However, it is difficult to pharmacologically
inhibit oncogenic signaling of some driver oncogenes. For example,
the development of mutant KRAS-targeted drugs has proven
problematic over the previous three decades (13). Although recently, treatment with
AMG510, a novel inhibitor against KRAS G12C, resulted in a
promising response rate in patients with lung cancer harboring this
specific type of mutation, development of drugs targeting other
types of KRAS mutations have not yet been successful
(14–16). In addition, mutations in driver
oncogenes in a number of types of human cancer have not been
identified (17). In such cases,
cancer results from non-oncogenes conferring various malignant
phenotypes, occasionally in a context-dependent manner (18) and these genes may serve as novel
therapeutic targets. For example, a study demonstrated that cancer
cells depend on non-oncogene Heat shock factor 1 (HSF1), which is
the master regulator of the heat shock response in eukaryotes, for
their proliferation and survival than their non-transformed
counterparts (19). To identify drug
target genes for cancer cells harboring oncogenes which are
difficult to pharmacologically inhibit, or do not have known
oncogenes, it is vital to perform an unbiased, large-scale
functional screening (20). Two
important gene modulating technologies, RNA interference (RNAi) and
clustered regularly interspaced short palindromic
repeats-associated protein 9 (CRISPR-Cas9) have emerged as powerful
tools for evaluating gene function (21). In addition, technologies in next
generation sequencing have improved. The combination of these
advanced technologies has allowed investigation of gene function at
genome-wide levels in a high-throughput manner.
Thus, functional screening based on cancer-specific
characteristics has been extensively conducted. In the majority of
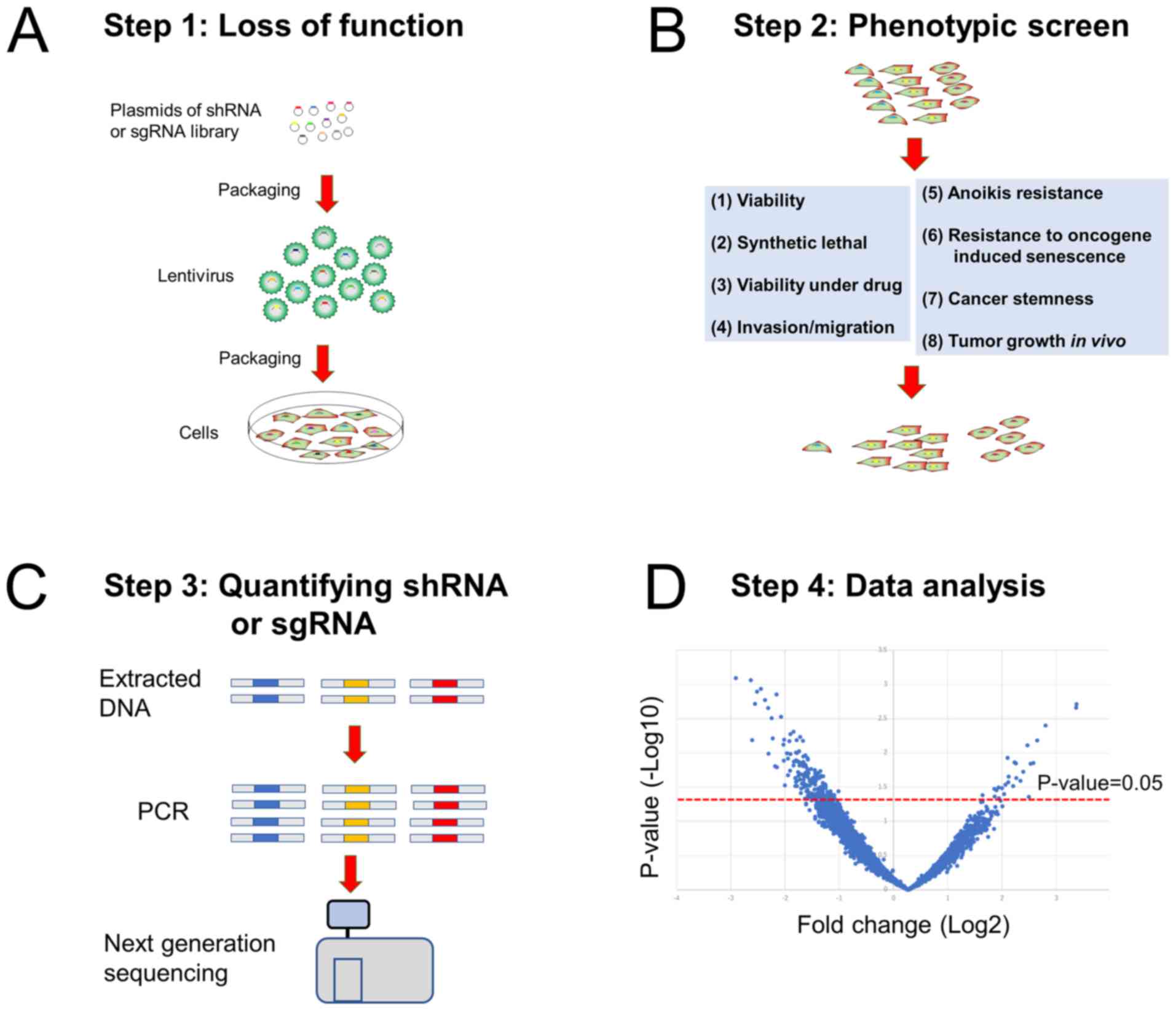
cases, functional screening is a four-step process: i) Inducing
loss-of-function via RNA interference (RNAi) or CRISPR-Cas9 in
cells; ii) evaluating the effects of the loss of the selected gene
on phenotypes critical to cancer cells; iii) quantifying short
hairpin RNAs (shRNAs) or single-guide RNAs (sgRNAs) via
next-generation sequencing or microarray hybridization; and iv)
data analysis (Fig. 1). Malignant
phenotypes used for functional screening include uncontrolled
promoted proliferation, drug resistance, invasiveness and the
ability to bypass oncogene-induced senescence (OIS). In the present
study, the recent advances in functional screening to identify
cancer drug target genes have been summarized, and current issues
and future perspectives have been discussed.
Using genome-wide methodologies to identify target
genes that substantially contribute to the uncontrolled
proliferation of cancer cells is a straightforward approach to
discovering cancer drug target genes for new drug development. This
type of assay is called dropout viability screening. Two pioneering
studies have conducted genome-wide dropout shRNA screening in
various human cancer cell lines and identified genes essential for
cancer cells (22,23). The Project Achilles study (launched
in 2011) systemically identified genes essential for proliferation
and/or survival in particular cancer cell types (genetic
vulnerabilities) by performing an integrative analysis involving
two steps: i) Conducting a pooled shRNA screen that targeted 11,194
genes in 102 (updated to 216 in the latest study) human cancer cell
lines, including ovarian, colon, pancreatic, esophageal and
non-small cell lung cancers; and ii) combining these results with
information on alterations of cancer genome through using publicly
available databases (24,25). By analyzing such diverse types of
cancer, the study identified a number of lineage-specific essential
genes. Another similar study used a pooled shRNA library comprised
of 72 breast, pancreatic and ovarian cancer cell lines (26). In addition, after a
CRISPR-Cas9-mediated gene-knockout technology became available in
the experimental cell biology field (27), two studies demonstrated the
feasibility of using lentiviral CRISPR-Cas9 libraries for
functional screening, with certain advantages over RNAi libraries
in efficacy and reliability (28,29). Via
negative screening with RNAi or CRISPR-Cas9, these studies
identified genes essential for proliferation in cancer cells, of
which certain genes were lineage-specific.
One critical issue resulting from the nature of
dropout viability screening is that such identified essential genes
for cancer cells may also be essential for normal cells; for
example, housekeeping genes involved in the ribosomal, proteasomal
and spliceosomal pathways (26).
Nevertheless, such essential genes may serve as promising
therapeutic targets, as cancer cells highly depend on them for
proliferation and/or survival compared with normal cells. One way
to identify general essential genes that are likely to serve as
cancer drug targets is to integrate results of genomic library
screening with gene expression data and copy number changes between
cancer and normal cells (20). This
helps identify the genes that are associated with proliferation
and/or survival in cancer cells (24). Using this approach, two housekeeping
genes have been identified, proteasome 20S subunit alpha 6
(PSMA6; a proteasomal catalytic subunit) and eukaryotic
translation initiation factor 2 subunit beta (eIF2β; a
subunit of translation-initiation factor EIF2), as promising
therapeutic targets for lung cancer (30,31).
Another way to identify essential genes that
contribute to oncogenic phenotypes is to reveal the genes which
cancer cells depend on in specific contexts; for example, with
certain types of driver oncogenes (32). This situation is referred to as
synthetic lethality and is described later. One study demonstrated
that an essential gene BUD31, a component of the spliceosome
is a potential therapeutic target specifically in MYC-driven
cancers (33).
A synthetic lethality refers to a phenomenon in
which inhibition of one of two genes has no significant effects on
cell viability but perturbation of both genes results in cell death
(32). Synthetic lethality has
attracted interest for the following reasons: i) If the synthetic
lethality specifically occurs in cancer cells, treatments targeting
genes involved in the synthetic lethality have a high therapeutic
index; and ii) if the synthetic lethality involves driver oncogenes
highly refractory to currently available treatment strategies,
synthetic lethal genes may serve as good targets in types of cancer
influenced by these oncogenes. A good example of such a gene is
oncogenic KRAS, the most frequently mutated oncogene,
although KRAS-targeted therapy is not used clinically
(14). Using RNAi library screening,
several studies have identified synthetic lethal genes in
KRAS-mutated cancers, such as STK33, TBK1, PLK1, SNAIL2,
CDK1 and GATA2 (34–39).
However, these identified genes rarely overlapped between studies
(40) and the identification of a
synthetic lethal effect caused by STK33 has not been
reproduced (41,42). A recently conducted large-scale
synthetic lethal RNAi screen, Project DRIVE, also failed to confirm
significant synthetic interactions of mutant KRAS with these
identified synthetic lethal genes (20). There are several possible reasons for
such inconsistent results, including differences in methods of gene
silencing (for example RNAi methodologies such as transient
transfection of siRNAs or shRNA, and difference in types of
library), and differences in types of cells used (for example
variable dependencies on KRAS signaling). In particular, the
latter seems to significantly influence screening results. Most
studies of KRAS synthetic screens used cancer cell lines
with or without mutant KRAS and/or isogenic cancer cell
lines transfected with or without mutant KRAS (34–39).
Cancer cell lines are highly variable in genetic changes (even
those with the same driver oncogenes), which may result in
inconsistent screening results (17,43).
Project DRIVE comprehensively assessed dependencies
and synthetic lethal relationships using 398 cancer cell lines from
different organs (20). To minimize
false-positive rates, an average of 20 shRNAs per gene were used
and, although synthetic lethal genes could not be confirmed for
mutant KRAS, a number of novel findings regarding synthetic
lethality which are translatable to developing novel therapeutics
were identified. For example, reduced expression levels of an
anti-apoptotic protein BCL2L1, and increased expression levels of
pro-apoptotic protein BIM, were the strongest predictors of the
growth-inhibiting effects following knockdown of anti-apoptotic
protein myeloid cell leukemia sequence 1 (MCL1).
Recently, via genome-wide CRISPR-Cas9 screening, two
independent groups identified WRN helicase as a synthetic lethal
target in microsatellite unstable cancer types (44,45).
Moreover, a small molecule inhibitor of WRN helicase (NSC617145)
has been revealed to exhibit cytotoxic effects in cells derived
from patients with Fanconi anemia, in a synthetic lethal manner
(46).
Dropout viability screening in the presence of
anti-cancer drugs is a powerful approach to identifying genes
responsible for drug resistance and several potentially
chemo-sensitizing targets have been reported (Table I). Using a genome-wide an arrayed
RNAi library, Whitehurst et al (50) identified several genes influencing
resistance to paclitaxel in a lung cancer cell line. Lin et
al (51) identified MCL1
as a potential drug target gene that sensitizes a small cell lung
cancer cell line to ABT-737, an inhibitor of the antiapoptotic
molecules Bcl-2, Bcl-X(L) and Bcl-w. After the development of
pooled RNAi library technology, numerous investigators began using
such libraries. For example, Prahallad et al (52) revealed genes responsible for
resistance to a BRAF inhibitor PLX4032 (vemurafenib) in
types of cancer harboring BRAF V600E mutations. It was
revealed that EGFR activation, which is rapidly induced by
vemurafenib treatment, induces resistance to vemurafenib treatment,
suggesting that combination therapy of vemurafenib and an
EGFR inhibitor may be beneficial. Previously, studies using
CRISPR-Cas9 libraries were published. Most of these studies used
the same type of genome-wide library, GeCKO CRISPR Library version
1 or 2, comprising of >120,000 sgRNAs targeting nearly the
entire genome (53–56). For example, Sustic et al
identified the endoplasmic reticulum to nucleus signaling 1
(ERN1)-JNK-JUN pathway as a potential target for improving
the anti-cancer effects of MET inhibitors in KRAS-mutated
colon cancer (56).
KRAS-targeted therapy has not been successfully developed
previously and, therefore, these findings are promising.
Immune therapy using immune checkpoint inhibitors
provides significant clinical benefit to patients with various
types of cancer, including melanoma, lymphoma, and lung cancer
(57). However, intrinsic or
acquired resistance inevitably occurs, limiting the clinical
benefits (49). Using genome-wide
CRISPR-Cas9 or siRNA libraries, two studies identified APLNR
(encoding the apelin receptor) and C-C motif chemokine receptor 9
as genes that may cause resistance to immune checkpoint inhibitors
(58,59).
Metastasis is significantly associated with a poor
patient prognosis, and patients with metastatic cancer exhibit poor
survival outcomes (60). Metastasis
comprises several sequential steps: i) Migration from a primary
site; ii) intravasation; iii) passage by blood flow; iv)
extravasation; v) and final settlement at distant sites. To
complete this process, cancer cells must acquire the ability to
invade and migrate and cancer cells exhibit these oncogenic
properties. Previous studies demonstrated that
epithelial-mesenchymal transition (EMT) significantly contributes
to metastasis in cancer cells (61,62).
EMT, and its reverse phenomenon MET, were initially identified
during embryonic development, in which embryonic cells transform
into terminally differentiated, specialized cells via several
cycles of EMT and MET (61). A
number of studies suggest a central role of EMT in metastasis
(63–65). Previous studies have identified
target genes for inhibiting migration and/or invasion ability of
cancer cells through library screening. Pavan et al
(66) developed a system combining
RNAi library screening with a microscopy-based high-throughput
quantitative analysis to identify a signaling pathway contributing
to EMT in breast cancer. The group identified 59 genes whose
inhibition suppressed transforming growth factor β-induced EMT in
immortalized epithelial normal murine mammary gland cells. In
addition, Pavan et al (66)
focused on MEK5 and ERK5 belonging to the same
signaling pathway and demonstrated the potential of targeting
MEK5 and ERK5 as an anti-metastatic mechanism.
Another study used migration ability as a phenotype for functional
screening, identifying genes contributing to migration in
glioblastoma, a highly invasive cancer (67). The authors performed a genome-wide
RNAi screening in glioblastoma cells with a functional selection of
cells able to migrate through Matrigel, identifying two genes
[KH-type splicing regulatory protein (KHSRP) and host cell
factor C1 (HCFC1)] as targets of invasion-suppressing
therapeutics for glioblastoma.
Upon detachment from the extracellular matrix or
neighboring cells, normal epithelial cells undergo a type of
apoptosis called anoikis (68).
Anoikis prevents normal epithelial cells from colonizing at
different organ sites, thereby maintaining the integrity of the
body (68). Most cancer cells
acquire resistance to anoikis, which is called
anchorage-independent growth (AIG). The ability of AIG allows
cancer cells to metastasize to different organs and is considered a
hallmark of cancer cells (64).
Several different molecular mechanisms underlying AIG have been
identified, including the induction of intrinsic and extrinsic
anti-apoptotic signaling, often triggered by changes in the
expression patterns of integrin family members (68,69). In
addition, previous studies have demonstrated the role of EMT in AIG
(68,70); however, the underlying molecular
mechanisms of AIG are yet to be elucidated.
The cancer stem cell (CSC) theory hypothesizes that
CSCs have the ability to self-renew and to differentiate into
phenotypically diverse cancer cells (84). Although the CSC concept has not been
demonstrated, accumulating evidence suggests that a number of types
of cancer harbor CSCs (84,85). Notably, CSCs are hypothesized to be
resistant to chemotherapy and irradiation (84). Therefore, the development of
CSC-targeted therapeutics is attracting attention because of its
potential to eradicate cancer cells. A functional library screening
based on the sphere-forming ability of breast cancer cell lines
identified ATG4 as a promotor of the breast CSC-like
phenotype (86). However, the
usefulness of a sphere-formation assay for evaluating the
self-renewal capacity is based on the assumption that the assay
developed for normal neural stem cells can be accurately used for
CSCs. Therefore, validation of genes identified as cancer stemness
genes by other assays, such as a transplantation assays and
lineage-tracing approaches, are required.
A phenotype of genomic instability facilitates
diverse oncogenic properties because it causes numerous mutations
resulting from the activation of oncogenic genes or inactivation of
tumor suppressive genes (87). A
previous study performed a genome-wide RNAi screen to identified
the pathways and specific genes mediating genomic stability
(88). A screen using elevation of
γH2A.X variant histone (H2AX; a marker of double strand DNA damage)
as an indicator for detecting DNA damage was conducted in HeLa
cancer cells, identifying genes involved in DNA replication,
checkpoint activation and DNA repair. The identified genes included
TIMELESS and TIPIN encoding proteins that form a
complex, leading to activation of the replication checkpoint. The
identified genes may serve as promising drug targets to restore
genomic stability in cancer cells (88).
Shortly after RNAi technology for gene knockdown was
developed in the laboratory, attempts to conduct large-scale
functional screenings with RNAi were initiated (93). In addition, a gene knockout
technique, CRISPR-Cas9 was also introduced for laboratory use
(94). For >10 years, researchers
have extensively conducted functional genomic screening to identify
better targets and to develop new therapeutics for cancer. The
present paper reviewed and summarized knowledge obtained by these
studies, which has the potential to be used for drug development.
Nevertheless, breakthroughs that can be immediately translated into
clinical use are yet to be made. In particular, despite many
reported studies, KRAS synthetic genes that have been
reproducibly confirmed have not been successfully identified;
therefore, development of KRAS-synthetic lethal drugs has
not been successful. Project DRIVE suggested that no single
synthetic lethal genes for KRAS exist. However, there may be
certain strategies potentially enabling the identification of true
KRAS synthetic genes; for example, one approach may be using
more realistic modeling systems to evaluate malignant phenotypes.
Such models may include a 3-D culture of cell lines and
patient-derived xenografts (95,96),
although such models are usually difficult to manage for
large-scale screening. Due to the large heterogeneity in coexisting
genomic alterations among KRAS-mutated tumors, studies using
cancer cells may suffer from the presence of high background of
noise during screening. Therefore, focus is needed on cancer cells
which have higher similarities in harbored genetic alterations in
addition to mutant KRAS.
An improvement in consistency of identified genes
from a genome-wide screen has been revealed in CRISPR-Cas9 knockout
compared with shRNA techniques (28). However, pharmacological inhibition of
gene function with compounds is usually incomplete; thus, target
genes identified through partial knockdown with RNAi represent
improved targets. Therefore, results from CRISPR-Cas9 and RNAi
screens need to be regarded as complementary.
In conclusion, advances in the technology of gene
silencing and next generation sequencing have enabled researchers
to conduct large-scale high-throughput phenotypic screenings,
resulting in the identification of numerous potential novel drug
targets for cancer. However, there are several issues, such as low
reproducibility in the identified genes (40). Thus, substantial effort is required
to adequately address these problems in order to identify novel
cancer drug target genes.
Not applicable.
The present work was supported by grants from
Grant-in-Aid for Scientific Research (grant no. 18H02819) and a
Challenging Research Exploratory grant from the Japan Society for
the Promotion of Science (grant no. 19K22617).
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
MS designed the review, researched the literature
and wrote the manuscript.
Not applicable.
Not applicable.
The author declares that he has no competing
interests.
|
1
|
Armitage P and Doll R: The age
distribution of cancer and a multi-stage theory of carcinogenesis.
Br J Cancer. 8:1–12. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vogelstein B and Kinzler KW: The multistep
nature of cancer. Trends Genet. 9:138–141. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chaffer CL and Weinberg RA: How does
multistep tumorigenesis really proceed? Cancer Discov. 5:22–24.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barretina J, Caponigro G, Stransky N,
Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV,
Sonkin D, et al: The cancer cell line encyclopedia enables
predictive modelling of anticancer drug sensitivity. Nature.
483:603–607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bailey MH, Tokheim C, Porta-Pardo E,
Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim
J, Reardon B, et al: Comprehensive characterization of cancer
driver genes and mutations. Cell. 173:371–385 e18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Garraway LA and Lander ES: Lessons from
the cancer genome. Cell. 153:17–37. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seshadri R, Matthews C, Dobrovic A and
Horsfall DJ: The significance of oncogene amplification in primary
breast cancer. Int J Cancer. 43:270–272. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sato M, Shames DS, Gazdar AF and Minna JD:
A translational view of the molecular pathogenesis of lung cancer.
J Thorac Oncol. 2:327–343. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murugan AK, Grieco M and Tsuchida N: RAS
mutations in human cancers: Roles in precision medicine. Semin
Cancer Biol. 59:23–35. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ryan MB and Corcoran RB: Therapeutic
strategies to target RAS-mutant cancers. Nat Rev Clin Oncol.
15:709–720. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Govindan R, Fakih M, Price T, Falchook G,
Desai J, Kuo J, Strickler J, Krauss J, Li B, Denlinger C, et al:
OA02.02 Phase 1 study of safety, tolerability, PK and efficacy of
AMG 510, a novel KRASG12C inhibitor, evaluated in NSCLC. J Thorac
Oncol. 14 (Suppl):S2082019. View Article : Google Scholar
|
|
16
|
Canon J, Rex K, Saiki AY, Mohr C, Cooke K,
Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al: The
clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity.
Nature. 575:217–223. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nagel R, Semenova EA and Berns A: Drugging
the addict: Non-oncogene addiction as a target for cancer therapy.
EMBO Rep. 17:1516–1531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai C, Whitesell L, Rogers AB and
Lindquist S: Heat shock factor 1 is a powerful multifaceted
modifier of carcinogenesis. Cell. 130:1005–1018. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McDonald ER III, de Weck A, Schlabach MR,
Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E,
Gampa K, et al: Project DRIVE: A compendium of cancer dependencies
and synthetic lethal relationships uncovered by large-scale, deep
RNAi screening. Cell. 170:577–592 e10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schuster A, Erasimus H, Fritah S, Nazarov
PV, van Dyck E, Niclou SP and Golebiewska A: RNAi/CRISPR Screens:
From a pool to a valid hit. Trends Biotechnol. 37:38–55. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schlabach MR, Luo J, Solimini NL, Hu G, Xu
Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, et al: Cancer
proliferation gene discovery through functional genomics. Science.
319:620–624. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Silva JM, Marran K, Parker JS, Silva J,
Golding M, Schlabach MR, Elledge SJ, Hannon GJ and Chang K:
Profiling essential genes in human mammary cells by multiplex RNAi
screening. Science. 319:617–620. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheung HW, Cowley GS, Weir BA, Boehm JS,
Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, et al:
Systematic investigation of genetic vulnerabilities across cancer
cell lines reveals lineage-specific dependencies in ovarian cancer.
Proc Natl Acad Sci USA. 108:12372–12377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cowley GS, Weir BA, Vazquez F, Tamayo P,
Scott JA, Rusin S, East-Seletsky A, Ali LD, Gerath WF, Pantel SE,
et al: Parallel genome-scale loss of function screens in 216 cancer
cell lines for the identification of context-specific genetic
dependencies. Sci Data. 1:1400352014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Marcotte R, Brown KR, Suarez F, Sayad A,
Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn
Y, Koh JLY, et al: Essential gene profiles in breast, pancreatic,
and ovarian cancer cells. Cancer Discov. 2:172–189. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 8:2281–2308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shalem O, Sanjana NE, Hartenian E, Shi X,
Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG and
Zhang F: Genome-scale CRISPR-Cas9 knockout screening in human
cells. Science. 343:84–87. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang T, Wei JJ, Sabatini DM and Lander ES:
Genetic screens in human cells using the CRISPR-Cas9 system.
Science. 343:80–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kakumu T, Sato M, Goto D, Kato T, Yogo N,
Hase T, Morise M, Fukui T, Yokoi K, Sekido Y, et al: Identification
of proteasomal catalytic subunit PSMA6 as a therapeutic target for
lung cancer. Cancer Sci. 108:732–743. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tanaka I, Sato M, Kato T, Goto D, Kakumu
T, Miyazawa A, Yogo N, Hase T, Morise M, Sekido Y, et al: eIF2β, a
subunit of translation-initiation factor EIF2, is a potential
therapeutic target for non-small cell lung cancer. Cancer Sci.
109:1843–1852. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
O'Neil NJ, Bailey ML and Hieter P:
Synthetic lethality and cancer. Nat Rev Genet. 18:613–623. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hsu TY, Simon LM, Neill NJ, Marcotte R,
Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al:
The spliceosome is a therapeutic vulnerability in MYC-driven
cancer. Nature. 525:384–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kumar MS, Hancock DC, Molina-Arcas M,
Steckel M, East P, Diefenbacher M, Armenteros-Monterroso E,
Lassailly F, Matthews N, Nye E, et al: The GATA2 transcriptional
network is requisite for RAS oncogene-driven non-small cell lung
cancer. Cell. 149:642–655. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luo J, Emanuele MJ, Li D, Creighton CJ,
Schlabach MR, Westbrook TF, Wong KK and Elledge SJ: A genome-wide
RNAi screen identifies multiple synthetic lethal interactions with
the Ras oncogene. Cell. 137:835–848. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Scholl C, Fröhling S, Dunn IF, Schinzel
AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S,
et al: Synthetic lethal interaction between oncogenic KRAS
dependency and STK33 suppression in human cancer cells. Cell.
137:821–834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Barbie DA, Tamayo P, Boehm JS, Kim SY,
Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al:
Systematic RNA interference reveals that oncogenic KRAS-driven
cancers require TBK1. Nature. 462:108–112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Y, Ngo VN, Marani M, Yang Y, Wright
G, Staudt LM and Downward J: Critical role for transcriptional
repressor Snail2 in transformation by oncogenic RAS in colorectal
carcinoma cells. Oncogene. 29:4658–4670. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Costa-Cabral S, Brough R, Konde A, Aarts
M, Campbell J, Marinari E, Riffell J, Bardelli A, Torrance C, Lord
CJ and Ashworth A: CDK1 is a synthetic lethal target for KRAS
mutant tumours. PLoS One. 11:e01490992016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Downward J: RAS synthetic lethal screens
revisited: Still seeking the elusive prize? Clin Cancer Res.
21:1802–1809. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Babij C, Zhang Y, Kurzeja RJ, Munzli A,
Shehabeldin A, Fernando M, Quon K, Kassner PD, Ruefli-Brasse AA,
Watson VJ, et al: STK33 kinase activity is nonessential in
KRAS-dependent cancer cells. Cancer Res. 71:5818–5826. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fröhling S and Scholl C: STK33 kinase is
not essential in KRAS-dependent cells-letter. Cancer Res.
71:7716author reply 7717. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Forbes SA, Beare D, Boutselakis H, Bamford
S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al:
COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids
Res. 45D:D777–D783. 2017. View Article : Google Scholar
|
|
44
|
Behan FM, Iorio F, Picco G, Gonçalves E,
Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et
al: Prioritization of cancer therapeutic targets using CRISPR-Cas9
screens. Nature. 568:511–516. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chan EM, Shibue T, McFarland JM, Gaeta B,
Ghandi M, Dumont N, Gonzalez A, McPartlan JS, Li T, Zhang Y, et al:
WRN helicase is a synthetic lethal target in microsatellite
unstable cancers. Nature. 568:551–556. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Aggarwal M, Banerjee T, Sommers JA,
Iannascoli C, Pichierri P, Shoemaker RH and Brosh RM Jr: Werner
syndrome helicase has a critical role in DNA damage responses in
the absence of a functional fanconi anemia pathway. Cancer Res.
73:5497–5507. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gottesman MM, Lavi O, Hall MD and Gillet
JP: Toward a better understanding of the complexity of cancer drug
resistance. Annu Rev Pharmacol Toxicol. 56:85–102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jackson CM, Choi J and Lim M: Mechanisms
of immunotherapy resistance: Lessons from glioblastoma. Nat
Immunol. 20:1100–1109. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Whitehurst AW, Bodemann BO, Cardenas J,
Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth
MG, et al: Synthetic lethal screen identification of
chemosensitizer loci in cancer cells. Nature. 446:815–819. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lin X, Morgan-Lappe S, Huang X, Li L,
Zakula DM, Vernetti LA, Fesik SW and Shen Y: ‘Seed’ analysis of
off-target siRNAs reveals an essential role of Mcl-1 in resistance
to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene.
26:3972–3979. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Prahallad A, Sun C, Huang S, Di
Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A
and Bernards R: Unresponsiveness of colon cancer to BRAF(V600E)
inhibition through feedback activation of EGFR. Nature.
483:100–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kurata M, Rathe SK, Bailey NJ, Aumann NK,
Jones JM, Veldhuijzen GW, Moriarity BS and Largaespada DA: Using
genome-wide CRISPR library screening with library resistant DCK to
find new sources of Ara-C drug resistance in AML. Sci Rep.
6:361992016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hou P, Wu C, Wang Y, Qi R, Bhavanasi D,
Zuo Z, Dos Santos C, Chen S, Chen Y, Zheng H, et al: A Genome-wide
CRISPR screen identifies genes critical for resistance to FLT3
inhibitor AC220. Cancer Res. 77:4402–4413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sun W, He B, Yang B, Hu W, Cheng S, Xiao
H, Yang Z, Wen X, Zhou L, Xie H, et al: Genome-wide CRISPR screen
reveals SGOL1 as a druggable target of sorafenib-treated
hepatocellular carcinoma. Lab Invest. 98:734–744. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sustic T, van Wageningen S, Bosdriesz E,
Reid RJD, Dittmar J, Lieftink C, Beijersbergen RL, Wessels LFA,
Rothstein R and Bernards R: A role for the unfolded protein
response stress sensor ERN1 in regulating the response to MEK
inhibitors in KRAS mutant colon cancers. Genome Med. 10:902018.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sharma P and Allison JP: The future of
immune checkpoint therapy. Science. 348:56–61. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Khandelwal N, Breinig M, Speck T, Michels
T, Kreutzer C, Sorrentino A, Sharma AK, Umansky L, Conrad H,
Poschke I, et al: A high-throughput RNAi screen for detection of
immune-checkpoint molecules that mediate tumor resistance to
cytotoxic T lymphocytes. EMBO Mol Med. 7:450–463. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Patel SJ, Sanjana NE, Kishton RJ,
Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM,
Yamamoto TN, et al: Identification of essential genes for cancer
immunotherapy. Nature. 548:537–542. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Steeg PS: Targeting metastasis. Nat Rev
Cancer. 16:201–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sato M, Shames DS and Hasegawa Y: Emerging
evidence of epithelial-to-mesenchymal transition in lung
carcinogenesis. Respirology. 17:1048–1059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Chaffer CL, San Juan BP, Lim E and
Weinberg RA: EMT, cell plasticity and metastasis. Cancer Metastasis
Rev. 35:645–654. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Heerboth S, Housman G, Leary M, Longacre
M, Byler S, Lapinska K, Willbanks A and Sarkar S: EMT and tumor
metastasis. Clin Transl Med. 4:62015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Pavan S, Meyer-Schaller N, Diepenbruck M,
Kalathur RKR, Saxena M and Christofori G: A kinome-wide
high-content siRNA screen identifies MEK5-ERK5 signaling as
critical for breast cancer cell EMT and metastasis. Oncogene.
37:4197–4213. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yang J, Fan J, Li Y, Li F, Chen P, Fan Y,
Xia X and Wong ST: Genome-wide RNAi screening identifies genes
inhibiting the migration of glioblastoma cells. PLoS One.
8:e619152013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Paoli P, Giannoni E and Chiarugi P:
Anoikis molecular pathways and its role in cancer progression.
Biochim Biophys Acta. 1833:3481–3498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Taddei ML, Giannoni E, Fiaschi T and
Chiarugi P: Anoikis: An emerging hallmark in health and diseases. J
Pathol. 226:380–393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Takeyama Y, Sato M, Horio M, Hase T,
Yoshida K, Yokoyama T, Nakashima H, Hashimoto N, Sekido Y, Gazdar
AF, et al: Knockdown of ZEB1, a master epithelial-to-mesenchymal
transition (EMT) gene, suppresses anchorage-independent cell growth
of lung cancer cells. Cancer Lett. 296:216–224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Eskiocak U, Kim SB, Ly P, Roig AI,
Biglione S, Komurov K, Cornelius C, Wright WE, White MA and Shay
JW: Functional parsing of driver mutations in the colorectal cancer
genome reveals numerous suppressors of anchorage-independent
growth. Cancer Res. 71:4359–4365. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wood LD, Parsons DW, Jones S, Lin J,
Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Simpson CD, Hurren R, Kasimer D, MacLean
N, Eberhard Y, Ketela T, Moffat J and Schimmer AD: A genome wide
shRNA screen identifies α/β hydrolase domain containing 4 (ABHD4)
as a novel regulator of anoikis resistance. Apoptosis. 17:666–678.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Larsson LG: Oncogene- and tumor suppressor
gene-mediated suppression of cellular senescence. Semin Cancer
Biol. 21:367–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gorgoulis VG and Halazonetis TD:
Oncogene-induced senescence: The bright and dark side of the
response. Curr Opin Cell Biol. 22:816–827. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Faget DV, Ren Q and Stewart SA: Unmasking
senescence: Context-dependent effects of SASP in cancer. Nat Rev
Cancer. 19:439–453. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell. 88:593–602.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Michaloglou C, Vredeveld LC, Soengas MS,
Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi
WJ and Peeper DS: BRAFE600-associated senescence-like cell cycle
arrest of human naevi. Nature. 436:720–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gray-Schopfer VC, Cheong SC, Chong H, Chow
J, Moss T, Abdel-Malek ZA, Marais R, Wynford-Thomas D and Bennett
DC: Cellular senescence in naevi and immortalisation in melanoma: A
role for p16? Br J Cancer. 95:496–505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
He S and Sharpless NE: Senescence in
health and disease. Cell. 169:1000–1011. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Vicent S, Chen R, Sayles LC, Lin C, Walker
RG, Gillespie AK, Subramanian A, Hinkle G, Yang X, Saif S, et al:
Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and
senescence in mouse and human models. J Clin Invest. 120:3940–3952.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kaplon J, Hömig-Hölzel C, Gao L, Meissl K,
Verdegaal EM, van der Burg SH, van Doorn R and Peeper DS:
Near-genomewide RNAi screening for regulators of
BRAF(V600E)-induced senescence identifies RASEF, a gene
epigenetically silenced in melanoma. Pigment Cell Melanoma Res.
27:640–652. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Tordella L, Khan S, Hohmeyer A, Banito A,
Klotz S, Raguz S, Martin N, Dhamarlingam G, Carroll T, González
Meljem JM, et al: SWI/SNF regulates a transcriptional program that
induces senescence to prevent liver cancer. Genes Dev.
30:2187–2198. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Batlle E and Clevers H: Cancer stem cells
revisited. Nat Med. 23:1124–1134. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Nassar D and Blanpain C: Cancer stem
cells: Basic concepts and therapeutic implications. Annu Rev
Pathol. 11:47–76. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wolf J, Dewi DL, Fredebohm J,
Müller-Decker K, Flechtenmacher C, Hoheisel JD and Boettcher M: A
mammosphere formation RNAi screen reveals that ATG4A promotes a
breast cancer stem-like phenotype. Breast Cancer Res. 15:R1092013.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Paulsen RD, Soni DV, Wollman R, Hahn AT,
Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero
DE, et al: A genome-wide siRNA screen reveals diverse cellular
processes and pathways that mediate genome stability. Mol Cell.
35:228–239. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Gargiulo G, Serresi M, Cesaroni M, Hulsman
D and van Lohuizen M: In vivo shRNA screens in solid tumors. Nat
Protoc. 9:2880–2902. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Singh M, Venugopal C, Tokar T, Brown KR,
McFarlane N, Bakhshinyan D, Vijayakumar T, Manoranjan B, Mahendram
S, Vora P, et al: RNAi screen identifies essential regulators of
human brain metastasis-initiating cells. Acta Neuropathol.
134:923–940. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lin L, Chamberlain L, Pak ML, Nagarajan A,
Gupta R, Zhu LJ, Wright CM, Fong KM, Wajapeyee N and Green MR: A
large-scale RNAi-based mouse tumorigenesis screen identifies new
lung cancer tumor suppressors that repress FGFR signaling. Cancer
Discov. 4:1168–1181. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Iorns E, Ward TM, Dean S, Jegg A, Thomas
D, Murugaesu N, Sims D, Mitsopoulos C, Fenwick K, Kozarewa I, et
al: Whole genome in vivo RNAi screening identifies the leukemia
inhibitory factor receptor as a novel breast tumor suppressor.
Breast Cancer Res Treat. 135:79–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Elbashir SM, Harborth J, Lendeckel W,
Yalcin A, Weber K and Tuschl T: Duplexes of 21-nucleotide RNAs
mediate RNA interference in cultured mammalian cells. Nature.
411:494–498. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang H, La Russa M and Qi LS: CRISPR/Cas9
in genome editing and beyond. Annu Rev Biochem. 85:227–264. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Nyga A, Cheema U and Loizidou M: 3D tumour
models: Novel in vitro approaches to cancer studies. J Cell Commun
Signal. 5:239–248. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hidalgo M, Amant F, Biankin AV, Budinská
E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Mælandsmo
GM, et al: Patient-derived xenograft models: An emerging platform
for translational cancer research. Cancer Discov. 4:998–1013. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Bartz SR, Zhang Z, Burchard J, Imakura M,
Martin M, Palmieri A, Needham R, Guo J, Gordon M, Chung N, et al:
Small interfering RNA screens reveal enhanced cisplatin
cytotoxicity in tumor cells having both BRCA network and TP53
disruptions. Mol Cell Biol. 26:9377–9386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lam LT, Davis RE, Ngo VN, Lenz G, Wright
G, Xu W, Zhao H, Yu X, Dang L and Staudt LM: Compensatory IKKalpha
activation of classical NF-kappaB signaling during IKKbeta
inhibition identified by an RNA interference sensitization screen.
Proc Natl Acad Sci USA. 105:20798–20803. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Xu Y, Karlsson A and Johansson M:
Identification of genes associated to 2′,2′-difluorodeoxycytidine
resistance in HeLa cells with a lentiviral short-hairpin RNA
library. Biochem Pharmacol. 82:210–215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Guerreiro AS, Fattet S, Kulesza DW, Atamer
A, Elsing AN, Shalaby T, Jackson SP, Schoenwaelder SM, Grotzer MA,
Delattre O and Arcaro A: A sensitized RNA interference screen
identifies a novel role for the PI3K p110γ isoform in
medulloblastoma cell proliferation and chemoresistance. Mol Cancer
Res. 9:925–935. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liu-Sullivan N, Zhang J, Bakleh A,
Marchica J, Li J, Siolas D, Laquerre S, Degenhardt YY, Wooster R,
Chang K, et al: Pooled shRNA screen for sensitizers to inhibition
of the mitotic regulator polo-like kinase (PLK1). Oncotarget.
2:1254–1264. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Fredebohm J, Wolf J, Hoheisel JD and
Boettcher M: Depletion of RAD17 sensitizes pancreatic cancer cells
to gemcitabine. J Cell Sci. 126:3380–3389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Milosevic N, Kühnemuth B, Mühlberg L,
Ripka S, Griesmann H, Lölkes C, Buchholz M, Aust D, Pilarsky C,
Krug S, et al: Synthetic lethality screen identifies RPS6KA2 as
modifier of epidermal growth factor receptor activity in pancreatic
cancer. Neoplasia. 15:1354–1362. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Wetterskog D, Shiu KK, Chong I, Meijer T,
Mackay A, Lambros M, Cunningham D, Reis-Filho JS, Lord CJ and
Ashworth A: Identification of novel determinants of resistance to
lapatinib in ERBB2-amplified cancers. Oncogene. 33:966–976. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
MacKay C, Carroll E, Ibrahim AFM, Garg A,
Inman GJ, Hay RT and Alpi AF: E3 ubiquitin ligase HOIP attenuates
apoptotic cell death induced by cisplatin. Cancer Res.
74:2246–2257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Maruyama Y, Miyazaki T, Ikeda K, Okumura
T, Sato W, Horie-Inoue K, Okamoto K, Takeda S and Inoue S: Short
hairpin RNA library-based functional screening identified ribosomal
protein L31 that modulates prostate cancer cell growth via p53
pathway. PLoS One. 9:e1087432014. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Sudo M, Mori S, Madan V, Yang H, Leong G
and Koeffler HP: Short-hairpin RNA library: Identification of
therapeutic partners for gefitinib-resistant non-small cell lung
cancer. Oncotarget. 6:814–824. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Prahallad A, Heynen GJ, Germano G, Willems
SM, Evers B, Vecchione L, Gambino V, Lieftink C, Beijersbergen RL,
Di Nicolantonio F, et al: PTPN11 is a central node in intrinsic and
acquired resistance to targeted cancer drugs. Cell Rep.
12:1978–1985. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Kobayashi H, Nishimura H, Matsumoto K and
Yoshida M: Identification of the determinants of 2-deoxyglucose
sensitivity in cancer cells by shRNA library screening. Biochem
Biophys Res Commun. 467:121–127. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Yamaguchi K, Iglesias-Bartolomé R, Wang Z,
Callejas-Valera JL, Amornphimoltham P, Molinolo AA, Cohen EE,
Califano JA, Lippman SM, Luo J and Gutkind JS: A
synthetic-lethality RNAi screen reveals an ERK-mTOR co-targeting
pro-apoptotic switch in PIK3CA+ oral cancers. Oncotarget.
7:10696–10709. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Yamanoi K, Matsumura N, Murphy SK, Baba T,
Abiko K, Hamanishi J, Yamaguchi K, Koshiyama M, Konishi I and
Mandai M: Suppression of ABHD2, identified through a functional
genomics screen, causes anoikis resistance, chemoresistance and
poor prognosis in ovarian cancer. Oncotarget. 7:47620–47636. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Combes E, Andrade AF, Tosi D, Michaud HA,
Coquel F, Garambois V, Desigaud D, Jarlier M, Coquelle A, Pasero P,
et al: Inhibition of ataxia-telangiectasia mutated and RAD3-related
(ATR) overcomes oxaliplatin resistance and promotes antitumor
immunity in colorectal cancer. Cancer Res. 79:2933–2946. 2019.
View Article : Google Scholar : PubMed/NCBI
|