Introduction
Like histone and DNA, the epigenetic modification of
RNA species has been extensively reported over the past few decades
(1). Since the 1950s, over 100
chemical modification types have been described in RNA,
particularly in rRNA and tRNA (2).
Of note, any micro-event during base modification can result in
potent influence on the metabolic pathways, as well as the
resulting organism phenotype alterations. As a result, the abnormal
alteration can result in the occurrence of abnormalities and
disease initiation like tumors (3,4).
The m6A modification has attracted wide
attention in the field of epitranscriptomics, which is associated
with the highest prevalence among transcripts (5,6). Thanks
to the developments of recent technology, N6-methyladenosine
(m6A) modifications in mRNA have been identified
(7,8). m6A modification reveals an
extensive, while rare, epitranscriptomic landscape, which
participates in various physiological processes, including cancer
(9).
There are 3 protein classes that can regulate
m6A modification, the ‘reader’ (m6A-binding
protein), ‘eraser’ (m6A demethylating enzyme) and
‘writer’ (adenosine methyltransferase) (3,10,11).
Specifically, m6A modification can be subjected to
reversible installment and removal by writers and erasers,
separately. This process is dynamic and reversible. However, the
deregulated m6A modification, which is associated with
abnormal expression levels or functions of the m6A
readers, erasers and writers, may result in cancer genesis and
progression (12).
Hepatocellular carcinoma (HCC), one of the most
frequently observed liver cancer types, is a severe worldwide
health problem (13,14). Nonetheless, no existing study has
comprehensively analyzed the expression levels of m6A
RNA methylation regulators among HCCs that have various clinical
and pathological features, or their role and prognosis significance
in the malignant development of HCC. The present study carried out
a systemic analysis on the expression levels of 13 extensively
identified m6A RNA regulators in HCCs, according to the
RNA sequencing information extracted from The Cancer Genome Atlas
(TCGA) (n=377) database. In addition, the expression profiles for
all 13 m6A modification regulators were provided based
on various clinical and pathological characteristics. According to
the present results, the expression levels of the m6A
RNA methylation regulators played an important role during HCC
malignant development. A signature was also constructed using 6
screened m6A RNA methylation regulators for HCC
prognosis stratification. The constructed signature was further
confirmed by the International Cancer Genome Consortium (ICGC)
database.
Materials and methods
Data extraction
Data were downloaded from the TCGA database. Gene
expression data and the clinical information of HCC patients
(https://tcga-data.nci.nih.gov/) were
downloaded using the Data Transfer Tool (provided by GDC Apps). A
total of 374 tumor and 50 normal samples from 377 HCC patients were
used in this study to analyze the differentially expressed
m6A RNA methylation regulators. Typically, the list of
the 13 m6A RNA methylation regulators was determined
with reference to published literature (4). All data were publicly available and
open-access; as a result, Ethics Committee approval was not
required. Data were processed in accordance with the data access
policies, as well as the TCGA Human Subject Protection system
formulated by the National Institutes of Health (NIH; http://cancergenome.nih.gov/publications/publicationguidelines).
The LIRI-JP project from the ICGC database was used as an
independent validation cohort (n=237).
Bioinformatic analysis
First, the expression patterns of m6A RNA
methylation regulators were compared between tumors and normal
samples, and Spearman's rank correlation coefficient was used for
correlation analysis among the regulators. In addition, the
interactions between m6A RNA methylation regulators
would be examined using the Search Tool for the Retrieval of
Interacting Genes/Proteins database (http://www.string-db.org/). To investigate the
function of m6A RNA methylation regulators in HCCs, HCCs
were clustered in various groups using the ‘ConsensusClusterPlus’
(http://www.bioconductor.org/). In
addition, gene expression profiles of the various HCC groups were
investigated using principal component analysis (PCA) as well as R
package. Moreover, the c2.cp.kegg.v6.2.symbols were examined based
on gene set enrichment analysis (GSEA) at 1,000 random sample
permutations using JAVA procedure (http://software.broadinstitute.org/gsea/index.jsp).
Construction of a signature based on
m6A RNA methylation regulators
The association between each m6A RNA
methylation regulator and patient overall survival (OS) was
calculated using the univariate Cox model. Subsequently, the
thirteen m6A RNA methylation regulators were screened
and verified by least absolute shrinkage and selection operator
(LASSO) regression using the ‘glmnet’ R software. Finally, the
regulator-based prognostic risk score was constructed through
linearly multiplying the expression level with the regression model
(β) according to the following formula: Risk=β
regulator1 × regulator1 expression + β
regulator2 × regulator2 expression + · ···· +
β regulatorn × regulatorn expression
(15,16).
Confirmation of the signature based on
m6A RNA methylation regulators
Patients, together with their survival information,
were distributed according to risk score. Furthermore, patients
were classified as high- or low-risk, according to their median
risk score value. Next, survival curves were drawn according to the
Kaplan-Meier method, which could predict the high or low risk of
patients. Subsequently, the sensitivity and specificity of survival
prediction were compared using risk score, and the time-dependent
receiver operating characteristic (ROC) curves were employed to
evaluate the accuracy of predicting the 5-year prognosis. In
addition, one-way analysis of variance or t-test were carried out
to compare risk scores among different cases stratified according
to their clinical and molecular pathological features, in order to
assess the signature risk score for HCC cases possessing various
clinical and pathological features. Univariate and multivariate Cox
proportional hazards regression analysis was then conducted to
examine whether the risk was predicted independently from other
clinical factors.
Statistical analysis
A two-sided P<0.05 was considered to indicate a
statistically significant difference. Prism 7 (GraphPad Software
Inc., La Jolla, CA, USA) and R software (version 3.4.1; R
Foundation, Vienna, Austria), were employed for all analyses.
Results
Expression difference in the
m6A RNA methylation regulators between HCCs and normal
tissues
The clinicopathological information of all patients
is summarized in Table I.
Considering the important biological functions of each
m6A RNA methylation regulator during tumorigenesis and
development, the differences in all m6A RNA methylation
regulators between HCCs and normal samples were comprehensively
examined. The expression level of each m6A RNA
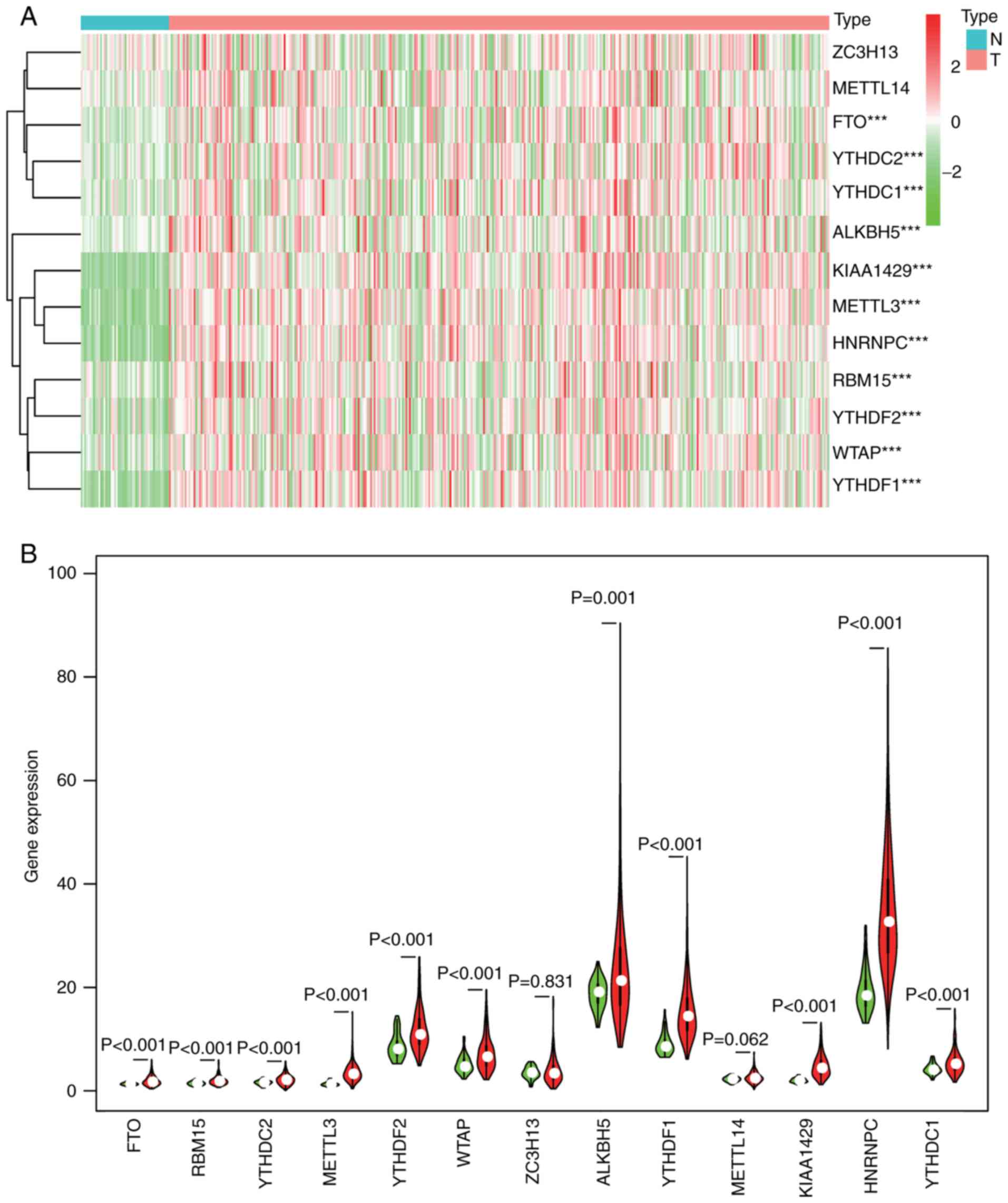
methylation regulator is presented in heatmaps (Fig. 1A) and violin plots (Fig. 1B), which showed that the expression
of most m6A RNA methylation regulators was markedly
upregulated in HCCs, namely ZC3H13 [not significant (NS)], METTL14
(NS), FTO (P<0.001), YTHDC2 (P<0.001), YTHDC1 (P<0.001),
ALKBH5 (P=0.001), KIAA1429 (P<0.001), METTL3 (P<0.001),
HNRNPC (P<0.001), RBM15 (P<0.001), YTHDF2 (P<0.001), WTAP
(P<0.001) and YTHDF1 (P<0.001).
 | Table I.Baseline patient characteristics. |
Table I.
Baseline patient characteristics.
|
Characteristics | Number | Percentage |
|---|
| Total | 377 | 100.0 |
| Median follow-up,
days (range) | 557 (0–3,675) |
|
| Age, years (mean ±
SD) | 59.5±13.5 |
|
| Sex |
|
Male | 255 | 67.6 |
|
Female | 122 | 32.4 |
| Ethnicity |
|
White | 235 | 62.3 |
|
Others | 142 | 37.7 |
| Grade |
| I | 55 | 14.6 |
| II | 180 | 47.7 |
|
III | 124 | 32.9 |
| IV | 13 | 3.4 |
|
Unknown | 5 | 1.3 |
| Stage |
| I | 175 | 46.4 |
| II | 87 | 23.1 |
|
III | 86 | 22.8 |
| IV | 5 | 1.3 |
|
Unknown | 24 | 6.4 |
| T stage |
| I | 185 | 49.1 |
| II | 95 | 25.2 |
|
III | 81 | 21.5 |
| IV | 13 | 3.4 |
|
Unknown | 3 | 0.8 |
| N |
| No | 257 | 68.2 |
|
Yes | 4 | 1.1 |
|
Unknown | 116 | 30.8 |
| M |
| No | 272 | 72.1 |
|
Yes | 4 | 1.1 |
|
Unknown | 101 | 26.8 |
Regulator correlation and
interaction
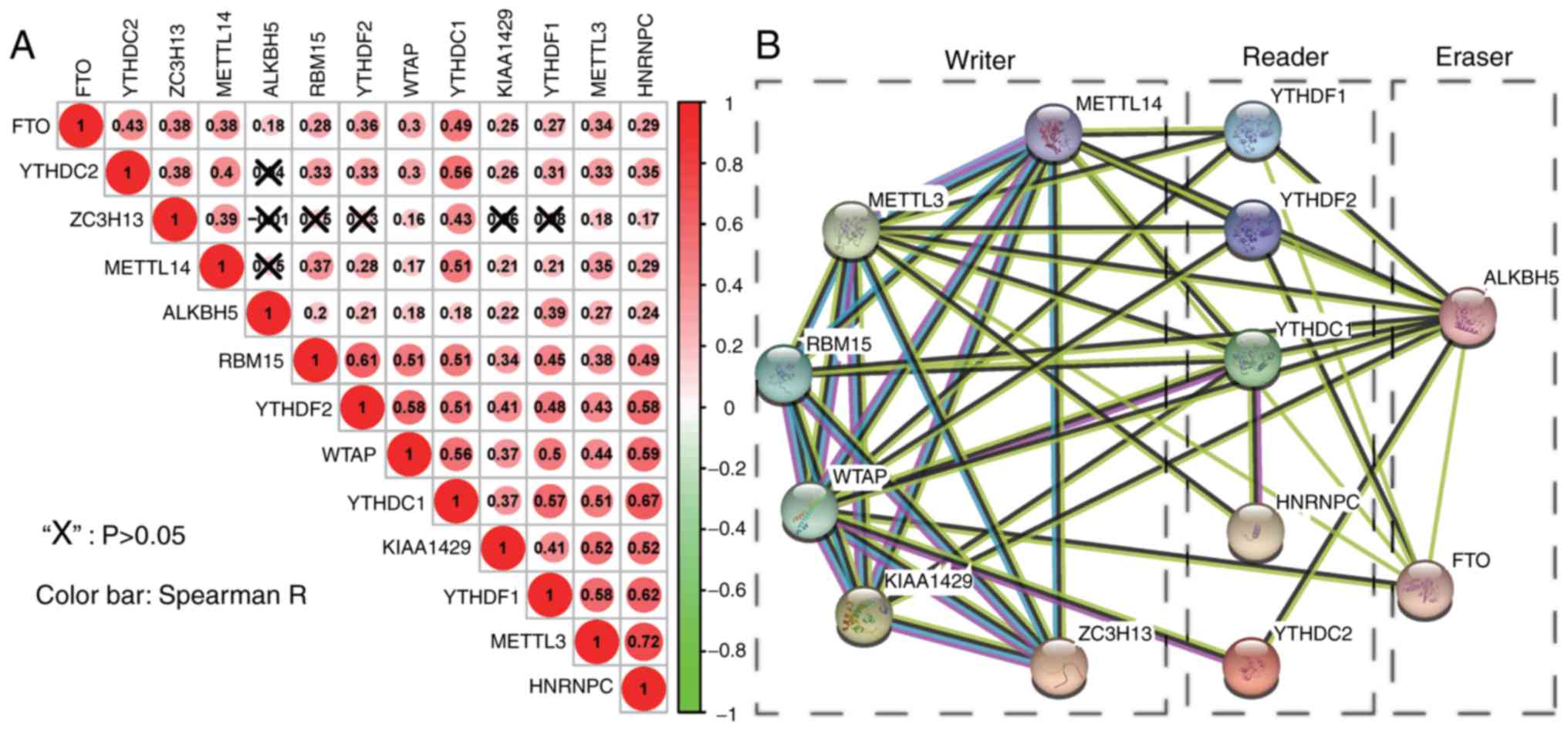
For a better understanding of interactions between
these 13 m6A RNA methylation regulators, the correlation
(Fig. 2A) and interaction (Fig. 2B) among them was also analyzed.
Clearly, ZC3H13 and ALKBH5 were negatively correlated, while the
other pairs were positively correlated. FTO, WTAP, YTHDC1, METTL3
and HNRNPC exhibited a significantly positive correlation with the
other 12 regulators. Of note, the correlation between HNRNPC and
METTL3 (0.72), YTHDC1 (0.67) and YTHDF1 (0.62), ranked top among
all correlations. In Fig. 2B, the
interactions between 2 regulators were supported by experimental
determination (pink lines), the existing databases (blue lines),
co-expression (black lines), or text mining (dark olive green
lines). In addition, there was a pink line connected to neither two
erasers (FTO and ALKBH5) nor two readers (YTHDF1 and YTHDF2),
suggesting that more experiments should be carried out on these 4
regulators to examine their interactions with other regulators.
m6A RNA methylation
regulator cluster analysis
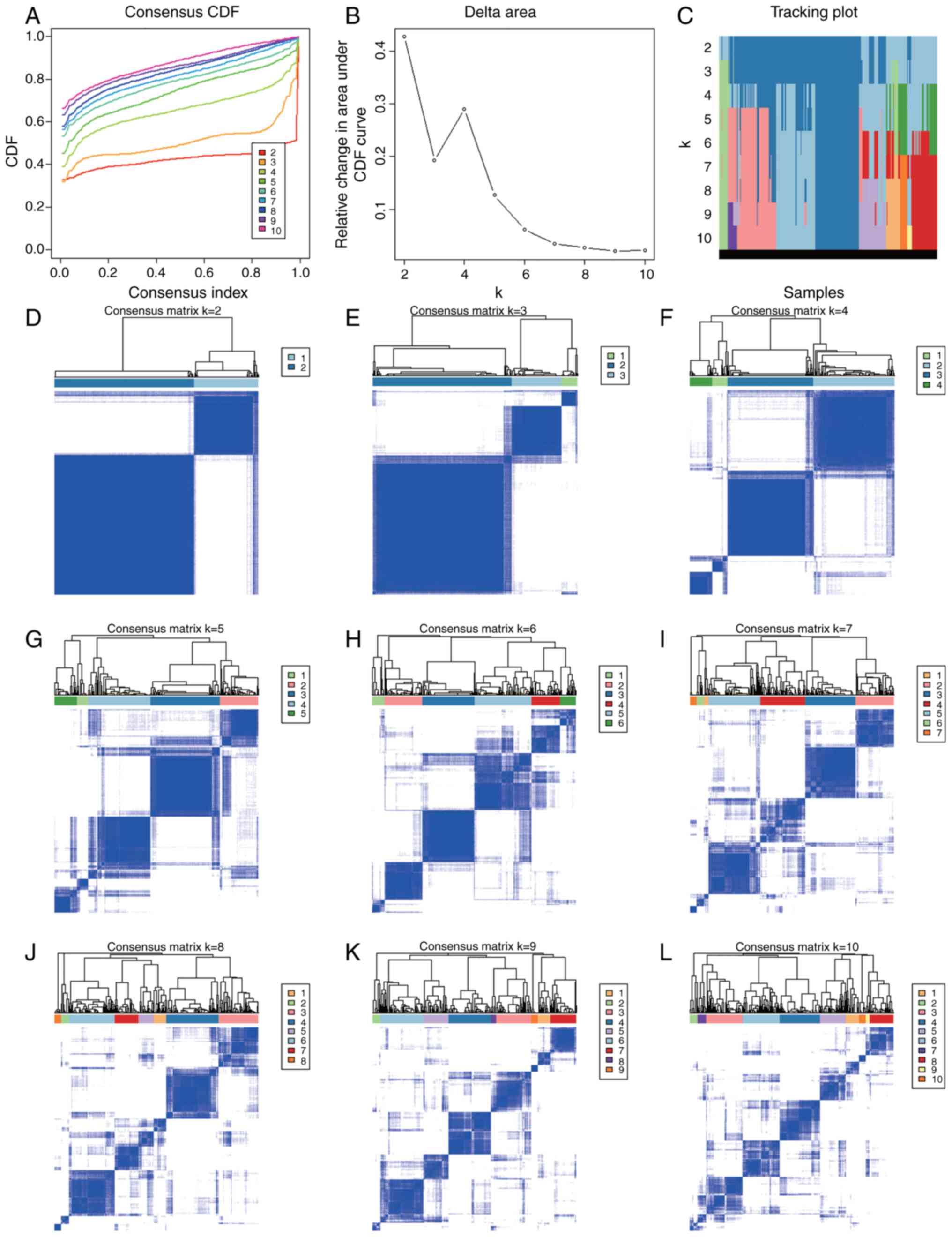
Based on the expression similarity of m6A
RNA methylation regulators, it appeared that k=2 was a sufficient
value from the clustering stability range of k=2-10 in the TCGA
datasets (Fig. 3A-L). Thereafter,
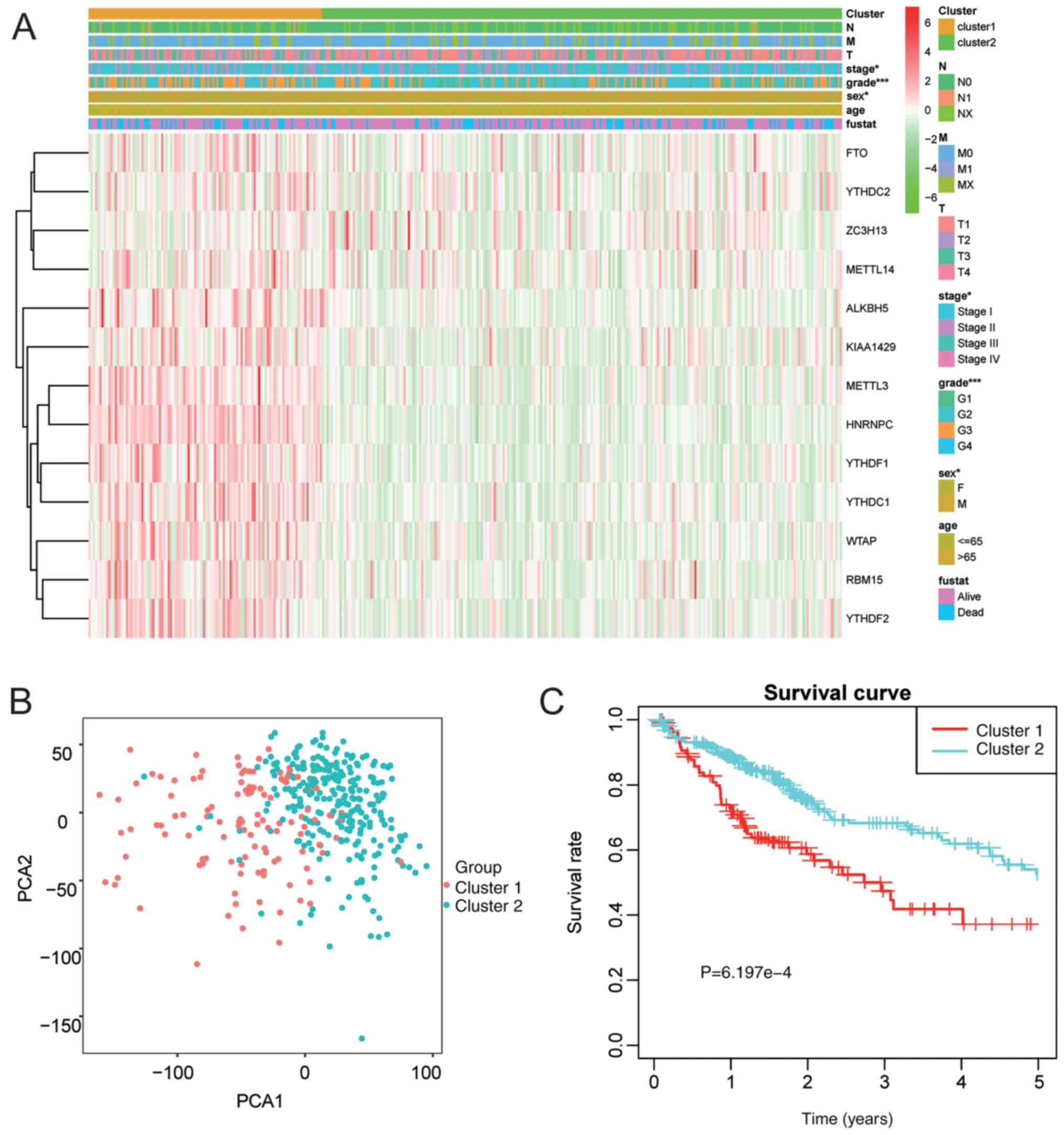
patients were clustered into one of the two subgroups. Therefore,
the clinical and pathological characteristics between the two
subgroups classified based on k=2 (clusters 1/2) were compared
(Fig. 4A). The cluster 1 subgroup
was markedly correlated with late stage at diagnosis (P<0.05)
and high frequency of grade III/IV (P<0.001). Furthermore, PCA
was also employed for comparing transcriptional patterns between
the two subgroups. Our findings indicated that these two subgroups
were distinctly different (Fig. 4B).
In addition, the cluster 1 subgroup had an evidently reduced OS, as
compared with that in the cluster 2 subgroup (P=6.197e-4) (Fig. 4C).
Prognosis of m6A RNA
methylation regulator, as well as construction and validation of
the risk signature
The prognostic value of the m6A RNA
methylation regulators in HCCs was also examined. Specifically,
gene expression in TCGA datasets was analyzed using the univariate
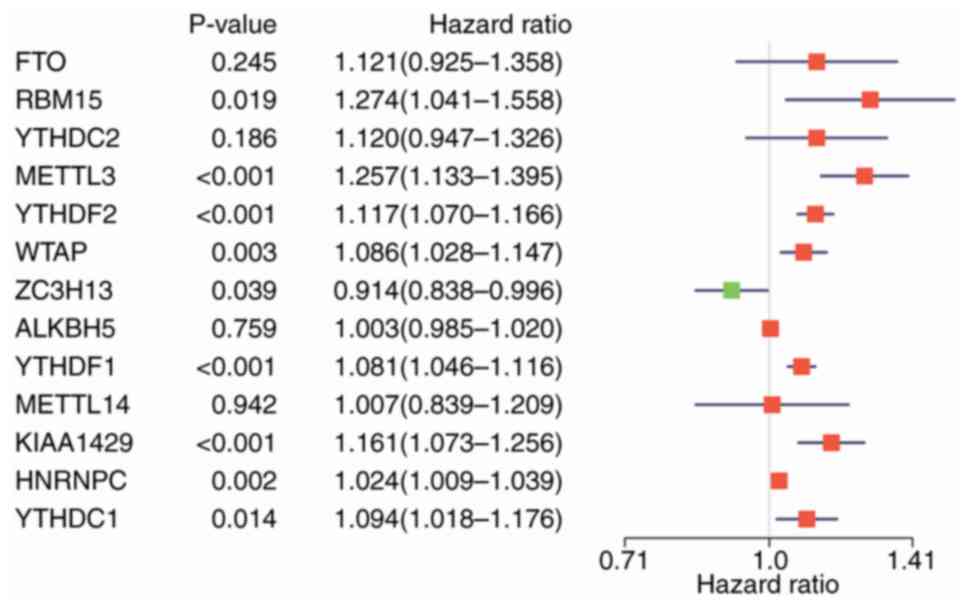
Cox regression model. According to the findings, 9/13 genes
examined in this study exhibited a marked correlation with OS
(P<0.05; Fig. 5). Among these 9
genes, YTHDF2, YTHDF1, METTL3, KIAA1429, HNRNPC, WTAP, YTHDC1 and
RBM15 were the risk genes with a hazard ratio (HR) of >1, while
ZC3H13 was the protective gene with a HR of <1.
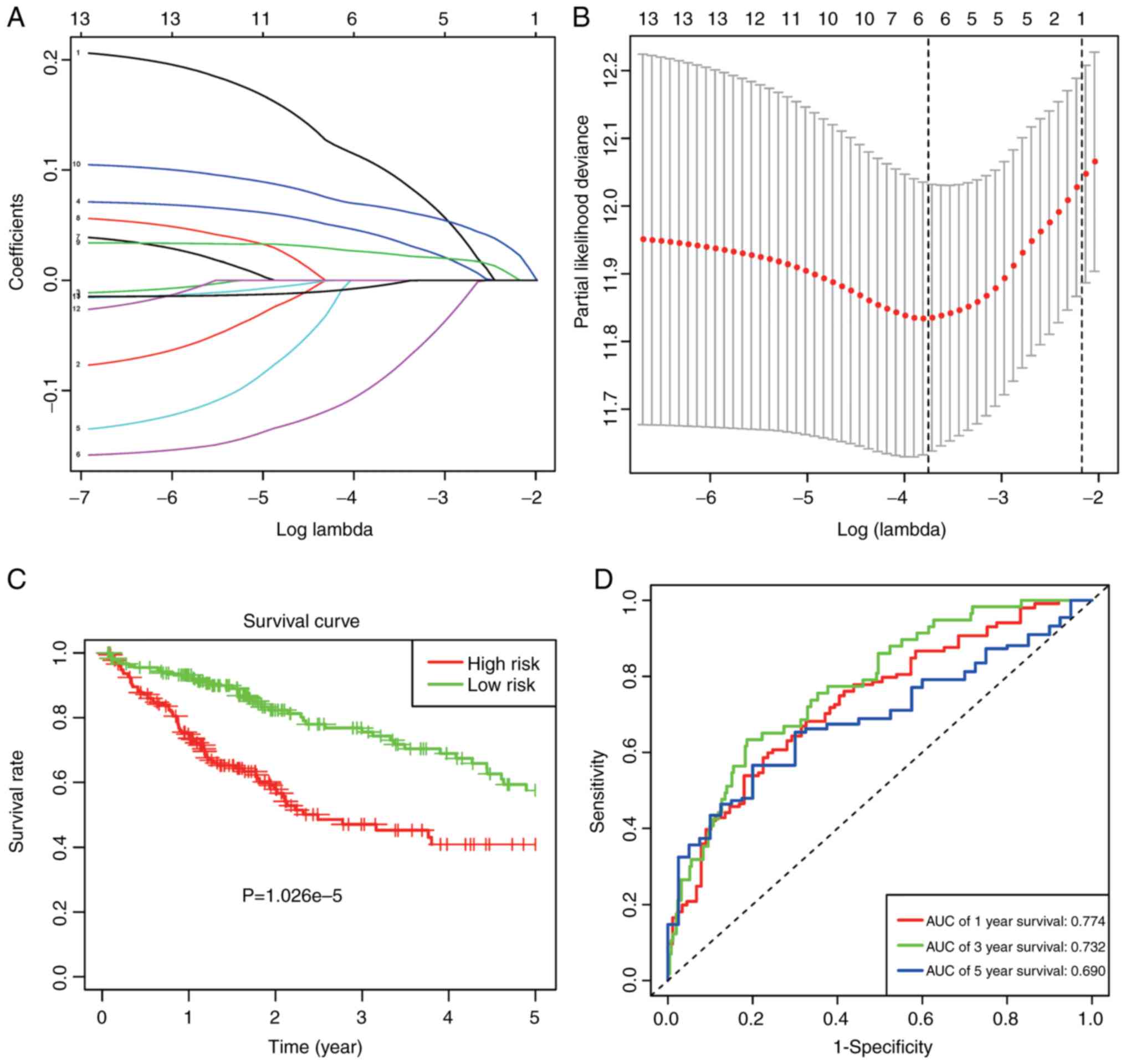
For a more precise prediction of HCC prognosis using
the m6A RNA methylation regulators, the Cox regression
algorithm LASSO was utilized (Fig. 6A
and B). Six genes, including METTL3, KIAA1429, ZC3H13, YTHDF1,
YTHDF2 and ALKBH5, were selected for the construction of a risk
signature, according to the minimal standards. In addition, the
associated coefficients were acquired based on the LASSO algorithm.
Risk was formulated as follows: Risk=0.105*METTL3 expression +
0.041*KIAA1429 expression - 0.094*ZC3H13 expression + 0.025*YTHDF1
expression + 0.067*YTHDF2 expression - 0.005*ALKBH5 expression.
HCC cases obtained from TCGA datasets were
classified as low- or high-risk, according to the median risk score
value of 3.266, and the distinct heterogeneities with regard to OS
were observed between these two subgroups, in order to examine the
value of the as-constructed signature in predicting prognosis
(P=1.062e-5; Fig. 6C). Furthermore,
the ROC curves verified that, prognosis prediction using the risk
signature could attain an area under the ROC curve (AUC) value of
0.774 (1 year), 0.732 (3 years) and 0.690 (5 years; Fig. 6D).
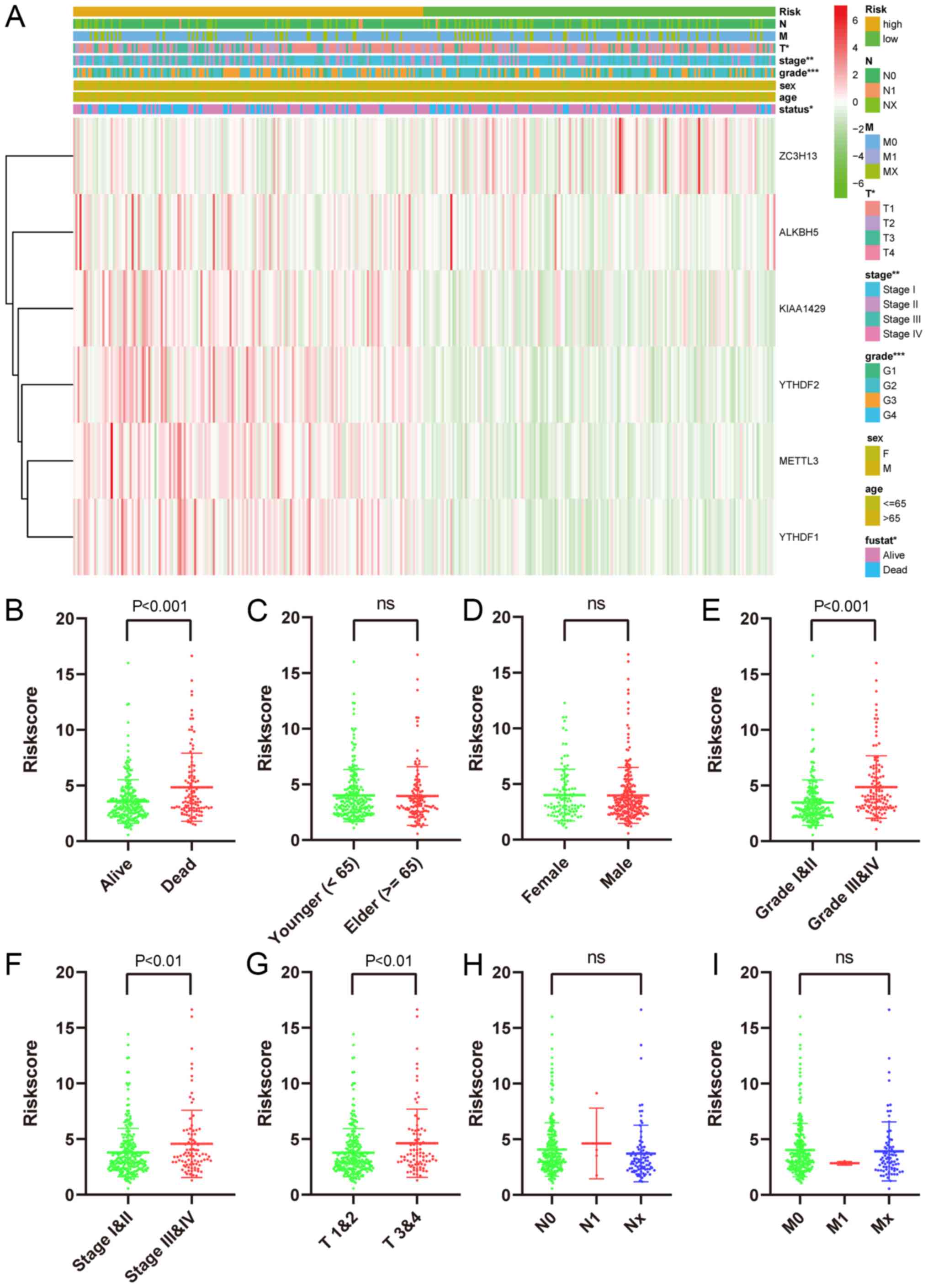
The risk signature showed a strong association
between clinicopathological features and OS. The expression levels
of 6 screened m6A RNA methylation regulators in patients
from the high- and low-risk groups within the TCGA dataset are
presented in the heatmap (Fig. 7A).
Clearly, differences in T stage (P<0.05), grade (P<0.001),
status (P<0.05) and stage (P<0.01) were statistically
significant between the two groups. Moreover, the association
between risk score and every clinicopathological characteristic was
examined, and it was found that differences in the risk scores
among patients were associated with T stage, stage, grade, and
status subgroups, but not age, gender, N stage and M stage
(Fig. 7B-I).
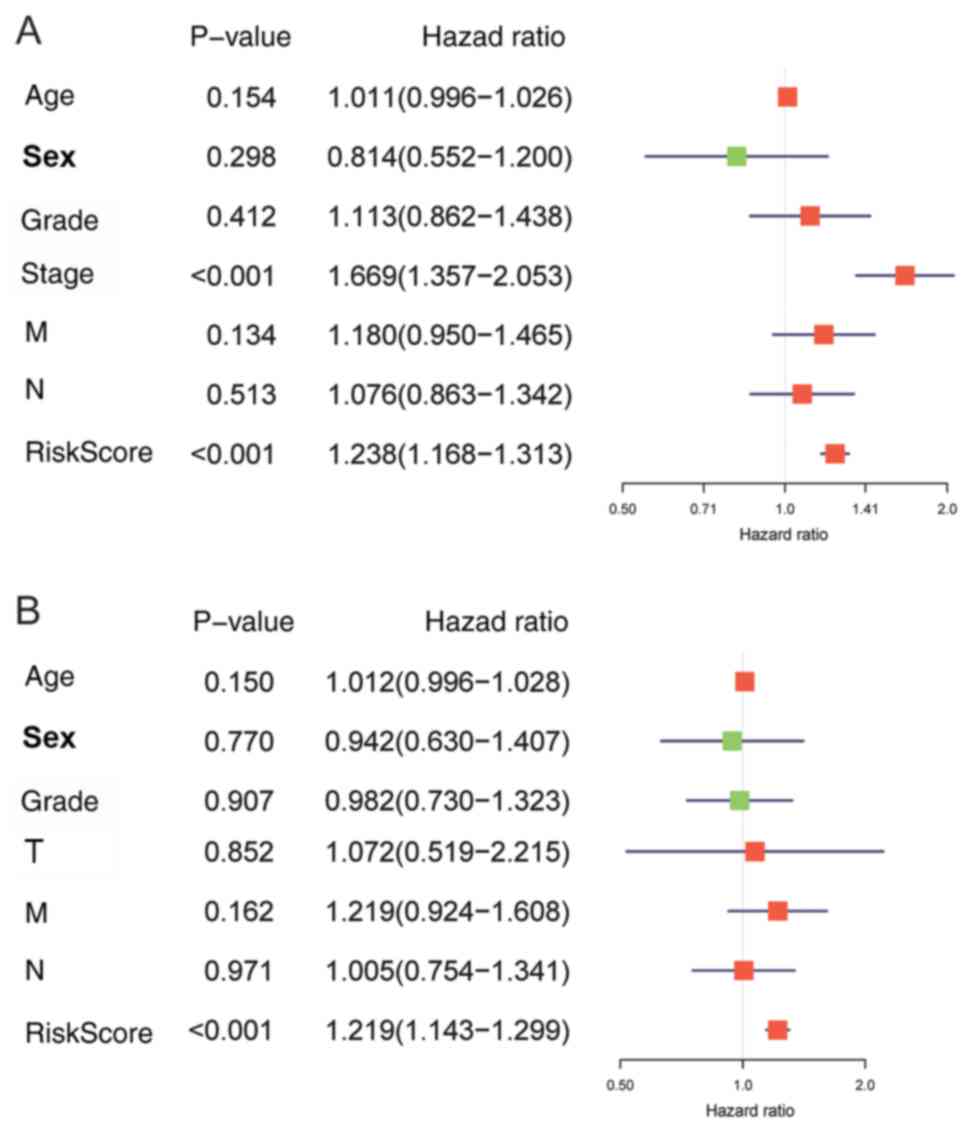
Meanwhile, the risk signature HR was 1.238 upon
univariate Cox proportional hazards regression [95% confidence
interval (CI): 1.168–1.313; P<0.001; Fig. 8A)]. In addition, the same results
could be obtained by multivariate Cox proportional hazards
regression analysis with adjusted clinical covariate (HR=1.219, 95%
CI: 1.143–1.299; P<0.001; Fig.
8B).
The above findings suggested that risk scores
determined based on the as-constructed signature were able to
precisely estimate the prognosis and clinicopathological
characteristics of HCC patients.
External validation of the prognostic
signature in the ICGC cohort
To confirm the external validity, the prognostic
signature was applied in the ICGC data. The expression levels of
the 6 regulators were compared between the high- and low-risk
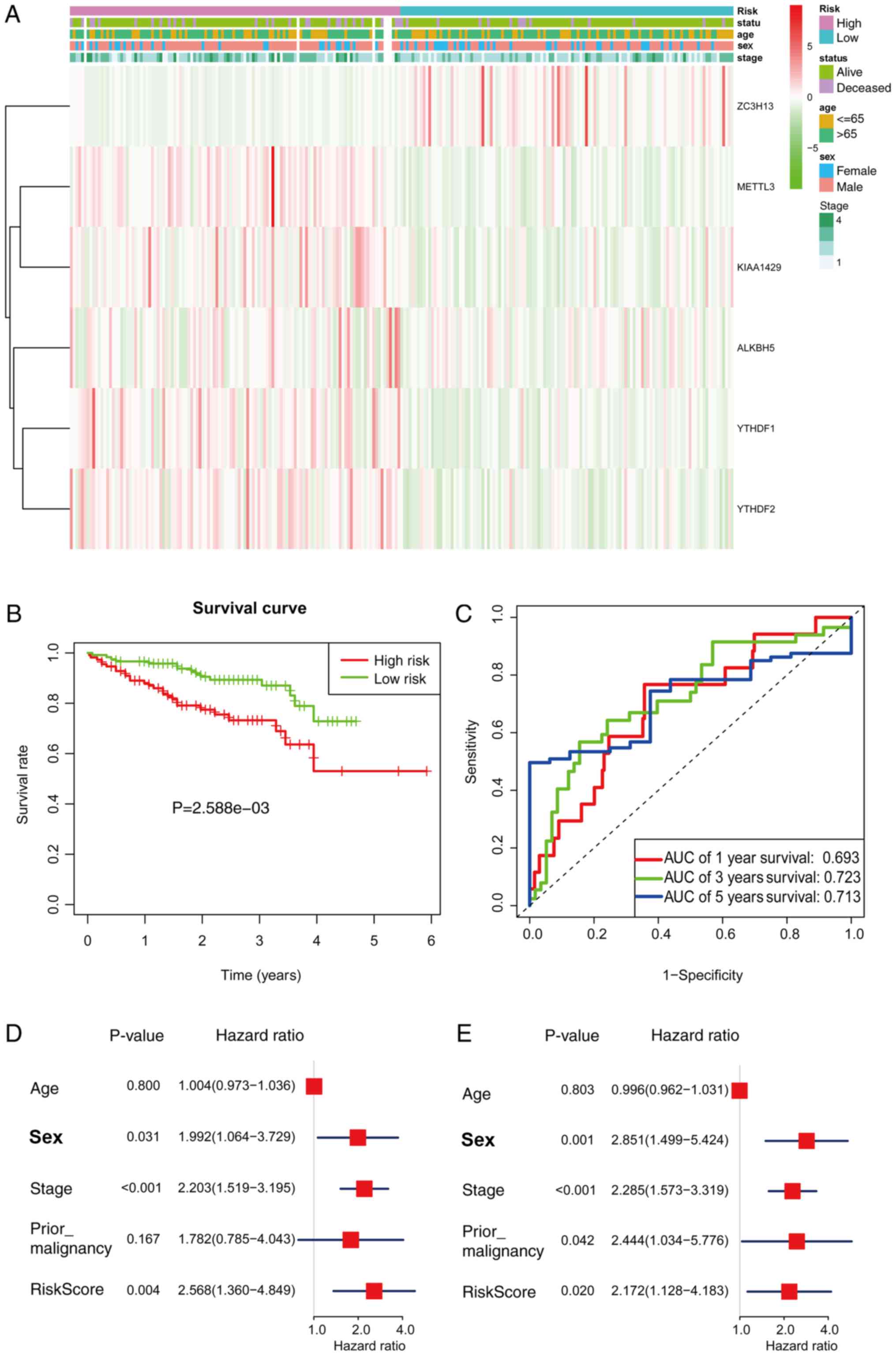
groups, and the heatmap is presented in Fig. 9A. The high-risk group had a
significantly shorter survival than the low-risk group in the ICGC
cohorts (P=2.588e-3; Fig. 9B). ROC
curve analysis showed that risk signature prognosis prediction
could attain an AUC value of 0.693 (1 year), 0.723 (3 years) and
0.713 of (5 years; Fig. 9C). Using
univariate (P=0.004) and multivariate (P=0.020) Cox regression
analysis, the signature was further confirmed as an independent
prognostic factor (Fig. 9D and
E).
Functional analysis
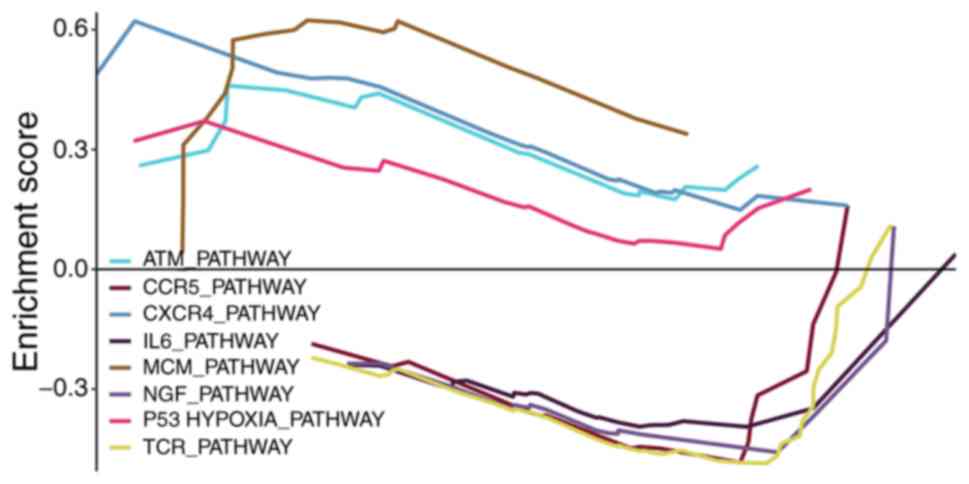
mRNAs associated with the m6A RNA
methylation regulators were applied into the GSEA for enrichment
analysis, in order to examine the potential biological functions.
As indicated in Fig. 10, the top
enrichments included ATM_PATHWAY, CCR5_PATHWAY, CXCR4_PATHWAY,
IL6_PATHWAY, MCM_PATHWAY, NGF_PATHWAY, P53HYPOXIA_PATHWAY and
TCR_PATHWAY.
Discussion
The present findings showed that the expression of
m6A RNA methylation regulators was closely associated
with malignant grade and prognosis for HCCs. In addition, two HCC
subgroups, namely cluster 1 and 2, were classified using consensus
clustering on the basis of m6A RNA methylation regulator
expression levels. Specifically, the cluster 1/2 subgroups affected
patient prognosis and exhibited a close correlation with
clinicopathological features. Furthermore, a risk signature for
prognosis was also constructed based on the 6 screened
m6A RNA methylation regulators, which could stratify
patient OS into high- or low-risk subgroups.
The present study displayed obvious advantages.
First, clustering analysis of m6A modification
regulators was carried out. Specifically, clusters were formed so
that patients in the same cluster were similar, while patients in
different clusters were distinct. Second, with regard to
methodology, the application of the LASSO-penalized regression
could boost the accuracy of the bioinformatics analysis. Different
from the conventional stepwise regression used in prior research,
the LASSO algorithm could analyze all independent factors
simultaneously, identifying the most significant variables
(17). Consequently, this
formulation approach displayed a higher accuracy than stepwise
regression using the multivariate Cox model, particularly in huge
datasets, such as genomics (18).
Thirdly, the results were validated in the ICGC dataset to check
the general applicability. Next, we comprehensively analyzed 13
regulators simultaneously, while previous published studies usually
focused on one regulator. Cheng et al (19) reported that KIAA1429 could regulate
HCC invasion and migration by changing the m6A
modification in ID2 mRNA. In addition, Chen et al (20) reported that METTL3 expression was
usually increased in human HCC, which contributed to the
progression of HCC, while the SOCS2 level in HCC was repressed by a
mechanism that depended on m6A-YTHDF2. Zhao et al
(21) discovered that YTHDF1 played
a vital role in the regulation of HCC metabolism, as well as cell
cycle development. Ma et al (22) reported that METTL14 could suppress
the metastatic capacity of HCC cells by regulating the primary
miRNA processing of m6A-dependent tumor suppressors. In
addition, it was found that YTHDF2 could modulate the
m6A level in HCC (23).
However, the aforementioned studies only focused on one
m6A RNA methylation regulator. Recently, Zhou et
al (24) reported the
m6A-related genes in HCC, and confirmed the independent
predictive value of both METTL3 and YTHDF1 in OS through
multivariate Cox regression analysis; therefore, patients were
further divided into three groups, based on METTL3 and YTHDF1
expression. Notably, no differential expression of ZC3H13 was
observed between tumor and non-tumor samples (exact data not
shown). However, ZC3H13 was a protective gene in univariate Cox
regression analysis, and further investigations are needed.
The present study revealed that the m6A
RNA methylation regulators are correlated with biological processes
during the malignant development of HCC. The RNA m6A
methylation function within the tumor was recently confirmed, and
certain biological processes were found to be affected by it,
including tumor stem cell growth, tumorigenesis and self-renewal
(25,26), as well as DNA damage response
secondary to radiotherapy or chemotherapy (27,28).
Herein, the expression levels of m6A RNA methylation
regulators in HCC were found to be correlated with HCC-related
biological processes, such as ATM_PATHWAY (29), CXCR4_PATHWAY (30) and IL6_PATHWAY (31).
The present results showed that the expression
levels of m6A RNA methylation regulators could serve as
prognostic markers. The overexpression of YTHDF1 was associated
with poor prognosis, which was consistent with the results of Zhao
et al (21). In this study
YTHDF2 overexpression was correlated with poor prognosis however
YTHDF2 suppressed cell proliferation and growth in the study by
Zhong et al (32). More
importantly, the as-constructed risk signature for the prognosis of
HCC based on the 6 selected m6A RNA methylation regulators was
proven valuable, and its significance in predicting the T stage,
stage, grade and survival status was determined. However, no
significant difference in risk score was identified between the N
and M stages, which might be partially due to the small number of
patients at these stages (Table I).
Moreover, risk significance was finally verified by multivariate
Cox analysis.
The study, however, had the following limitations:
First, more data are necessary to confirm these findings. Second,
these 13 regulators, as well as others, require further
investigation. Third, consensus clustering analysis was conducted
based on the m6A RNA methylation regulator expression
levels rather than writers, readers or erasers.
In conclusion, the present study comprehensively
illustrated the expression patterns, possible role and prognostic
significance of m6A RNA methylation regulators in HCC.
Typically, the expression levels of m6A RNA methylation
regulators exhibited a strong association with malignant clinical
and pathological characteristics in HCCs, as well as with
upregulated gene expression involved in biological processes to
accelerate the malignant development of HCC. The present study
provided critical support for future research into RNA
m6A methylation function in HCCs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81801804).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WL, QFC, PW and ZLH conceived and designed the
study. WL, QFC, TH, PW and LS analyzed the data. WL, TH, PW and LS
wrote the paper. WL, QFC, PW and ZLH reviewed and edited the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
m6A
|
N6-methyladenosine
|
|
HCC
|
hepatocellular carcinoma
|
|
TCGA
|
The Cancer Genome Atlas
|
|
ICGC
|
International Cancer Genome
Consortium
|
|
NS
|
not significant
|
|
PCA
|
principal component analysis
|
|
OS
|
overall survival
|
|
HR
|
hazard ratio
|
|
LASSO
|
least absolute shrinkage and selection
operator
|
|
ROC
|
receiver operating characteristic
|
|
AUC
|
area under the ROC curve
|
|
CI
|
confidence interval
|
|
KM
|
Kaplan-Meier
|
|
GSEA
|
gene set enrichment analysis
|
References
|
1
|
He C: Grand challenge commentary: RNA
epigenetics? Nat Chem Biol. 6:863–865. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boccaletto P, Machnicka MA, Purta E,
Piatkowski P, Baginski B, Wirecki TK, de Crécy-Lagard V, Ross R,
Limbach PA, Kotter A, et al: MODOMICS: A database of RNA
modification pathways. 2017 update. Nucleic Acids Res.
46:D303–D307. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ji P, Wang X, Xie N and Li Y:
N6-methyladenosine in RNA and DNA: An epitranscriptomic and
epigenetic player implicated in determination of stem cell fate.
Stem Cells Int. 2018:32565242018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chai RC, Wu F, Wang QX, Zhang S, Zhang KN,
Liu YQ, Zhao Z, Jiang T, Wang YZ and Kang CS: m(6)A RNA methylation
regulators contribute to malignant progression and have clinical
prognostic impact in gliomas. Aging (Albany NY). 11:1204–1225.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meyer KD and Jaffrey SR: Rethinking m(6)A
readers, writers, and erasers. Annu Rev Cell Dev Biol. 33:319–342.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roundtree IA and He C: RNA
epigenetics-chemical messages for posttranscriptional gene
regulation. Curr Opin Chem Biol. 30:46–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dominissini D, Moshitch-Moshkovitz S,
Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K,
Jacob-Hirsch J, Amariglio N, Kupiec M, et al: Topology of the human
and mouse m6A RNA methylomes revealed by m6A-seq. Nature.
485:201–206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li X, Xiong X and Yi C: Epitranscriptome
sequencing technologies: Decoding RNA modifications. Nat Methods.
14:23–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W,
Li H, Qiu M, Xu Y, Xia W, et al: Roles of RNA methylation by means
of N(6)-methyladenosine (m(6)A) in human cancers. Cancer Lett.
408:112–120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang J and Yin P: Structural insights
into N(6)-methyladenosine (m(6)A) modification in the
transcriptome. Genomics Proteomics Bioinformatics. 16:85–98. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luo J, Liu H, Luan S, He C and Li Z:
Aberrant regulation of mRNA m(6)A modification in cancer
development. Int J Mol Sci. 19:E25152018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deng X, Su R, Feng X, Wei M and Chen J:
Role of N(6)-methyladenosine modification in cancer. Curr Opin
Genet Dev. 48:1–7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen QF, Jia ZY, Yang ZQ, Fan WL and Shi
HB: Transarterial chemoembolization monotherapy versus combined
transarterial chemoembolization-microwave ablation therapy for
hepatocellular carcinoma tumors </=5 cm: A propensity analysis
at a single center. Cardiovasc Intervent Radiol. 40:1748–1755.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen QF, Huang T, Shen L and Li W:
Predictive value of a nomogram for hepatocellular carcinoma with
brain metastasis at initial diagnosis: A population-based study.
PLoS One. 14:e02092932019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen QF, Li W, Wu P, Shen L and Huang ZL:
Alternative splicing events are prognostic in hepatocellular
carcinoma. Aging (Albany NY). 11:4720–4735. 2019.PubMed/NCBI
|
|
16
|
Chen QF, Li W, Wu PH, Shen LJ and Huang
ZL: Significance of tumor-infiltrating immunocytes for predicting
prognosis of hepatitis B virus-related hepatocellular carcinoma.
World J Gastroenterol. 25:5266–5282. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Friedman J, Hastie T and Tibshirani R:
Regularization paths for generalized linear models via coordinate
descent. J Stat Softw. 33:1–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McNeish DM: Using lasso for predictor
selection and to assuage overfitting: A method long overlooked in
behavioral sciences. Multivariate Behav Res. 50:471–484. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng X, Li M, Rao X, Zhang W, Li X, Wang
L and Huang G: KIAA1429 regulates the migration and invasion of
hepatocellular carcinoma by altering m6A modification of ID2 mRNA.
OncoTargets Ther. 12:3421–3428. 2019. View Article : Google Scholar
|
|
20
|
Chen M, Wei L, Law CT, Tsang FH, Shen J,
Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, et al: RNA
N6-methyladenosine methyltransferase-like 3 promotes liver cancer
progression through YTHDF2-dependent posttranscriptional silencing
of SOCS2. Hepatology. 67:2254–2270. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao X, Chen Y, Mao Q, Jiang X, Jiang W,
Chen J, Xu W, Zhong L and Sun X: Overexpression of YTHDF1 is
associated with poor prognosis in patients with hepatocellular
carcinoma. Cancer Biomark. 21:859–868. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH,
Wang F, Wang TT, Xu QG, Zhou WP and Sun SH: METTL14 suppresses the
metastatic potential of hepatocellular carcinoma by modulating
N6 -methyladenosine-dependent primary MicroRNA
processing. Hepatology. 65:529–543. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang
S, Liu Y, Ye L, Li Y and Zhang X: MicroRNA-145 modulates
N6-methyladenosine levels by targeting the
3′-untranslated mRNA Region of the N6-methyladenosine
binding YTH domain family 2 protein. J Biol Chem. 292:3614–3623.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou Y, Yin Z, Hou B, Yu M, Chen R, Jin H
and Jian Z: Expression profiles and prognostic significance of RNA
N6-methyladenosine-related genes in patients with hepatocellular
carcinoma: Evidence from independent datasets. Cancer Manag Res.
11:3921–3931. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pan Y, Ma P, Liu Y, Li W and Shu Y:
Multiple functions of m6A RNA methylation in cancer. J
Hematol Oncol. 11:482018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun
G, Lu Z, Huang Y, Yang CG, et al: m6A RNA methylation
regulates the self-renewal and tumorigenesis of glioblastoma stem
cells. Cell Rep. 18:2622–2634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dai D, Wang H, Zhu L, Jin H and Wang X:
N6-methyladenosine links RNA metabolism to cancer progression. Cell
Death Dis. 9:1242018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiang Y, Laurent B, Hsu CH, Nachtergaele
S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al: RNA
m6A methylation regulates the ultraviolet-induced DNA
damage response. Nature. 543:573–576. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao SB, Li KL, Qiu H, Zhu LY, Pan CB, Zhao
Y, Wei SH, Shi S, Jin GH and Xue LX: Enhancing chemotherapy
sensitivity by targeting PcG via the ATM/p53 pathway. Am J Cancer
Res. 7:1874–1883. 2017.PubMed/NCBI
|
|
30
|
Meng YM, Liang J, Wu C, Xu J, Zeng DN, Yu
XJ, Ning H, Xu L and Zheng L: Monocytes/Macrophages promote
vascular CXCR4 expression via the ERK pathway in hepatocellular
carcinoma. Oncoimmunology. 7:e14087452017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cheng Y, Li H, Deng Y, Tai Y, Zeng K,
Zhang Y, Liu W, Zhang Q and Yang Y: Cancer-associated fibroblasts
induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster
immune suppression in hepatocellular carcinoma. Cell Death Dis.
9:4222018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhong L, Liao D, Zhang M, Zeng C, Li X,
Zhang R, Ma H and Kang T: YTHDF2 suppresses cell proliferation and
growth via destabilizing the EGFR mRNA in hepatocellular carcinoma.
Cancer Lett. 442:252–261. 2019. View Article : Google Scholar : PubMed/NCBI
|