Introduction
Renal cell carcinoma (RCC) is the most common
subtype of kidney cancer that accounts for 3% of all malignancies
in adults in the United States (1).
RCC incidence rates among men in 2012 varied from ~1 in Africa to
>15 in Europe (cases per 100,000 standard population) (2). RCC is resistant to radiotherapy and
chemotherapy, and surgical resection remains the primary
therapeutic technique for early-localized RCC. Between 25–30% of
patients have metastatic disease at the time of RCC diagnosis, and
patients with metastatic RCC have a very poor prognosis (3). Although a great deal of progress has
been made in targeted molecular therapy for the treatment of
metastatic RCC, their medicinal performance remains less than
satisfactory.
Resveratrol (Res), a polyphenolic compound, is
widely distributed in a variety of plants (4). Since the first study reported its
inhibitory effect on carcinogenesis in a mouse skin cancer model in
1997 (5), a large number of studies
have demonstrated that Res can inhibit multiple types of cancer
in vitro. Furthermore, Res also possesses antitumor effects
in vivo (6). However, the
antitumor effect of Res on RCC remains largely unknown due to its
complex pharmacological activities. The present study aimed to
investigate the underlying molecular mechanism of Res in RCC.
The 786-O cell line possesses numerous
characteristics of RCC, including mutations in the VHL gene
(7) and high activation of vascular
endothelial growth factor (VEGF) (8), and is widely used in RCC research. The
present study revealed that in 786-O cells, Res damaged
mitochondria, activated caspase 3 and induced apoptosis through
reactive oxygen species (ROS). Furthermore, Res activated c-Jun
N-terminal kinase (JNK) via ROS to induce autophagy, while
inhibition of autophagy further exacerbated Res-induced
apoptosis.
Materials and methods
Reagents and antibodies
Res was purchased from Selleck Chemicals. A Cell
Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular
Technologies, Inc. Z-VAD-FMK was purchased from Santa Cruz
Biotechnology, Inc. Chloroquine (CQ) was supplied by Enzo Life
Sciences, Inc. N-acetyl cysteine (NAC) and
2′,7′-dichlorofluorescin-diacetate (DCFH-DA) were purchased from
Beyotime Institute of Biotechnology. SB203580 and SP600125 were
obtained from MedChemExpress. Antibodies against PARP (1:1,000;
catalog no. 9532), GAPDH (1:2,000; catalog no. 5714), AMPKα
(1:1,000; catalog no. 5831), p-AMPKα (1:1,000; catalog no. 2535),
S6 (1:1,000; catalog no. 2317), p-S6 (1:1,000; catalog no. 4858),
p38 (1:1,000; catalog no. 8690), p-p38 (1:1,000; catalog no. 4511),
JNK (1:1,000; catalog no. 9252), p-JNK (1:1,000; catalog no. 4668),
ERK (1:1,000; catalog no. 4695), p-ERK (1:1,000; catalog no. 4370),
BCL2 (1:1,000; catalog no. 4223) and p-BCL2 (1:1,000; catalog no.
2827) were all purchased from Cell Signaling Technology, Inc. LC3B
antibody (1:1,000; catalog no. ab192890) was purchased from Abcam,
and Beclin 1 antibody (1:500; catalog no. sc-48341) was purchased
from Santa Cruz Biotechnology, Inc.
Cell culture
The 786-O cell line was purchased from the Shanghai
Institute of Cell Biology, Chinese Academy of Sciences. Cells were
maintained in RPMI-1640 medium (HyClone; GE Healthcare Life
Sciences) supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin and streptomycin, at 37°C in a
humidified atmosphere containing 5% CO2 until they
reached 80–90% confluence.
Cell viability assay
Cell viability assay was performed using CCK-8
reagent (Dojindo Molecular Technologies), according to the
manufacturer's protocol. The 786-O cells were seeded at a density
of 4×103 cells/well into 96-well plates. Following
overnight incubation at 37°C, the cells were treated with the
indicated concentrations of Res (10, 20, 40 and 80 µM) for 24 or 48
h. Following Res treatment, CCK-8 reagent was added into every
well, followed by incubation at 37°C for 1 h in the dark.
Subsequently, the optical density was determined using a microplate
reader (Bio-Rad Laboratories, Inc.), at a wavelength of 450 nm.
Cell apoptosis assay
Cell apoptosis was assessed using an
AnnexinV-FITC-propidium iodide (PI) double staining kit
(MultiSciences Biotech, Co., Ltd.), according to the manufacturer's
protocol. Briefly, cells were treated with 10, 20 µM Res for 48,
and 40 µM of Res for 24 or 48 h. For some experiments, cells were
treated with 40 µM Res for 48 h in the presence or absence of 50 µM
Z-VAD-FMK, 10 mM NAC or 50 µM CQ. Following treatment, cells were
harvested and washed twice with PBS. Subsequently, cells were
incubated in buffer containing Annexin V-FITC and PI at room
temperature for 5 min in the dark. Apoptotic cells were identified
using a BD FACSCanto II flow cytometer (BD Biosciences) and data
were analyzed using FACSDiVa software (version 7.0; BD
Biosciences).
ROS assay
Cells were harvested, washed twice with PBS, and
then incubated in serum-free RPMI-1640 medium containing DCFH-DA at
37°C for 20 min. Cells were re-washed twice with PBS and
intracellular ROS was detected via the aforementioned flow
cytometry method.
Caspase 3 activity assay
Caspase 3 activity was determined using a caspase 3
activity assay kit (ApexBio Technology), according to the
manufacturer's protocol. Briefly, cells were washed twice with PBS
and incubated in staining buffer containing FITC-DEVD-FMK probe at
37°C for 30 min. Cells were re-washed twice with PBS and harvested,
and caspase 3 activity was detected via the aforementioned flow
cytometry method.
Mitochondrial membrane potential (ΔΨm)
assay
The ΔΨm assay was performed using a JC1
mitochondrial membrane potential assay kit (Beijing Solarbio
Science & Technology Co., Ltd.), according to the
manufacturer's protocol. In brief, cells were washed twice with PBS
and incubated in fresh RPMI-1640 medium containing JC1 regent at
37°C for 30 min. Cells were re-washed twice with PBS and harvested,
and ΔΨm was determined via the aforementioned flow cytometry
method. ΔΨm was calculated as the ratio of red to green
fluorescence.
Cyto-ID autophagy detection assay
A CYTO-ID autophagy detection kit (Enzo Life
Sciences, Inc.) was utilized in the present study. Briefly, cells
were washed twice with PBS and then incubated in PBS containing
CYTO-ID probe and 5% FBS at 37°C for 20 min in the dark. Following
the incubation, cells were re-washed twice with PBS and observed
under a fluorescence microscope (Olympus Corporation;
magnification, ×200). In order to evaluate autophagy with flow
cytometry, cells were harvested following incubation with CYTO-ID
probe at 37°C for 20 min in the dark. Subsequently, autophagy was
determined via the aforementioned flow cytometry method.
Western blotting
Cells were treated with 40 µM Res for 24 or 48 h.
For some experiments, 10 mM NAC, 20 µM SP600125 or 20 µM SB203580
were used. In order to evaluate autophagic flux, cells were treated
with 40 µM Res for 48 h in the presence or absence of 50 µM CQ.
Cells were lysed using a total protein extraction
kit (Nanjing KeyGen Biotech Co., Ltd.), according to the
manufacturer's protocol. Protein concentrations were determined
using a bicinchoninic acid assay kit (Nanjing KeyGen Biotech Co.,
Ltd.) and 30 µg protein/lane was separated via SDS-PAGE on a 10–12%
gel. The separated proteins were subsequently transferred onto a
polyvinylidene difluoride membrane and blocked with 5% BSA 1 h at
room temperature. The membranes were incubated with the
aforementioned primary antibodies, overnight at 4°C. The membranes
were then washed three times with TBST and incubated with
corresponding anti-rabbit or anti-mouse HRP-conjugated secondary
antibodies (1:5,000; catalog no. GAR007 and GAM007; MultiSciences
Biotech, Co., Ltd.) at room temperature for 1 h. Protein bands were
visualized using an ECL system (EMD Millipore) and densitometric
analysis was performed using ImageJ software (version 1.48v;
National Institutes of Health).
Small interfering (si)RNA and
transfection
Beclin 1 (catalog no. sc-29797) and scrambled
control siRNA (catalog no. sc-37007) were purchased from Santa Cruz
Biotechnology, Inc. The Beclin 1 siRNA sequences were as follows:
Forward, 5′-CAGCUCAACGUCACUGAAATT-3′ and reverse,
5′-UUUCAGUGACGUUGAGCUGTT-3′. The control siRNA sequences were not
available. Briefly, 100 nM of Beclin 1 or control siRNA was
transfected into cells using Lipofectamine® 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol, and non-transfected cells were set as a
blank control. After 24 h, cells were treated with 40 µM Res for an
additional 48 h and subsequently analyzed via western blotting as
previously described.
Statistical analysis
All experiments were performed in triplicate, and
the data are presented as the mean ± standard deviation. Unpaired
Student's t-test was used for comparisons between two groups.
One-way analysis of variance followed by post hoc comparisons using
Tukey's test was used for comparisons between three groups or more.
P<0.05 was considered to indicate a statistically significant
difference.
Results
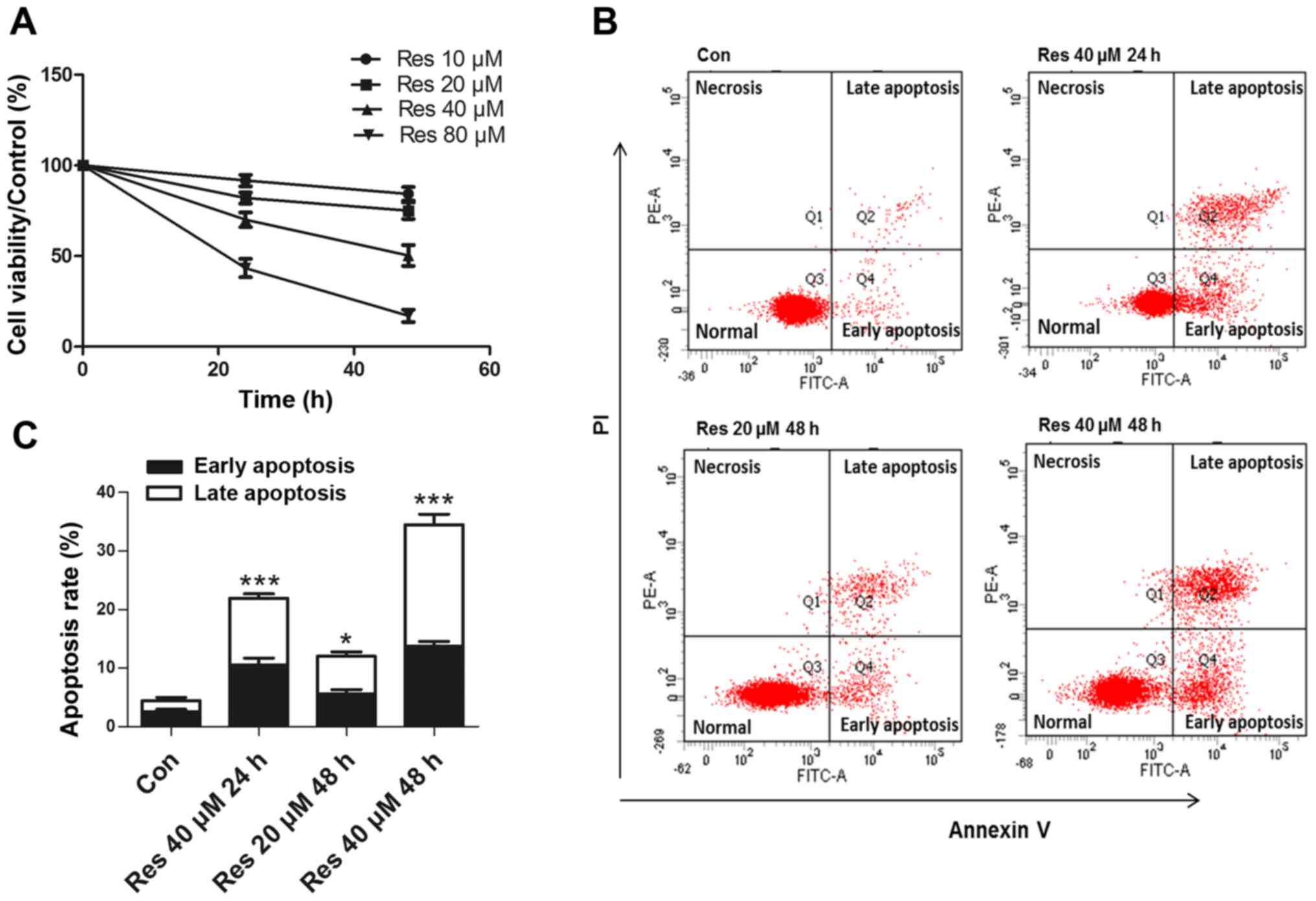
Res inhibits cell viability and
induces apoptosis of 786-O cells
The 786-O cells were exposed to various
concentrations of Res (10, 20, 40 and 80 µM) for 24 and 48 h. The
CCK-8 assay revealed that Res inhibited the cell viability in a
time- and dose-dependent manner (Fig.
1A). Since a previous study (9)
reported that high concentrations (≥50 µM) of Res exhibited
significant toxicity to normal renal epithelial cells, the present
study performed further experiments at lower concentrations. The
flow cytometric analysis revealed that Res at 20 and 40 µM induced
apoptosis (Fig. 1B and C), while 10
µM Res had no effect on apoptosis (data not shown).
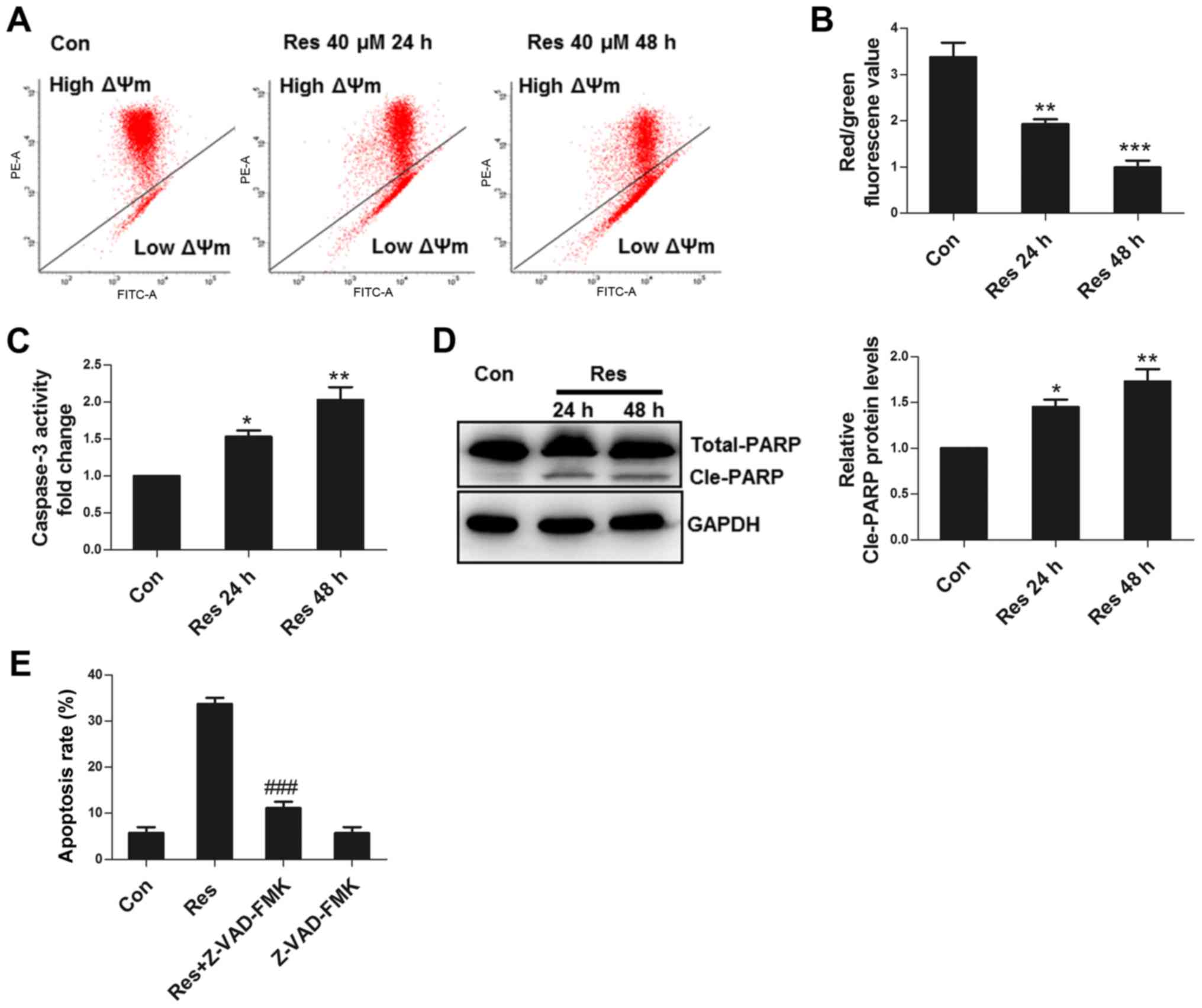
Res damages mitochondria and activates
caspase to induce apoptosis
Mitochondria are the key elements for apoptotic
execution, and damaged mitochondria can release pro-apoptotic
factors that activate caspase to induce apoptosis. ΔΨm is an
important parameter in the evaluation of mitochondrial function
(10). The present study observed
that Res decreased ΔΨm (Fig. 2A and
B), activated caspase 3 (Fig.
2C), and induced cleavage of PARP, a substrate of caspase 3
(Fig. 2D). Furthermore, Z-VAD-FMK, a
pan-caspase inhibitor, significantly inhibited apoptosis (Fig. 2E), demonstrating that Res induced
apoptosis in 786-O cells primarily through the activation of
caspase.
ROS is responsible for Res-induced
apoptosis
As an important signaling factor, normal ROS levels
can maintain cell homeostasis, while excessive ROS can cause cell
injury (11). Res significantly
increased the ROS level in 786-O cells (Fig. 3A and B). NAC, an antioxidant,
attenuated ROS production (Fig. 3C),
improved ΔΨm (Fig. 3D), decreased
caspase 3 activity (Fig. 3E),
impaired apoptosis (Fig. 3F) and
decreased the cleavage of PARP (Fig.
3G), suggesting that Res could damage mitochondria, activate
caspase 3 and cause apoptosis through ROS in 786-O cells.
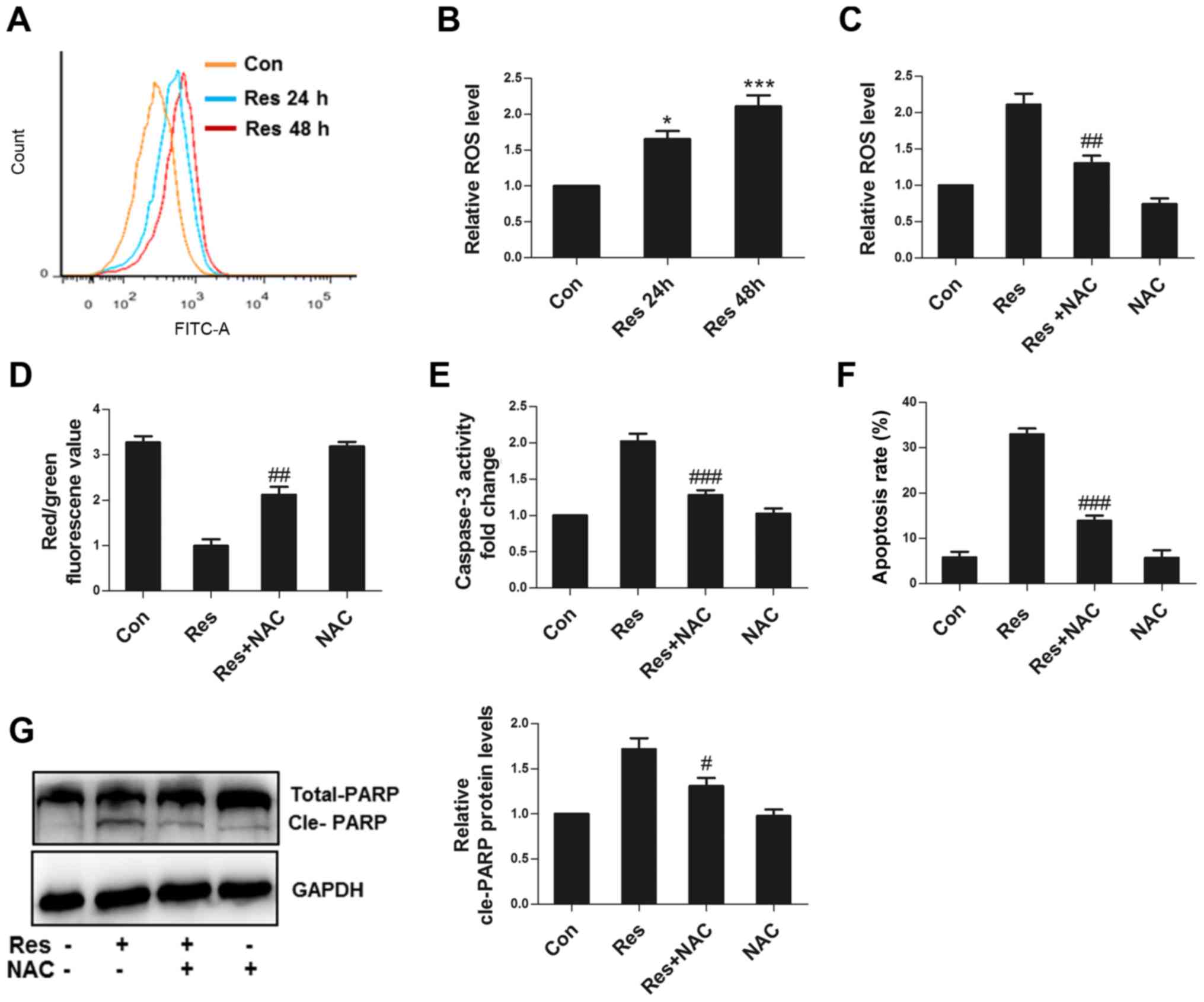 | Figure 3.ROS are responsible for apoptosis
induced by Res. (A) Following treatment with 40 µM Res for 24 or 48
h, representative images of flow cytometric assay for ROS. (B)
Following treatment with 40 µM Res for 24 or 48 h, quantitative
analysis of ROS detected by flow cytometry was performed. Following
treatment with 40 µM Res in the presence or absence of 10 mM NAC
for 48 h, quantitative analysis of (C) ROS, (D) JC1 red/green
fluorescence value, (E) caspase 3 activity, (F) apoptosis and (G)
western blot analysis of PARP expression were detected by flow
cytometry. *P<0.05, ***P<0.001 vs. control group;
#P<0.05, ##P<0.01,
###P<0.001 vs. Res group. ROS, reactive oxygen
species; Res, resveratrol; NAC, N-acetyl cysteine. |
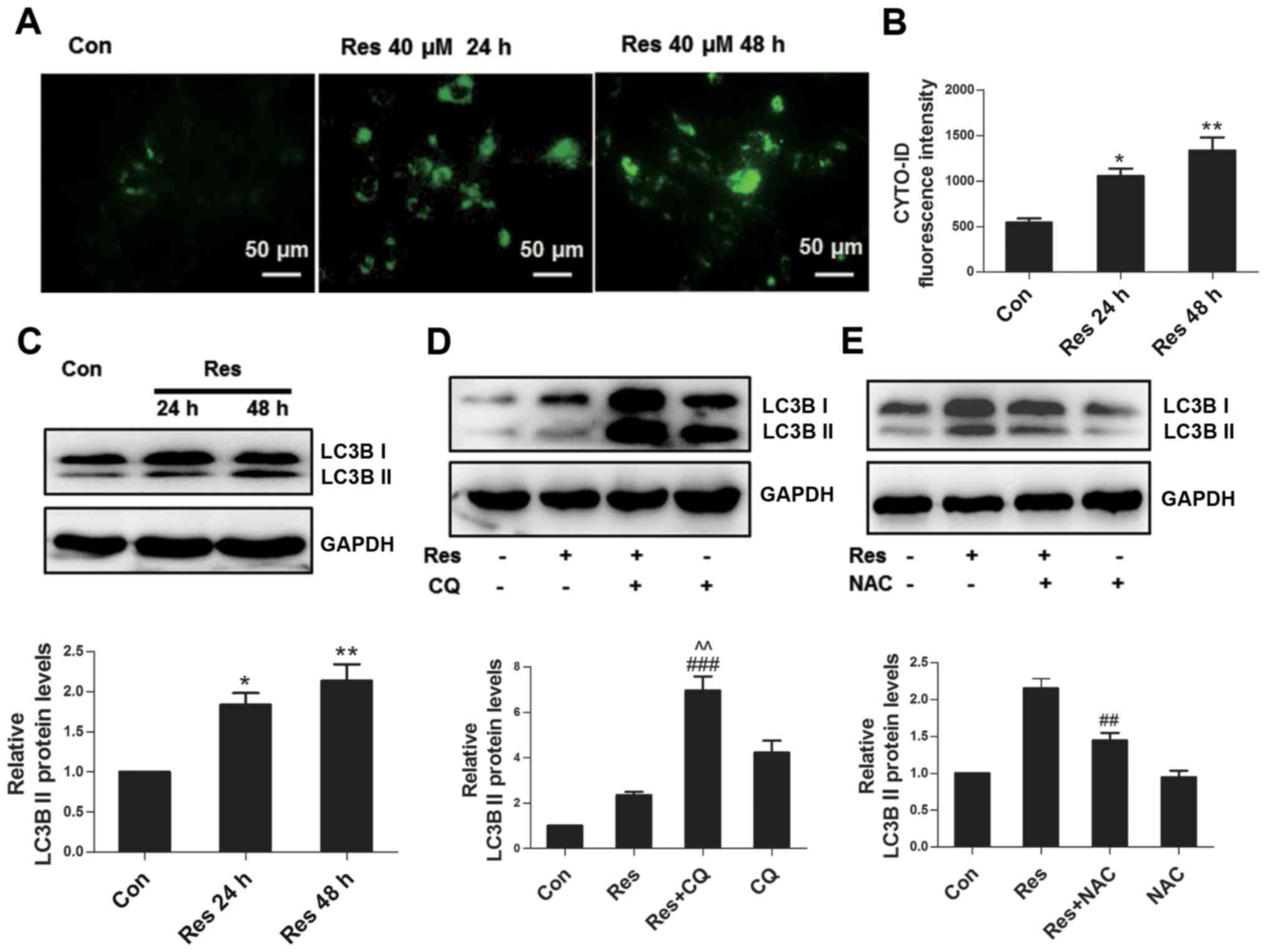
Res induces autophagy partially
through ROS
As a conservative mechanism, autophagy can be
activated in multiple environments, including nutritional
deficiency, hormone imbalance and oxidative stress, among others
(12). The Cyto-ID fluorescent probe
specifically labels autophagosomes in live cells (13), which was used to assess whether Res
initiated autophagy in the present study. The results revealed that
Res stimulated the formation of autophagosomes in 786-O cells
(Fig. 4A), and the flow cytometric
assay further confirmed these findings (Fig. 4B). Furthermore, Res increased the
expression level of autophagy marker protein LC3B II (Fig. 4C). Since the upregulation of LC3B II
can reflect activated autophagy or impaired autophagic flux, the
present study inhibited the degradation of autophagy using
autophagy inhibitor CQ, which further upregulated the expression of
LC3B II at the protein level (Fig.
4D), indicating that Res induced complete autophagic flux. ROS
has been well known as a signaling molecule regulating autophagy
(14); therefore, the present study
subsequently assessed whether ROS participated in Res-induced
autophagy in 786-O cells. As expected, inhibition of ROS by NAC
attenuated the expression of LC3B II (Fig. 4E), suggesting that Res induced
autophagy partially through ROS.
JNK activated by ROS is required for
Res-induced autophagy
ROS can induce autophagy through multiple signaling
pathways, including AMPK-mammalian target of rapamycin (mTOR)
(15), p53 (16) and MAPK (17). Since the status of p53 remains
controversial in 786-O cells (18),
the present study did not investigate its role here. Res had no
effect on the expression levels of p-AMPK and p-S6 protein, a
downstream protein in the mTOR signaling pathway (Fig. 5A), indicating that the AMPK-mTOR
signaling pathway was not involved in the autophagic process in the
experiments performed in the present study. The present study
further investigated the MAPK family-associated proteins (ERK, JNK
and p38) and revealed that Res inhibited ERK, and activated JNK and
p38 (Fig. 5B). Notably, NAC reversed
the effects of Res on ERK, JNK and p38 (Fig. 5C). SB203580 (a p38 inhibitor)
increased the expression of LC3B II, while in contrast, SP600125 (a
JNK inhibitor) attenuated the expression of LC3B II (Fig. 5D), suggesting that Res activated JNK
via ROS to induce autophagy. JNK phosphorylates the BCL2 protein,
resulting in the dissociation of Beclin 1 from BCL2 to activate
autophagy (19). Indeed, it was
revealed in the present study that SP600125 decreased the
Res-induced upregulation of phosphorylated BCL2 in 786-O cells
(Fig. 5E).
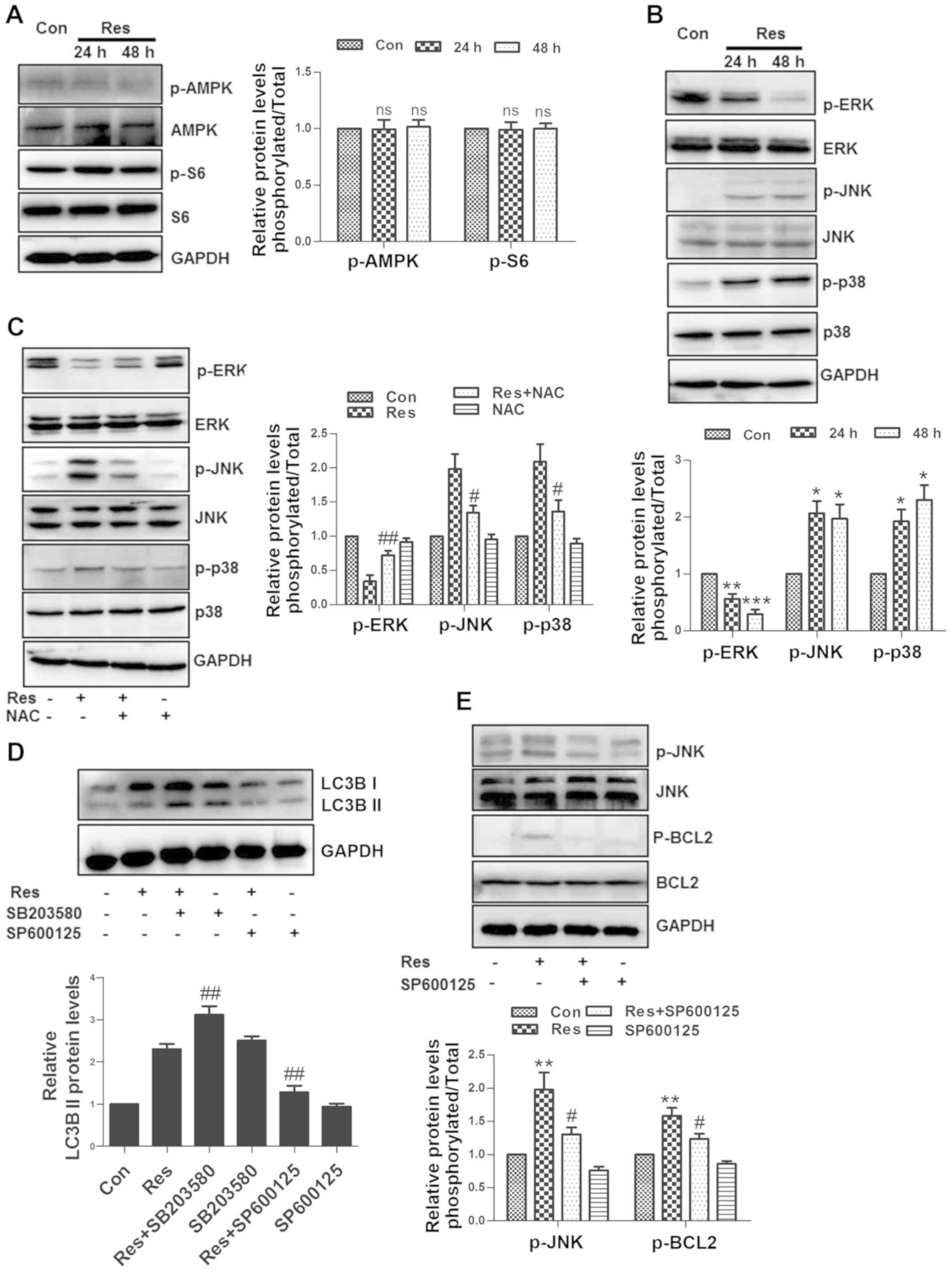 | Figure 5.JNK activated by ROS is required for
Res-induced autophagy. Following treatment with 40 µM Res for 24 h
or 48 h, (A) western blot analysis of p-AMPK and p-S6, and (B)
p-ERK, p-p38, p-JNK, ERK, p38 and JNK was performed. (C) Following
treatment with 40 µM Res in the presence or absence of 10 mM NAC
for 48 h, western blot analysis of p-ERK, p-p38 and p-JNK was
performed. (D) Following treatment with 40 µM Res in the presence
of 20 µM SB203580 or 20 µM SP600125 for 48 h, western blot analysis
of LC3B was performed. (E) Following treatment with 40 µM Res in
the presence or absence of 20 µM SP600125 for 48 h, western blot
analysis of p-JNK and p-BCL2. NS, *P<0.05, **P<0.01,
***P<0.001 vs. control group; #P<0.05,
##P<0.01 vs. Res group. JNK, c-Jun N-terminal kinase;
ROS, reactive oxygen species; Res, resveratrol; NAC, N-acetyl
cysteine; NS, not significant. |
Inhibition of autophagy enhances
Res-induced apoptosis
Since autophagy can promote cell survival or death
in different environments (20), the
present study used CQ to inhibit autophagy and it was observed that
CQ exacerbated Res-induced apoptosis in 786-O cells (Fig. 6A). In addition, CQ further activated
caspase 3 (Fig. 6B) and promoted the
cleavage of PARP (Fig. 6C). However,
CQ alone did not affect the amount of apoptosis, caspase 3 activity
and cleavage of PARP. Beclin 1, a conserved protein, initiates
autophagosome formation, thus playing a central role during the
autophagic process (21).
siRNA-mediated Beclin 1 knockdown increased the level of cleaved
PARP (Fig. 6D), further confirming
that autophagy could suppress Res-induced apoptosis in 786-O
cells.
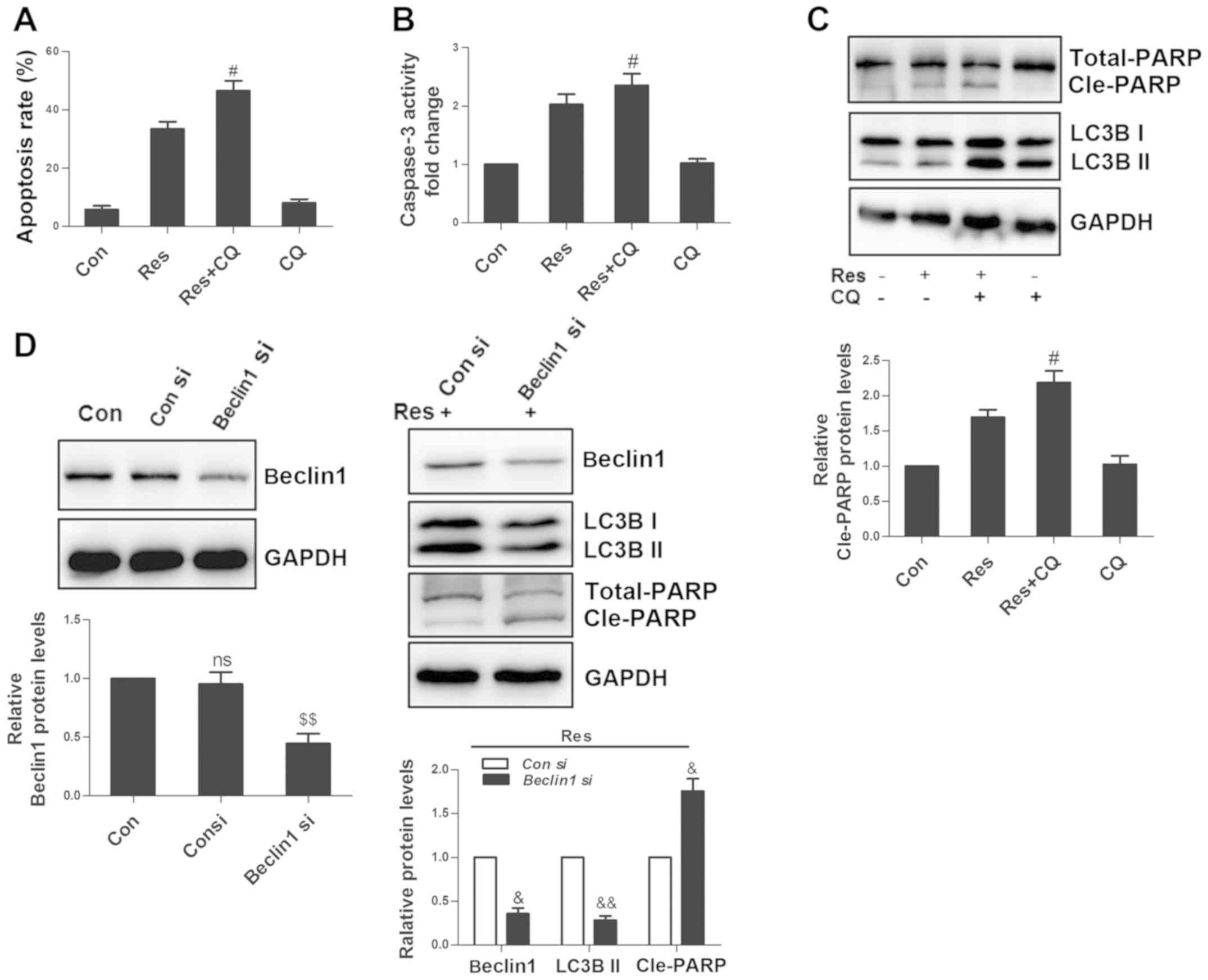 | Figure 6.Inhibition of autophagy enhances
Res-induced apoptosis. Following treatment with 40 µM Res in the
presence or absence of 50 µM CQ for 48 h, quantitative analysis of
(A) apoptosis and (B) caspase 3 activity were detected by flow
cytometry, and (C) via western blot analysis of PARP and LC3B. (D)
Cells were transfected with 100 nM Beclin 1 siRNA or control siRNA,
and non-transfected cells were set as a blank control. After 24 h,
cells were treated with 40 µM Res for an additional 48 h, and
western blotting was used for the analysis of Beclin 1, LC3B and
PARP expressions. NS vs. control group; #P<0.05 vs.
Res group; $$P<0.01 vs. con siRNA group;
&P<0.05, &&P<0.01 vs. con
siRNA+Res group. NS, not significant; Res, resveratrol; CQ,
chloroquine; siRNA, small interfering RNA. |
Discussion
Mitochondria play an important role in the process
of cell death. Damaged mitochondria can release cytochrome c to
activate caspase 3 for the execution of apoptosis, or translocate
apoptosis-inducing factor into the nucleus, leading to
caspase-independent apoptosis (22).
ΔΨm disruption is a key event in apoptosis (23). It was observed in the present study
that Res decreased the ΔΨm, activated caspase 3 and caused the
cleavage of PARP. Furthermore, caspase inhibitor Z-VAD-FMK
significantly inhibited Res-induced apoptosis. All the
aforementioned findings suggested that Res induced apoptosis via
mitochondria in a caspase-dependent manner in 786-O cells.
Res acts as an antioxidant or pro-oxidant depending
on the environment and cell type (24). Previous studies have demonstrated
that Res causes tumor cell death through ROS (25,26), and
that Res promotes the production of ROS in 786-O cells (27). However, the authors did not
investigate the association between ROS and cell death further.
Indeed, in certain circumstances, elevated ROS is only an event
that accompanies cell death, and even enhanced ROS production can
play a protective role in decreasing the amount of cell death
(28,29). The present study clearly revealed
that inhibition of ROS by NAC significantly attenuated apoptosis,
suggesting that ROS was responsible for Res-induced apoptosis in
786-O cells.
Previous studies have demonstrated that Res can
activate (30,31) or inhibit autophagy (32,33). The
present study confirmed that Res activated autophagy and induced
complete autophagic flux in 786-O cells. Furthermore, Res induced
autophagy partially through ROS. AMPK-mTOR is a classic regulatory
signaling pathway in autophagy (34). However, in the experiments performed
in the present study, Res did not affect AMPK and mTOR, indicating
that Res induced non-classical autophagy. Previous research has
reported that p38 inhibits autophagy (35) and that JNK can promote autophagy by
phosphorylating BCL2 (19). The
present study revealed that Res activated JNK and p38 through ROS,
while inhibition of p38 or JNK promoted or attenuated autophagy,
respectively. Furthermore, inhibition of JNK decreased the
increased amount of BCL2 phosphorylation, suggesting that JNK was
involved in the autophagic process. It was also noted that Res
inhibited ERK activity. ERK can activate HIF-1α to promote VEGF
transcription (36). Notably, high
expression of VEGF is a common feature of RCC, and the association
between Res and ERK deserves further investigation.
Under oxidative stress, the association between
autophagy and apoptosis is complicated. On the one hand, autophagy
can capture damaged proteins and organs (such as damaged
mitochondria) for degradation, maintaining cell survival. On the
other hand, extreme autophagy promotes cell death (37). In the experiments performed in the
present study, autophagy inhibitor CQ and Beclin 1 siRNA further
promoted apoptosis, demonstrating that autophagy exerted a
protective effect on Res-induced apoptosis in 786-O cells. To the
best of our knowledge, only one study (38) has reported the role of Res-induced
autophagy in RCC, demonstrating that Res induces autophagy via the
AMPK-mTOR signaling pathway and that autophagy, in turn, promotes
apoptosis. This difference may be attributed to the different cell
lines used. Furthermore, the previous study only detected the
expression levels of autophagy-associated proteins and genes to
ascertain their effect on Res-mediated apoptosis. In the absence of
autophagic flux studies, their evidence remains unconvincing.
Overall, ROS was involved in the process of
Res-induced apoptosis in 786-O cells. On the one hand, ROS damaged
mitochondria and activated caspase to execute apoptosis. On the
other hand, it induced autophagy through JNK, and autophagy
suppressed apoptosis. Therefore, a combination of Res and autophagy
inhibitor could enhance the inhibitory effect of Res on RCC.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Natural Science
Foundation of Jiangsu Province (grant no. BK20151180), the Applied
Basic Research of Changzhou City (grant no. CJ20159014) and the
Major Science and Technology Project of Changzhou Health Bureau
(grant no. ZD201405).
Availability of data and materials
The datasets used and/or analysed in the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
HY and MF interpreted the data and drafted the
initial manuscript. XH designed the experiments and revised the
initial manuscript and HY performed the experiments. All authors
read and approved the final manuscript
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Simard EP, Ward EM, Siegel R and Jemal A:
Cancers with increasing incidence trends in the United States: 1999
through 2008. CA Cancer J Clin. 62:118–128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Znaor A, Lortet-Tieulent J, Laversanne M,
Jemal A and Bray F: International variations and trends in renal
cell carcinoma incidence and mortality. Eur Urol. 67:519–530. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Motzer RJ, Agarwal N, Beard C, Bhayani S,
Bolger GB, Carducci MA, Chang SS, Choueiri TK, Hancock SL, Hudes
GR, et al: Kidney cancer. J Natl Compr Canc Netw. 9:960–977. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Novelle MG, Wahl D, Diéguez C, Bernier M
and de Cabo R: Resveratrol supplementation: Where are we now and
where should we go? Ageing Res Rev. 21:1–15. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jang M, Cai L, Udeani GO, Slowing KV,
Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta
RG, et al: Cancer chemopreventive activity of resveratrol, a
natural product derived from grapes. Science. 275:218–220. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carter LG, D'Orazio JA and Pearson KJ:
Resveratrol and cancer: Focus on in vivo evidence. Endocr Relat
Cancer. 21:R209–R225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iliopoulos O, Kibel A, Gray S and Kaelin
WG Jr: Tumour suppression by the human von Hippel-Lindau gene
product. Nat Med. 1:822–826. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kucejova B, Peña-Llopis S, Yamasaki T,
Sivanand S, Tran TA, Alexander S, Wolff NC, Lotan Y, Xie XJ,
Kabbani W, et al: Interplay between pVHL and mTORC1 pathways in
clear-cell renal cell carcinoma. Mol Cancer Res. 9:1255–1265. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khan MA, Chen HC, Wan XX, Tania M, Xu AH,
Chen FZ and Zhang DZ: Regulatory effects of resveratrol on
antioxidant enzymes: A mechanism of growth inhibition and apoptosis
induction in cancer cells. Mol Cells. 35:219–225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen LB: Mitochondrial membrane potential
in living cells. Annu Rev Cell Biol. 4:155–181. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dröge W: Free radicals in the
physiological control of cell function. Physiol Rev. 82:47–95.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dewaele M, Maes H and Agostinis P:
ROS-mediated mechanisms of autophagy stimulation and their
relevance in cancer therapy. Autophagy. 6:838–854. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chan LL, Shen D, Wilkinson AR, Patton W,
Lai N, Chan E, Kuksin D, Lin B and Qiu J: A novel image-based
cytometry method for autophagy detection in living cells.
Autophagy. 8:1371–1382. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pajares M, Jiménez-Moreno N, Dias IHK,
Debelec B, Vucetic M, Fladmark KE, Basaga H, Ribaric S, Milisav I
and Cuadrado A: Redox control of protein degradation. Redox Biol.
6:409–420. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu J, Wu Y, Lu G, Xie S, Ma Z, Chen Z,
Shen HM and Xia D: Importance of ROS-mediated autophagy in
determining apoptotic cell death induced by physapubescin B. Redox
Biol. 12:198–207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xie X, Le L, Fan Y, Lv L and Zhang J:
Autophagy is induced through the ROS-TP53-DRAM1 pathway in Response
to mitochondrial protein synthesis inhibition. Autophagy.
8:1071–1084. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hung AC, Tsai CH, Hou MF, Chang WL, Wang
CH, Lee YC, Ko A, Hu SC, Chang FR, Hsieh PW and Yuan SS: The
synthetic β-nitrostyrene derivative CYT-Rx20 induces breast cancer
cell death and autophagy via ROS-mediated MEK/ERK pathway. Cancer
Lett. 371:251–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsao CC and Corn PG: MDM-2 antagonists
induce p53-dependent cell cycle arrest but not cell death in renal
cancer cell lines. Cancer Biol Ther. 10:1315–1325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gurkar AU, Chu K, Raj L, Bouley R, Lee SH,
Kim YB, Dunn SE, Mandinova A and Lee SW: Identification of ROCK1
kinase as a critical regulator of Beclin1-mediated autophagy during
metabolic stress. Nat Commun. 4:21892013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Otera H, Ohsakaya S, Nagaura Z, Ishihara N
and Mihara K: Export of mitochondrial AIF in response to
proapoptotic stimuli depends on processing at the intermembrane
space. EMBO J. 24:1375–1386. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brenner C and Kroemer G: Apoptosis.
Mitochondria-the death signal integrators. Science. 289:1150–1151.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Muqbil I, Beck FW, Bao B, Sarkar FH,
Mohammad RM, Hadi SM and Azmi AS: Old wine in a new bottle: The
warburg effect and anticancer mechanisms of resveratrol. Curr Pharm
Des. 18:1645–1654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miki H, Uehara N, Kimura A, Sasaki T, Yuri
T, Yoshizawa K and Tsubura A: Resveratrol induces apoptosis via
ROS-triggered autophagy in human colon cancer cells. Int J Oncol.
40:1020–1028. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gu S, Chen C, Jiang X and Zhang Z:
ROS-mediated endoplasmic reticulum stress and mitochondrial
dysfunction underlie apoptosis induced by resveratrol and arsenic
trioxide in A549 cells. Chem Biol Interact. 245:100–109. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim C, Baek SH, Um JY, Shim BS and Ahn KS:
Resveratrol attenuates constitutive STAT3 and STAT5 activation
through induction of PTPε and SHP-2 tyrosine phosphatases and
potentiates sorafenib-induced apoptosis in renal cell carcinoma.
BMC Nephrol. 17:192016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moriyama M, Moriyama H, Uda J, Kubo H,
Nakajima Y, Goto A, Morita T and Hayakawa T: BNIP3 upregulation via
stimulation of ERK and JNK activity is required for the protection
of keratinocytes from UVB-induced apoptosis. Cell Death Dis.
8:e25762017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Luo F and Zhao H: Paraquat-induced
reactive oxygen species inhibit neutrophil apoptosis via a p38
MAPK/NF-κB-IL-6/TNF-α positive-feedback circuit. PLoS One.
9:e938372014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kumar B, Iqbal MA, Singh RK and Bamezai
RN: Resveratrol inhibits TIGAR to promote ROS induced apoptosis and
autophagy. Biochimie. 118:26–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan HW, Hu WX, Zhang JY, Wang Y, Xia K,
Peng MY and Liu J: Resveratrol induces human K562 cell apoptosis,
erythroid differentiation, and autophagy. Tumour Biol.
35:5381–5388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alayev A, Sun Y, Snyder RB, Berger SM, Yu
JJ and Holz MK: Resveratrol prevents rapamycin-induced upregulation
of autophagy and selectively induces apoptosis in TSC2-deficient
cells. Cell Cycle. 13:371–382. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alayev A, Berger SM, Kramer MY, Schwartz
NS and Holz MK: The combination of rapamycin and resveratrol blocks
autophagy and induces apoptosis in breast cancer cells. J Cell
Biochem. 116:450–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Simon HU, Friis R, Tait SW and Ryan KM:
Retrograde signaling from autophagy modulates stress responses. Sci
Signal. 10(pii): eaag27912017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
de la Cruz-Morcillo MA, Valero ML,
Callejas-Valera JL, Arias-González L, Melgar-Rojas P, Galán-Moya
EM, García-Gil E, García-Cano J and Sánchez-Prieto R: P38MAPK is a
major determinant of the balance between apoptosis and autophagy
triggered by 5-fluorouracil: Implication in resistance. Oncogene.
31:1073–1085. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Masoud GN and Li W: HIF-1α pathway: Role,
regulation and intervention for cancer therapy. Acta Pharm Sin B.
5:378–389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kaminskyy VO and Zhivotovsky B: Free
radicals in cross talk between autophagy and apoptosis. Antioxid
Redox Signal. 21:86–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu Q, Fang Q, Ji S, Han Z, Cheng W and
Zhang H: Resveratrol-mediated apoptosis in renal cell carcinoma via
the p53/AMP-activated protein kinase/mammalian target of rapamycin
autophagy signaling pathway. Mol Med Rep. 17:502–508.
2018.PubMed/NCBI
|