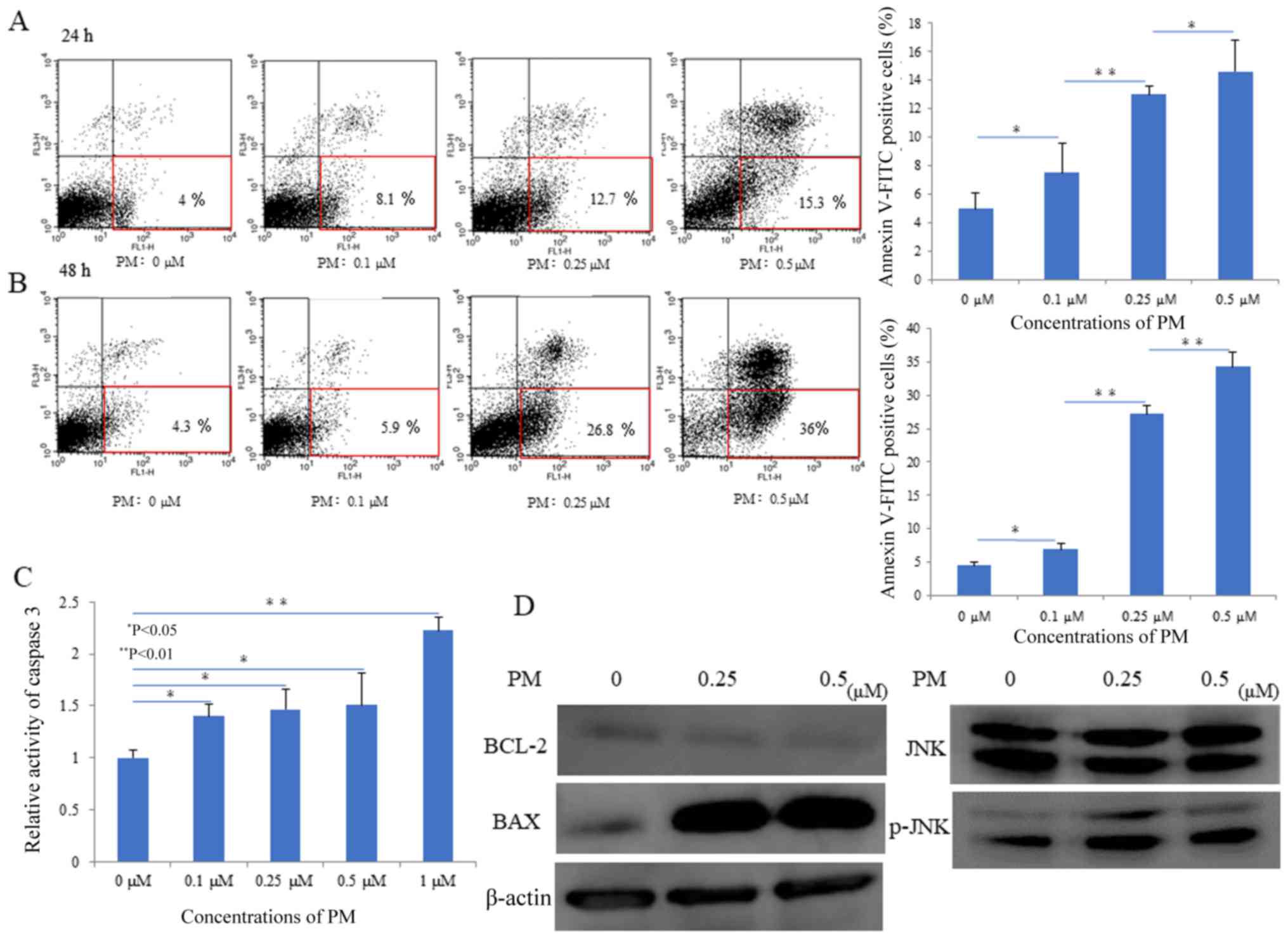
As shown in Fig. 2A and B, PM
treatment increased the percentage of early apoptotic in HT1080
cells in a concentration and time-dependent manner. However, no
significant apoptosis was induced in cells at the concentrations
≤0.1 µM.
PM induces apoptosis by activation of
caspases
In order to investigate if PM administration induced
apoptosis via activation of caspases, we performed a caspase-3
assay. As shown in Fig. 2C, PM
administration exhibited a concentration increase in caspase-3
activity after 6 h of treatment. Furthermore, western blot analysis
was performed to evaluate the expression of apoptosis-related
proteins. As shown in Fig. 2D,
treatment with PM increased the levels of phosphorylated JNK in a
concentration-dependent manner. Moreover, PM administration
decreased the expression of BCL-2, an apoptosis inhibiting factor,
and increased the expression of BAX, an apoptosis promoting
factor.
PM inhibits cell proliferation by
inhibition of AKT and MAPK signaling
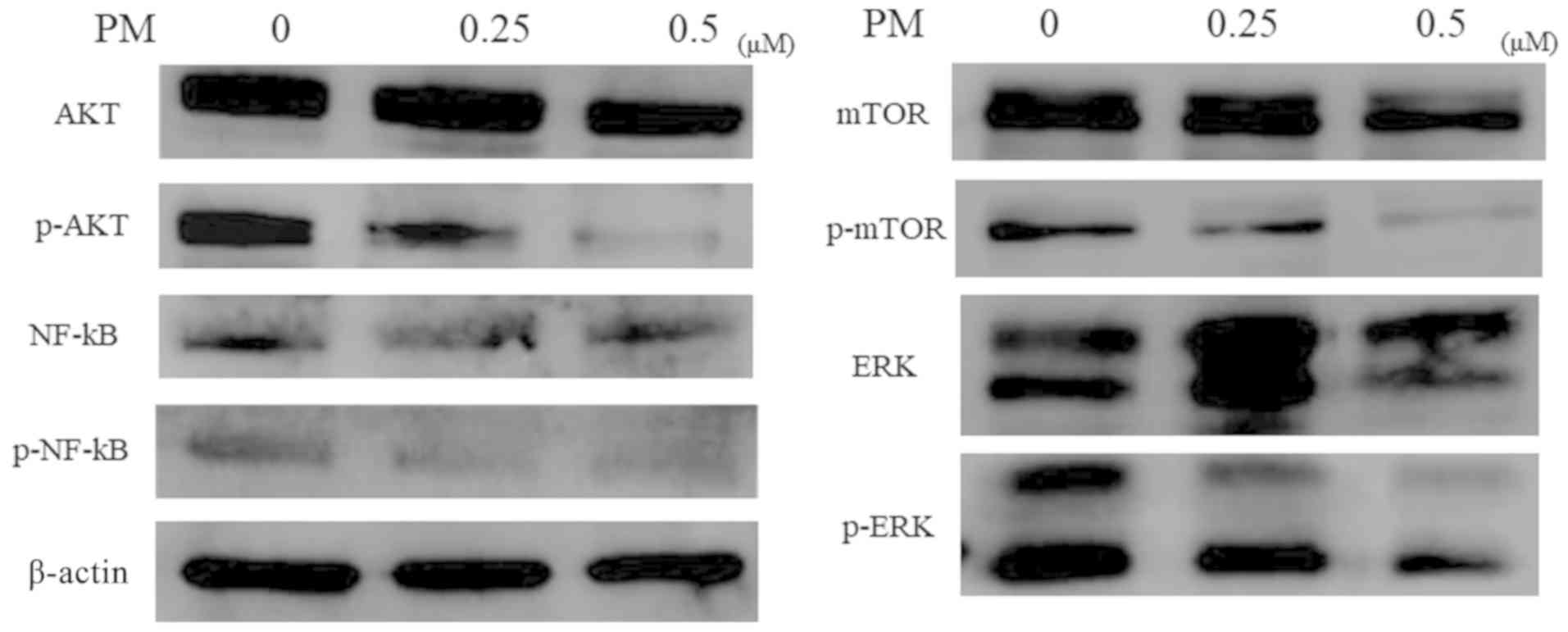
The correlation between PM treatment and AKT pathway
[AKT (cell survival signaling), mTOR, NF-κB] and the MAPK pathway
(ERK) was evaluated by western blot analysis. PM treatment
decreased the levels of phosphorylated p-AKT, p-NF-κB, and p-mTOR
in a concentration-dependent manner. Further, PM administration
decreased the phosphorylation of ERK, which is involved in cell
proliferation in a concentration-dependent manner (Fig. 3).
PM inhibits the proliferation of tumor
cells in vivo
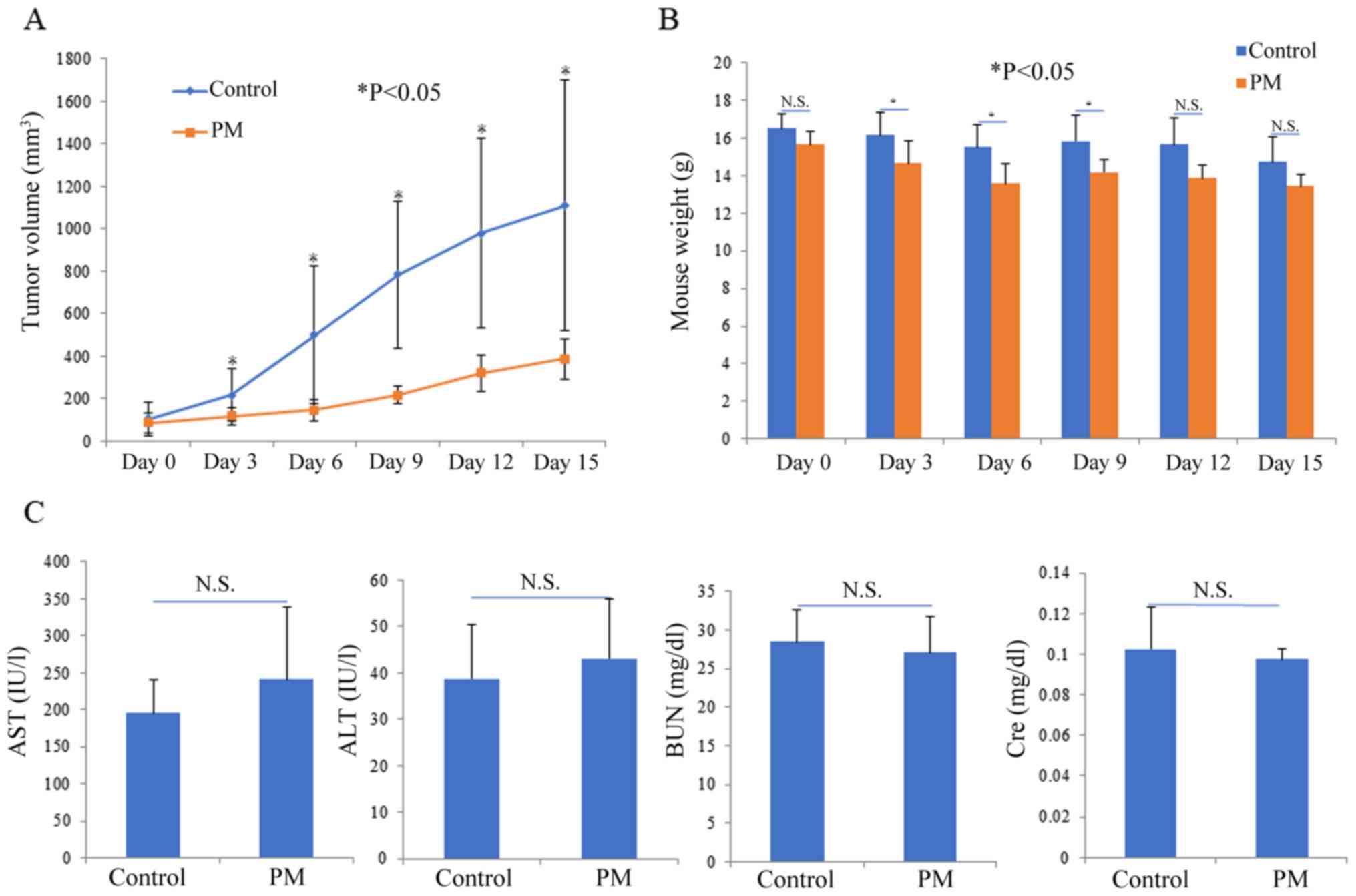
Mice were injected with subcutaneous grafts
comprising human fibrosarcoma cells, and the effect of PM
administration was analyzed. As shown in Fig. 4A and B, increase in tumor volume was
significantly reduced in the PM dose group as compared to the
control group. The increase in tumor volume was significantly
inhibited after 3 days of PM administration. Meanwhile, a
significant difference in body weight of the mice was observed in
the PM group immediately after PM administration, as compared to
the control group, but no significant difference was observed after
15 days of PM treatment. In an arterial blood draw, aspartate
transferase (AST) and alanine transferase (ALT) values, indicators
of hepatic impairment, were comparable in both the control group
and PM dose group. blood urea nitrogen (BUN) and creatinine
clearance (Cre) values, indicators of renal function, were also
normal in both groups.
Discussion
Fibrosarcoma is frequently difficult to treat
because of its poor sensitivity to anticancer drugs. Therefore, the
development of a novel drug is urgently needed. In recent years,
drugs derived from natural organic compounds have been used
clinically and are gaining attention. PM, a drug extracted from
plants, is a natural organic compound, and is reported to have
anti-inflammatory and anti-tumor effects (14–18).
PM has been reported to have anti-tumor effects on
human breast and lung cancer cells (21). We have previously shown that PM has
an anti-tumor effect on human osteosarcomas (22). In the present study, PM exhibited a
concentration- and time-dependent inhibition of cell viability in
human fibrosarcoma cells. In particular, at concentrations ≤0.25
µM, PM treatment reduced the viability of human fibrosarcoma cells
by >50%, but did not affect the cell viability of normal human
fibroblasts. Wu et al, reported that the IC50 values for
human breast cancer cell lines, MCF-10A and MDA-MB-231, were
1.4–1.6 and 0.5–0.6 µM, respectively, after 24 h of PM
administration and 1.0–1.2 and 0.4–0.6 µM, respectively, at 48 h.
Wu et al, also reported that the IC50 values for human lung
epithelial carcinoma cells, A549 and human liver cancer cell lines,
HepG2 and Hep3B, were 0.4–0.6 µM at 72 h of PM administration
(21). Mori et al, reported
that the IC50 value for human osteosarcoma cell lines, MNNG and
143B, were 0.8–0.9 and 0.5–0.6 µM, respectively, at 24 h of PM
administration, and 0.3–0.4 and 0.3–0.4 µM, respectively, at 48 h.
Meanwhile, the IC50 values for human osteoblasts were reported to
be ≥0.5 µM, at both 24 and 48 h of PM administration (22). In this study, The IC50 values for
human fibrosarcoma cells were 0.16 µM at 24 h of PM administration,
and 0.13 µM at 48 h of PM administration, whereas the IC50 values
for human fibroblasts were 0.59 µM at 24 h of PM administration,
and 0.32 µM at 48 h post PM administration. The IC50 of PM for
human fibrosarcoma cells was significantly lower than that for
human fibroblasts and human carcinoma cells; hence PM was believed
to have high sensitivity for human sarcoma cells.
Cell viability is regulated mainly by apoptosis and
proliferation (23). Apoptosis is a
form of programmed cell death that primarily occurs to preserve the
homeostasis of the individual (24).
PM is reported to have an anti-tumor effect, since it induces
apoptosis in human osteosarcoma and other carcinomas, such as
colorectal cancer, ovarian cancer, and leukemia (15,21,22,25–27).
Apoptosis is marked by structural changes in the cell membrane,
with progressive condensation of the nucleus, DNA fragmentation,
and changes in cell morphology (28). Structural changes in the cell
membrane are mainly detected by Annexin V staining. Annexin V is
cellular protein with strong affinity for phosphatidylserine (PS),
which is mainly found on the inner leaflet of the cell membrane in
healthy cells. Apoptosis induces structural changes in the symmetry
of the cell membrane flipping PS from the inner side of lipid
bilayer to the outer side. Hence, Annexin V, which binds to PS, can
be used to detect early-stage apoptosis. In the present study, PM
treatment caused a concentration-dependent increase in the number
of Annexin V-positive human fibrosarcoma cells. In addition, PM
administration increased the number of Annexin V-positive cells in
a time-dependent manner at concentrations of ≥0.25 µM. Therefore,
PM is believed to have an anti-tumor effect by inducing early
apoptosis in human fibrosarcoma cells in a concentration- and
time-dependent manner. The major pathway by which PM induces
apoptosis is via activation of caspases. Pro-apoptosis signals via
caspases are divided into initiator caspase group (caspases-2, −8
and −9) and a downstream effector caspase group (caspase-3 and −7),
and activation of the effector caspases adversely affects the cell
membrane. In the present study, that activity of caspases was
evaluated, with focus on the effector caspase, caspase-3. PM
activated caspase-3 after 6 h of administration. Docetaxel, which
also belongs to the terpenoid family, has been observed to activate
caspase-3, 24 h later after drug administration (29). In the present study, caspase-3
activity at 6 h after PM administration was found to be comparable
to that of docetaxel, however, PM treatment induced a faster
response. While the molecular weight of docetaxel is 808 kDa, the
molecular weight of PM is 465 kDa. PM is believed to initiate
apoptosis in a shorter time span than docetaxel, because cell
penetration ability is inversely proportional to molecular weight
(20).
JNK, BCL-2, and BAX are signaling proteins that are
components of the caspase pathway (26). JNK is activated by phosphorylation,
which causes the downstream inhibition of BCL-2, which in turn
increases the BAX/BCL-2 ratio, and induces apoptosis by acting on
caspase-9, an effector of the intrinsic caspase pathway (26). We hypothesize that PM treatment
increased the levels of phosphorylated JNK and BAX, and decreased
the levels of BCL-2, thereby activating caspase-3 and inducing
apoptosis.
Signal transduction pathways related to cell
proliferation include the PI3K/AKT/mTOR pathway and MAPK pathways
(30–33). In carcinomas and sarcomas, these
pathways are closely associated with cell proliferation and
anticancer drug resistance. PM has been shown to induce apoptosis
in human osteosarcomas and carcinomas, such as colorectal cancer,
ovarian cancer, and leukemia, via inhibition of AKT and MAPK
signaling (15,22,25,26,34).
Phosphorylation of AKT induces the downstream phosphorylation of
NF-κB and mTOR, thereby activating them. mTOR signaling integrates
environmental cues, such as growth factors, and nutrition/energy
status inside and outside of the cell and increases ribosome
production and protein synthesis, thereby, promoting cell growth
and proliferation. NF-κB is a key transcription factor that
regulates the cellular stress response, inflammation response,
cancer cell proliferation, and apoptosis resistance (35–39).
Therefore, mTOR and NF-κB are important therapeutic targets of
cancer treatment. In the present study, PM produced an anti-tumor
effect by decreasing the cellular levels of phosphorylated AKT,
NF-κB, and mTOR in a concentration-dependent manner. Moreover, MAPK
pathway has also been shown to be involved in transcription, cell
proliferation, cell mobility, and cell survival (40,41). In
particular, phosphorylated ERK acts on cyclin D1, inducing protein
synthesis and promoting transcription and cell proliferation
(42). In the present study, PM
treatment inhibited the phosphorylation of ERK, AKT, and hence MAPK
signaling, which suggests that PM administration affects cell
proliferation. PM suppressed an increase in tumor volume in
vivo. The growth inhibitory effect was equivalent to or better
than that of colorectal cancer and breast cancer (26,43). PM
treatment is believed to produce anti-tumor effect in vitro
and in vivo by inducing apoptosis and inhibiting cell
proliferation in human fibrosarcoma cells.
Generally, the biological effects of drugs are
mainly evaluated by the presence of anorexia or variation in body
weight (44). However, only a few
reports have analyzed body weight in mouse models. In the present
study, we observed a significant difference in body weight between
the PM dose group and the control group mice; hence we conclude
that PM exhibits low toxicity.
Liver and kidneys are the major organs involved in
drug metabolism pathways. AST, ALT, BUN, and creatine levels are
often used to evaluate hepatic and renal function. We have
previously reported that PM administration in mice does not
decrease hepatic function (22), but
we did not study the effects of PM administration on renal
function. In the present study, PM treatment did not cause either
hepatic or renal dysfunction. In addition, the tumor growth was
suppressed to the same level or higher even though the total number
of doses was reduced by reducing the number of administrations
compared to other carcinomas (26,43). We
thought that reducing the total dose of PM could more reliably
reduce the side effects of the drug. Therefore, we speculate that
PM can be safely administered with few side effects on the
body.
In conclusion, PM produced anti-tumor effects by
inducing apoptosis in human fibrosarcoma cells via activation of
the caspases, and inhibited cell proliferation by inhibiting the
AKT and MAPK signaling. Therefore, we conclude that PM is safe and
could prove to be an effective treatment drug for fibrosarcoma.
Acknowledgements
This study was funded by JSPS Grants-In-Aid for
Scientific Research JP17K10975 and JP18K09115. The authors would
like to thank Dr Masaharu Shin-Ya (Department of Immunology, Kyoto
Prefectural University of Medicine, Kyoto, Japan) who helped with
the research.
Funding
The present study was supported by the JSPS KAKENHI
(grant nos. 17K10975 and 18K09115).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TS, RT, ST, NM, YA, OM, TK and DH designed
experiments. YM, DH and NM performed the research and acquired
data. TS, RT, ST, YA, OM and TK interpreted the data and provided
valuable advice on the manuscript. DH wrote the manuscript. TS, RT,
YA, OM and TK proofread the manuscript and revised it critically.
All authors read the final manuscript and did a final check of the
version to publish. TK agreed to be accountable for all aspects of
the work in ensuring that questions related to the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
All in vivo procedures were performed in
accordance with the NIH Guide for the Care and Use of Laboratory
Animals. The present study using mice was approved The Experimental
Animals Committee, Kyoto Prefectural University of Medicine
(approval no. M30-528).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
IC50
|
50% inhibitory concentration
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
mTOR
|
mammalian target of rapamycin
|
|
NF-κB
|
nuclear factor-κB
|
|
MAPK
|
mitogen-activated protein kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
TBST
|
tris-buffered saline tablets
|
|
JSPS
|
Japan Society for the Promotion of
Science
|
References
|
1
|
Yonemori K, Kodaira M, Satoh T, Kudo T,
Takahashi S, Nakano K, Ando Y, Shimokata T, Mori J, Inoue K, et al:
Phase 1 study of olaratumab plus doxorubicin in Japanese patients
with advanced soft-tissue sarcoma. Cancer Sci. 109:3962–3970. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jain S, Xu R, Prieto VG and Lee P:
Molecular classification of soft tissue sarcomas and its clinical
applications. Int J Clin Exp Pathol. 3:416–428. 2010.PubMed/NCBI
|
|
3
|
Pervaiz N, Colterjohn N, Farrokhyar F,
Tozer R, Figueredo A and Ghert M: A systematic meta-analysis of
randomized controlled trials of adjuvant chemotherapy for localized
resectable soft-tissue sarcoma. Cancer. 113:573–581. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eilber FR, Giuliano AE, Huth JF and Morton
DL: A randomized prospective trial using postoperative adjuvant
chemotherapy (adriamycin) in high-grade extremity soft-tissue
sarcoma. Am J Clin Oncol. 11:39–45. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bramwell VH, Mouridsen HT, Santoro A,
Blackledge G, Somers R, Verwey J, Dombernowsky P, Onsrud M, Thomas
D, Sylvester R, et al: Cyclophosphamide versus ifosfamide: Final
report of a randomized phase II trial in adult soft tissue
sarcomas. Eur J Cancer Clin Oncol. 23:311–321. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mytilinaiou M, Nikitovic D, Berdiaki A,
Papoutsidakis A, Papachristou DJ, Tsatsakis A and Tzanakakis GN:
IGF-I regulates HT1080 fibrosarcoma cell migration through a
syndecan-2/Erk/ezrin signaling axis. Exp Cell Res. 361:9–18. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ratan R and Patel SR: Chemotherapy for
soft tissue sarcoma. Cancer. 122:2952–2960. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun Y, Gao LL, Tang MY, Feng BM, Pei YH
and Yasukawa K: Triterpenoids from Euphorbia maculata and their
anti-inflammatory effects. Molecules. 23:E21122018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patlolla JM and Rao CV: Triterpenoids for
cancer prevention and treatment: Current status and future
prospects. Curr Pharm Biotechnol. 13:147–155. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Akihisa T, Tokuda H, Ichiishi E, Mukainaka
T, Toriumi M, Ukiya M, Yasukawa K and Nishino H: Anti-tumor
promoting effects of multiflorane-type triterpenoids and cytotoxic
activity of karounidiol against human cancer cell lines. Cancer
Lett. 173:9–14. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Banno N, Akihisa T, Yasukawa K, Tokuda H,
Tabata K, Nakamura Y, Nishimura R, Kimura Y and Suzuki T:
Anti-inflammatory activities of the triterpene acids from the resin
of Boswellia carteri. J Ethnopharmacol. 107:249–253. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sassa H, Kogure K, Takaishi Y and Terada
H: Structural basis of potent antiperoxidation activity of the
triterpene celastrol in mitochondria: Effect of negative membrane
surface charge on lipid peroxidation. Free Radic Biol Med.
17:201–207. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dirsch VM, Kiemer AK, Wagner H and Vollmar
AM: The triterpenoid quinonemethide pristimerin inhibits induction
of inducible nitric oxide synthase in murine macrophages. Eur J
Pharmacol. 336:211–217. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee JS, Yoon IS, Lee MS, Cha EY, Thuong
PT, Diep TT and Kim JR: Anticancer activity of pristimerin in
epidermal growth factor receptor 2-positive SKBR3 human breast
cancer cells. Biol Pharm Bull. 36:316–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang H, Landis-Piwowar KR, Lu D, Yuan P,
Li L, Reddy GP, Yuan X and Dou QP: Pristimerin induces apoptosis by
targeting the proteasome in prostate cancer cells. J Cell Biochem.
103:234–244. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan YY, Bai JP, Xie Y, Yu JZ and Ma CG:
The triterpenoid pristimerin induces U87 glioma cell apoptosis
through reactive oxygen speciesmediated mitochondrial dysfunction.
Oncol Lett. 5:242–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Byun JY, Kim MJ, Eum DY, Yoon CH, Seo WD,
Park KH, Hyun JW, Lee YS, Lee JS, Yoon MY and Lee SJ: Reactive
oxygen species-dependent activation of Bax and poly(ADP-ribose)
polymerase-1 is required for mitochondrial cell death induced by
triterpenoid pristimerin in human cervical cancer cells. Mol
Pharmacol. 76:734–744. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Costa PM, Ferreira PM, Bolzani Vda S,
Furlan M, de Freitas Formenton Macedo Dos Santos VA, Corsino J, de
Moraes MO, Costa-Lotufo LV, Montenegro RC and Pessoa C:
Antiproliferative activity of pristimerin isolated from Maytenus
ilicifolia (Celastraceae) in human HL-60 cells. Toxicol In Vitro.
22:854–863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Okuno S, Edmonson J, Mahoney M, Buckner
JC, Frytak S and Galanis E: Phase II trial of gemcitabine in
advanced sarcomas. Cancer. 94:3225–3229. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Keller F, Wilms H, Schultze G, Offerman G
and Molzahn M: Effect of plasma protein binding, volume of
distribution and molecular weight on the fraction of drugs
eliminated by hemodialysis. Clin Nephrol. 19:201–205.
1983.PubMed/NCBI
|
|
21
|
Wu CC, Chan ML, Chen WY, Tsai CY, Chang FR
and Wu YC: Pristimerin induces caspase-dependent apoptosis in
MDA-MB-231 cells via direct effects on mitochondria. Mol Cancer
Ther. 4:1277–1285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mori Y, Shirai T, Terauchi R, Tsuchida S,
Mizoshiri N, Hayashi D, Arai Y, Kishida T, Mazda O and Kubo T:
Antitumor effects of pristimerin on human osteosarcoma cells in
vitro and in vivo. Onco Targets Ther. 10:5703–5710. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wyllie AH, Kerr JF and Currie AR: Cell
death: The significance of apoptosis. Int Rev Cytol. 68:251–306.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lu Z, Jin Y, Chen C, Li J, Cao Q and Pan
J: Pristimerin induces apoptosis in imatinib-resistant chronic
myelogenous leukemia cells harboring T315I mutation by blocking
NF-kappaB signaling and depleting Bcr-Abl. Mol Cancer. 9:1122010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yousef BA, Hassan HM, Guerram M, Hamdi AM,
Wang B, Zhang LY and Jiang ZZ: Pristimerin inhibits proliferation,
migration and invasion, and induces apoptosis in HCT-116 colorectal
cancer cells. Biomed Pharmacother. 79:112–119. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xie G, Yu X, Liang H, Chen J, Tang X, Wu S
and Liao C: Pristimerin overcomes adriamycin resistance in breast
cancer cells through suppressing Akt signaling. Oncol Lett.
11:3111–3116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Murgia M, Pizzo P, Sandoná D, Zanovello P,
Rizzuto R and Di Virgilio F: Mitochondrial DNA is not fragmented
during apoptosis. J Biol Chem. 267:10939–10941. 1992.PubMed/NCBI
|
|
29
|
Tamaki H, Harashima N, Hiraki M, Arichi N,
Nishimura N, Shiina H, Naora K and Harada M: Bcl-2 family
inhibition sensitizes human prostate cancer cells to docetaxel and
promotes unexpected apoptosis under caspase-9 inhibition.
Oncotarget. 5:11399–11412. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brown JS and Banerji U: Maximising the
potential of AKT inhibitors as anti-cancer treatments. Pharmacol
Ther. 172:101–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Burris HA III: Overcoming acquired
resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR
pathway. Cancer Chemother Pharmacol. 71:829–842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mu X, Shi W, Sun L, Li H, Jiang Z and
Zhang L: Pristimerin, a triterpenoid, inhibits tumor angiogenesis
by targeting VEGFR2 activation. Molecules. 17:6854–6868. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brasier AR: The NF-kappaB regulatory
network. Cardiovasc Toxicol. 6:111–130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Connolly JL, Rodgers SE, Clarke P, Ballard
DW, Kerr LD, Tyler KL and Dermody TS: Reovirus-induced apoptosis
requires activation of transcription factor NF-kappaB. J Virol.
74:2981–2989. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu G, Wang X, Han Y and Wang P: Protein
arginine methyltransferase 5 promotes bladder cancer growth through
inhibiting NF-κB dependent apoptosis. EXCLI J. 17:1157–1166.
2018.PubMed/NCBI
|
|
39
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Potthoff RF and George SL: Flexible phase
I clinical trials: Allowing for nonbinary toxicity response and
removal of other common limitations. Stat Biopharm Res. 1:213–228.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cevatemre B, Erkısa M, Aztopal N, Karakas
D, Alper P, Tsimplouli C, Sereti E, Dimas K, Armutak EII, Gurevin
EG, et al: A promising natural product, pristimerin, results in
cytotoxicity against breast cancer stem cells in vitro and
xenografts in vivo through apoptosis and an incomplete autopaghy in
breast cancer. Pharmacol Res Mar. 129:500–514. 2018. View Article : Google Scholar
|
|
44
|
Ravasco P, Monteiro-Grillo I, Vidal PM and
Camilo ME: Cancer: Disease and nutrition are key determinants of
patients' quality of life. Support Care Cancer. 12:246–252. 2004.
View Article : Google Scholar : PubMed/NCBI
|