Introduction
Precision molecular targeted agents in non-small
cell lung cancer (NSCLC) have improved survival of patients
harboring driver gene mutations. Epidermal growth factor receptor
(EGFR)-tyrosine kinase inhibitor (TKI) improves progression-free
survival (PFS) of NSCLC patients with EGFR mutations
compared with traditional platinum-based doublet chemotherapy
(1,2). Furthermore, osimertinib, a
third-generation EGFR-TKI, is promising as first-line treatment for
EGFR mutant NSCLC (3,4). Although good responses to EGFR-TKI
therapy have been shown, tumor cells can acquire resistance through
several methods, in particular, secondary gene mutations that cause
structural changes in the ATP binding site of the EGFR tyrosine
kinase domain. EGFR T790M mutation occurs in almost half of
patients following first- or second-generation EGFR-TKI therapy
(5), and EGFR C797S mutation
is the most common mechanism of acquired resistance against
third-generation EGFR-TKIs (6).
Approximately 0.4–8% of NSCLC patients harboring
de novo or germline T790M mutations are resistant to first-
or second-generation EGFR-TKIs (7).
However, the frequency of EGFR C797S gene mutation remains
unclear. To the best of our knowledge, only one case of an NSCLC
patient harboring concurrent C797S and L858R mutations prior to
receiving EGFR-TKI treatment has been reported (8).
Several other mechanisms of resistance against all
generation EGFR-TKIs have been identified including tertiary gene
mutations other than EGFR C797S mutation (9–11),
activation of bypass signaling by gene amplification (e.g.,
ERBB2 (12) and MET
(13,14), driver gene mutations (e.g., RAS,
RAF and PIK3CA) (3,15), gene
alteration in cell cycle genes (14), and transformation to mesenchyme,
small cell carcinoma (SCC), or squamous cell carcinoma (SqCC)
(2,16,17).
These described EGFR-TKI resistance mechanisms may also be
expressed during the pre-TKI NSCLC state (6,15) and
can be a challenge for cancer treatment of NSCLC patients with
EGFR mutations.
In this retrospective study, we assessed potential
resistance against third-generation EGFR-TKI therapy, such as
EGFR C797S mutation, and gene amplification in
EGFR-TKI-naïve surgical specimens from patients harboring known
EGFR mutations.
Materials and methods
Patient selection
Consecutive patients who underwent initial lung
resection or surgical tumor biopsy in Fukushima Medical University
Hospital and were diagnosed with NSCLC harboring a known
EGFR gene activating mutation (e.g., exon 19 deletion,
L858R, T790M, S768I, G719X and L861Q) at the time samples were
collected, and whose specimens were available for gene examination
described below, were included in this study. Patients who had
received systemic treatment or irradiation therapy before surgery
were excluded.
Ethics statement
This study was conducted with approval of the ethics
board at Fukushima Medical University (approval no. 2955). Human
rights and welfare of participants were protected in accordance
with the Declaration of Helsinki, and written informed consent was
obtained from participants.
Preparation of genomic DNA
Tumor DNA was extracted from macro-dissected tumor
tissue of formalin-fixed paraffin-embedded surgical specimens using
the QIAamp DNA FFPE Tissue kit (Qiagen) according to the
manufacturer's instructions. Tumor DNA quantity was assessed using
the Qubit dsDNA HS Assay kit (Thermo Fisher Scientific, Waltham,
MA, USA) and Qubit 2.0 Fluorometer (Thermo Fisher Scientific).
Peptide nucleic acid-locked nucleic
acid PCR clamp method
Premix Ex Taq (Takara Bio Inc.), 200 nM of primer,
100 nM of mutation LNA probe, 100 nM of total probe, 250–2,500 nM
of clamp probe, and sample DNA were mixed in a total reaction
volume of 25 µl and analyzed as described previously (18). Real-time PCR was performed in 50
cycles using Light Cycler480 II (Roche; denaturation: 5 sec at
95°C; annealing and extension: 30 sec at 62°C). EGFR C797S
and T790M mutations were judged using LightCycler 480 software
(Roche).
Multiplex ligation-dependent probe
amplification (MLPA) Assay
Gene amplification was analyzed according to the
standard protocol for MLPA (19)
using SALSA MLPA Probemix P175 Tumor Gain (MRC-Holland). ERBB2,
MET, EGFR, ALK, BRAF, FGFR1, MYC, RET, CCND1, CCND2, CDK4, CDK6,
MDM2 and MDM4 gene amplification was assessed. Fragments
of PCR products were analyzed using the 3130×l Genetic Analyzer
(Thermo Fisher Scientific) and amplification was judged by
Coffalyser Data analysis software (MRC-Holland).
Statistical analysis
Statistical analysis was performed using SPSS 21.0
(IBM; SPSS). Continuous variables were compared by two-tailed
t-tests or one-way ANOVA, and categorical variables were
compared by the chi-squared test or Fisher's exact test.
Multivariate analyses using a binary logistic regression model were
performed to evaluate independent predictors, and hazard ratio (HR)
and confidence interval (CI) were calculated. PFS was estimated
using the Kaplan-Meier method, and survival curves were compared
using log-rank tests. P-values of less than 0.05 were considered
statistically significant. Some of the statistical analysis was
performed by AC Medical Inc. (Tokyo, Japan).
Results
Patient characteristics
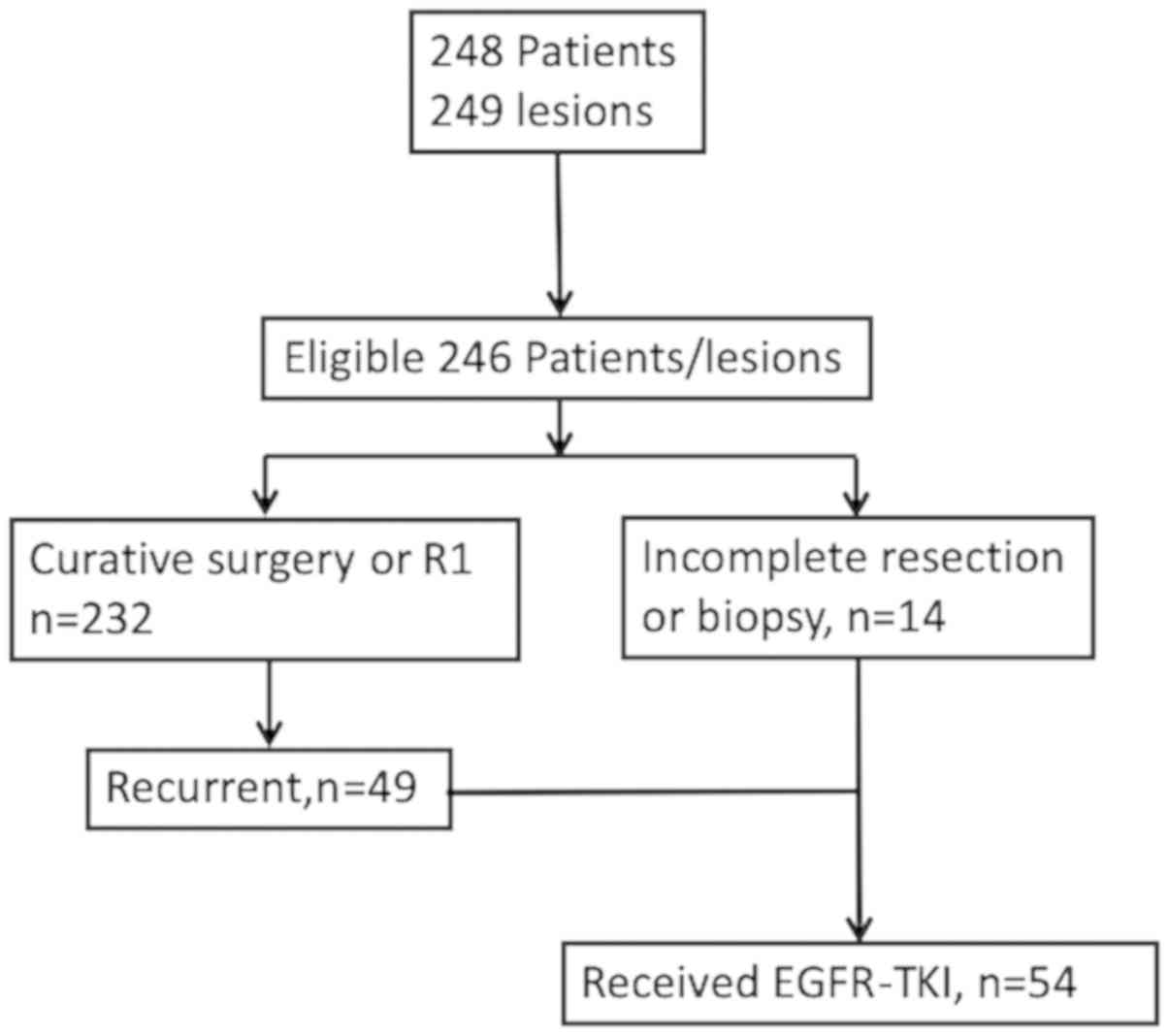
Between January 2007 and December 2015, we
identified 248 patients, of which 246 patients were eligible for
this study and included in the analyses. Of the 232 patients who
had undergone complete or microscopically incomplete resection,
recurrence was found in 49 patients. Surgery with incomplete
resection or tumor biopsy was performed in 14 patients. Of the 63
advanced or recurrent NSCLC patients, 54 patients received EGFR-TKI
therapy (Fig. 1). Demographic data
and clinicopathological characteristics of patients are presented
in Table I: Age at surgery or biopsy
ranged from 37 to 90 years (median age: 67 years). There were 159
(64.6%) women, 166 (67.4%) never smokers, 240 (97.6%) patients with
adenocarcinoma, and 193 (78.4%) patients with pathological stage I
(Table I).
 | Table I.Clinicopathologic, genetic and
histologic characteristics of patients. |
Table I.
Clinicopathologic, genetic and
histologic characteristics of patients.
| A,
Clinicopathologic characteristic |
|---|
|
|---|
| Characteristic | Total (n=246) | Received EGFR-TKI
therapy(n=44) |
|---|
| Sex |
|
|
|
Male | 87 (35.4) | 14 (31.8) |
| Age (y) |
|
|
| Median
(range) | 67.0 (37–90) | 65.5 (37–82) |
| Smoking status |
|
|
|
Never | 166 (67.4) | 30 (68.2) |
|
Former | 67 (27.2) | 11 (25.0) |
|
Current | 13 (5.3) | 3 (6.8) |
| Pathological
subtypes |
|
|
|
Adeno | 240 (97.6) | 42 (95.5) |
|
AdSq | 2 (0.8) | 2 (4.5) |
| Sq | 4 (1.6) | 0 (0.0) |
| p-Stage at initial
surgery |
|
|
| I | 193 (78.4) |
|
| II | 16 (6.5) |
|
|
III | 24 (9.7) |
|
| IV | 12 (4.9) |
|
|
| B, EGFR
mutation |
|
|
Characteristic | Total
(n=246) | Received
EGFR-TKI therapy(n=44) |
|
| C797S | 0 (0.0) | 0 (0.0) |
| Ex19del | 86 (35.0) | 21 (47.7) |
| Ex19del+T790M | 1 (0.4) | 0 (0.0) |
| L858R | 134 (54.5) | 20 (45.5) |
| L858R+G719S | 1 (0.4) | 1 (2.3) |
| L858R+T790M | 4 (1.6) | 0 (0.0) |
| T790M | 7 (2.8) | 0 (0.0) |
| G719A | 3 (1.2) | 0 (0.0) |
| G719C | 1 (0.4) | 0 (0.0) |
| L861Q | 1 (0.4) | 1 (2.3) |
| G719A+S768I | 2 (0.8) | 0 (0.0) |
| G719A+L861Q | 2 (0.8) | 1 (2.3) |
| G719C+L861Q | 1 (0.4) | 0 (0.0) |
|
| C, Gene
amplification |
|
|
Characteristic | Total
(n=246) | Received
EGFR-TKI therapy (n=44) |
|
| ERBB2 | 1 (0.4) | 1 (2.3) |
| MET | 0 (0.0) | 0 (0.0) |
| EGFR | 5 (2.0) | 3 (6.8) |
| ALK | 0 (0.0) | 0 (0.0) |
| RET | 0 (0.0) | 0 (0.0) |
| BRAF | 0 (0.0) | 0 (0.0) |
| FGFR1 | 1 (0.4) | 0 (0.0) |
| MYC | 2 (0.8) | 1 (2.3) |
| CCND1 | 2 (0.8) | 2 (4.5) |
| CCND2 | 1 (0.4) | 1 (2.3) |
| CDK4 | 15 (6.0) | 4 (9.1) |
| MDM2 | 17 (6.8) | 6 (13.6) |
| MDM4 | 2 (0.8) | 1 (2.3) |
EGFR C797S and T790M mutations
No patients harbored concurrent C797S mutation with
known EGFR mutations. T790M mutation was identified in 12
patients (4.9%); 5 patients had the deletion in exon19 or L858R and
7 patients had T790M mutation alone (Table I).
Gene amplification
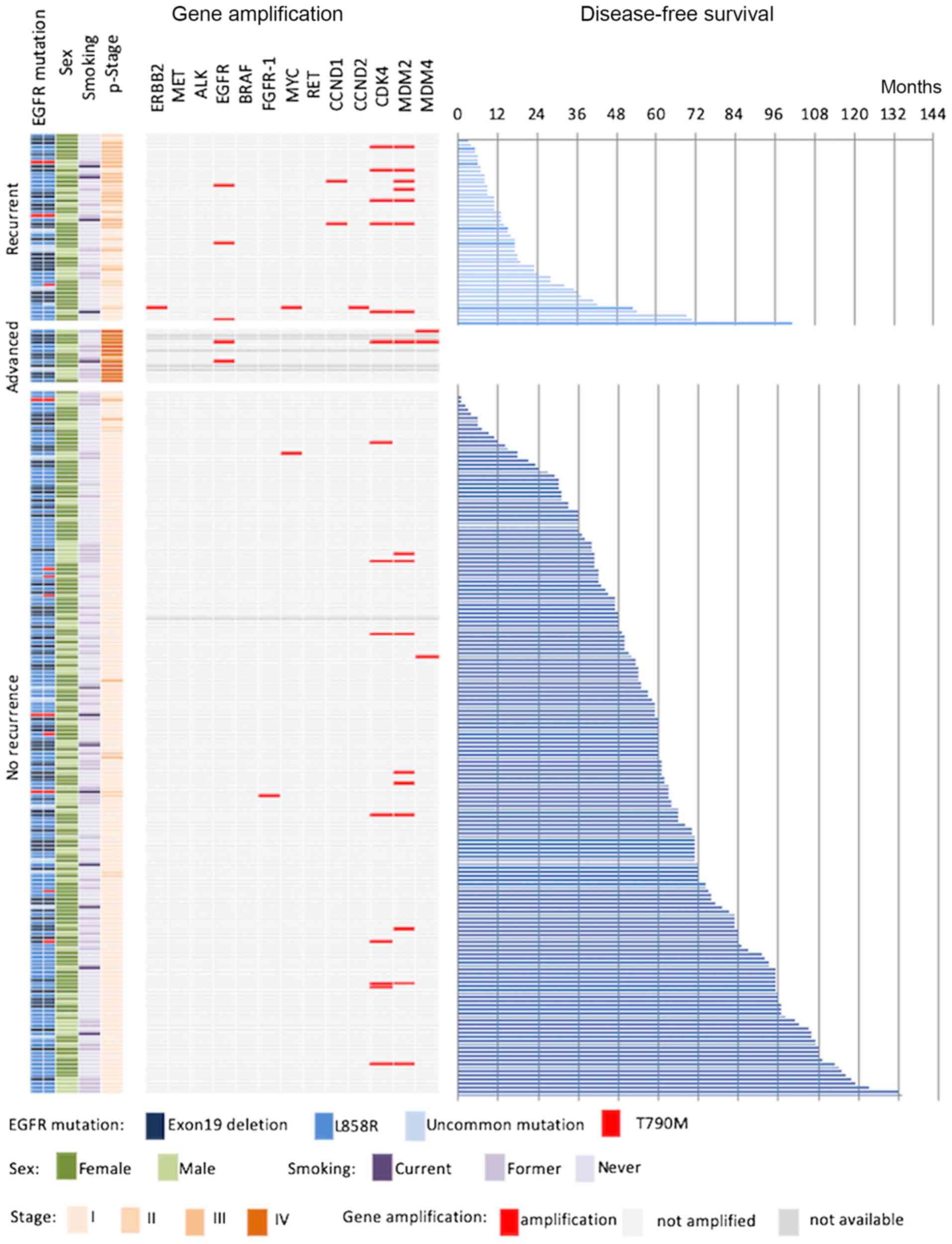
EGFR gene amplification was found in five
patients. All patients with EGFR gene amplification had
advanced disease or recurrence (9.8 vs. 0.0%, P<0.01).
ERBB2 gene amplification was found in only one patient
(0.4%) who developed recurrence. No patients harbored MET
gene amplification. MDM2 gene amplification was found in 17
patients (7.1%) and CDK4 gene amplification in 14 patients
(5.8%); duplication of both MDM2 and CDK4 was
observed in 11 patients (4.6%). MDM2 gene amplification was
significantly associated with tumor recurrence (16.7 vs. 5.1%,
P=0.032). Patients with tumors with any gene amplification had
significantly more advanced disease or developed recurrence
compared with patients without recurrence after surgery (33.3 vs.
8.9%, P=0.002) (Fig. 2).
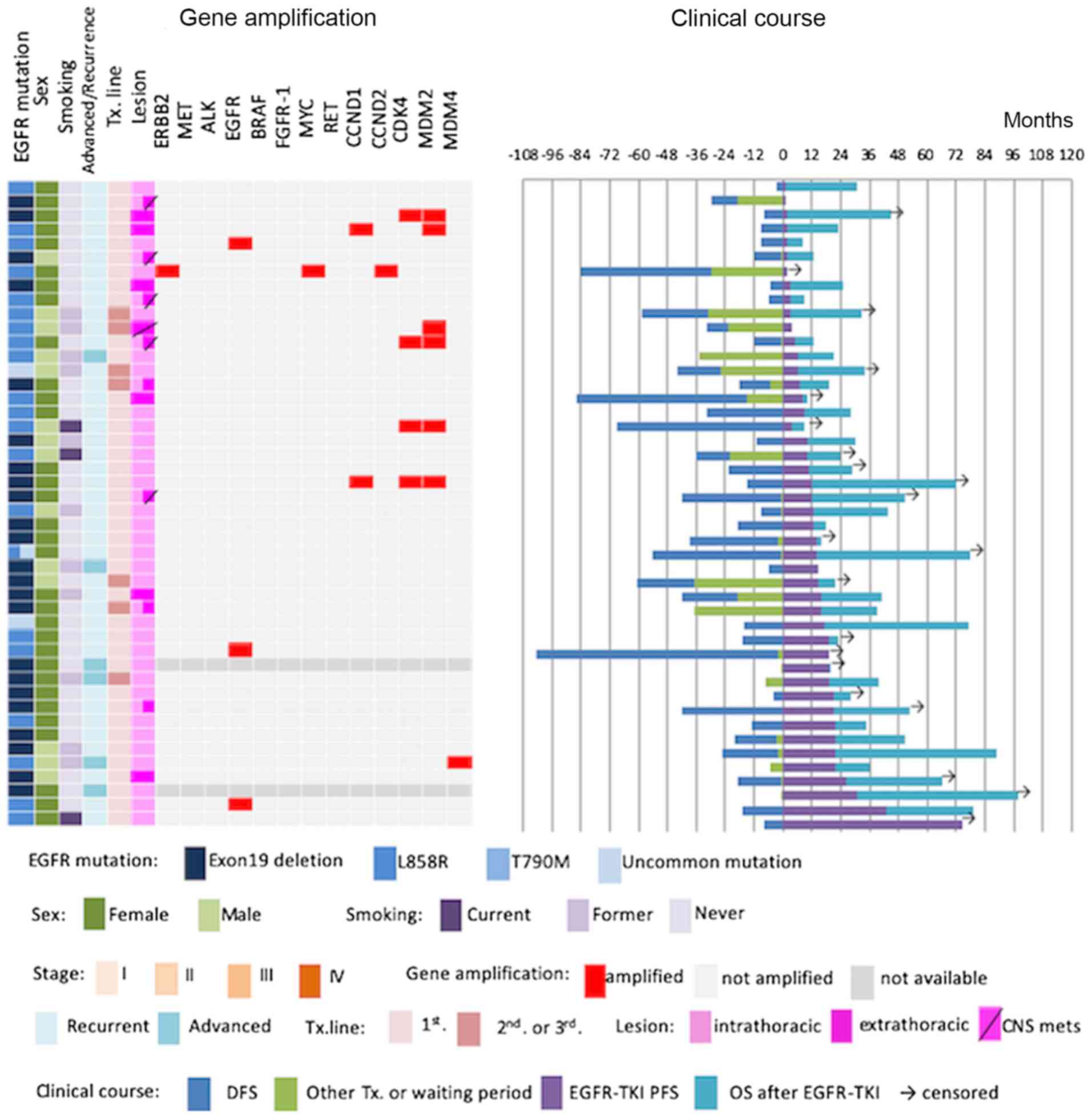
We next evaluated associations between
clinicopathological parameters and/or gene amplification and
progress of first- and second-generation EGFR-TKI treatment.
Fifty-four patients had received EGFR-TKI therapy, and 44 patients
underwent gene amplification analysis and were followed up.
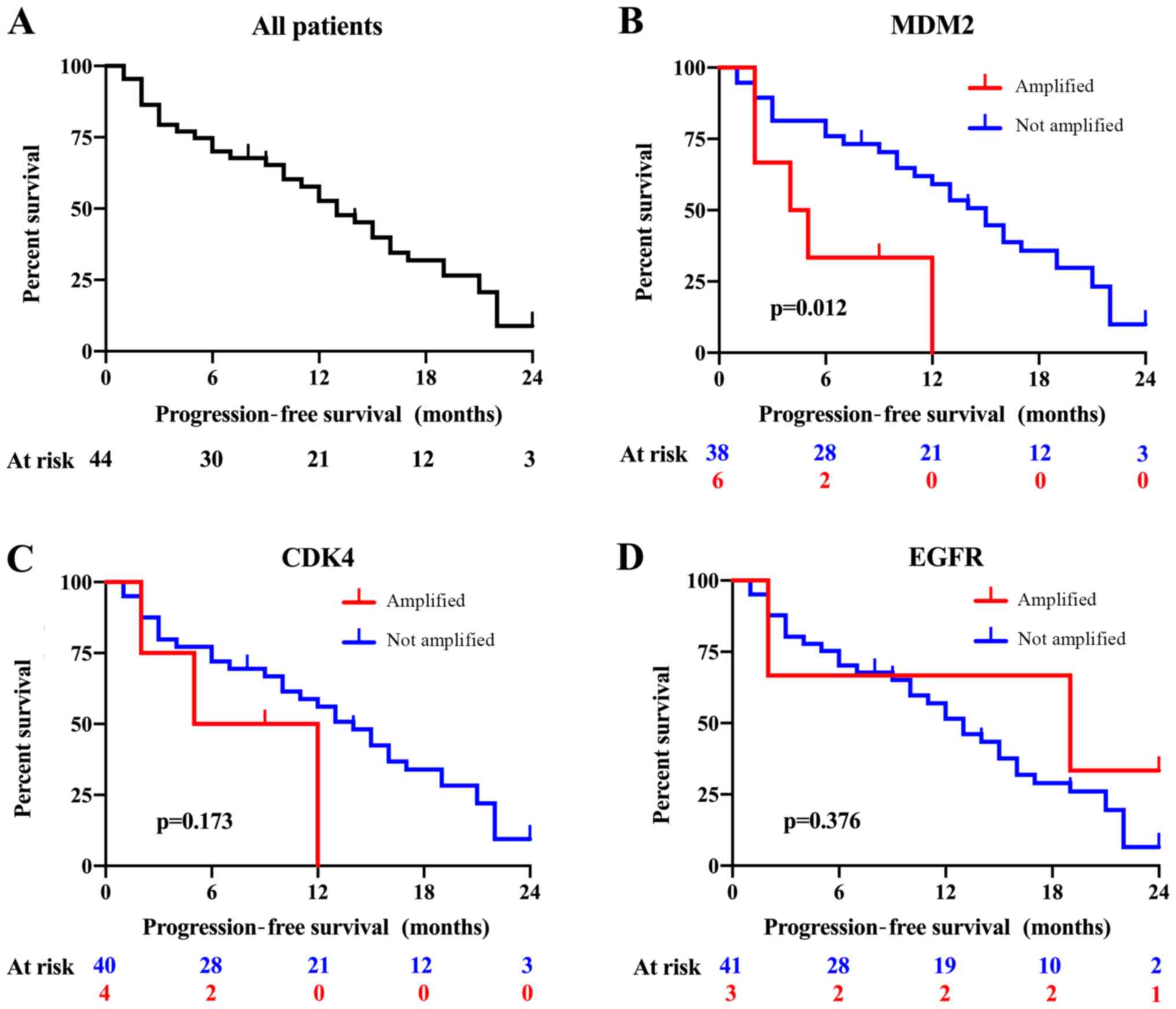
Patients who developed central nervous system metastasis (HR,
2.259, 95% CI, 1.150–4.436) and had MDM2 gene amplification
(HR, 3.405, 95% CI, 1.209–9.591) exhibited significantly shorter
PFS (Figs. 3,4). Patients who had EGFR gene
amplification (HR, 0.656, 95% CI, 0.195–2.210), ERBB2 gene
amplification (HR, 0.801, 95% CI, incalculable), CDK4 gene
amplification (HR, 0.194, 95% CI, 0.660–7.802) (Figs. 3,4),
CCND1 gene amplification (HR, 2.823 95% CI, 0.653–12.203),
CCND2 gene amplification (HR, 0.801, 95% CI, incalculable),
CDK4 gene amplification (HR, 0.194, 95% CI, 0.660–7.802),
and MDM4 gene amplification (HR, 0.465, 95% CI, 0.063–3.441)
(data not shown) showed no significant difference in PFS (Figs. 3,4).
Discussion
We herein showed that in our study cohort no
EGFR-TKI-naïve NSCLC patients harbored concurrent EGFR C797S
mutation with known EGFR gene mutation. Amplification of
several genes was found before EGFR-TKI therapy, and MDM2
gene amplification was associated with resistance to
first-generation EGFR-TKIs.
C797S mutation, which leads to acquired resistance
to third-generation EGFR TKIs, occurs in 5.3–40.0% of NSCLC
patients following osimertinib treatment (2,3,6,11,20) and
is the most common resistance mechanism against osimertinib. No
concurrent C797S mutation was found in this study, and, to our
knowledge, only one case of de novo somatic L858R and C797S
mutations has been reported (8).
C797S mutation is an important challenge for EGFR mutant
NSCLC therapy. A recent report suggests that therapeutic efficacy
is dependent on allelic context of common EGFR mutations,
C797S and T790M. If T790M and C797S mutations occur on separate
alleles, then combination therapy comprising first- and
third-generation EGFR-TKIs can restore EGFR inhibition in
vitro (21). Unfortunately,
these mutations almost always occur on the same allele (22). Conversely, combination therapy
composed of osimertinib, bevacizumab, and brigatinib is effective
for NSCLC with T790M and C797S mutations occurring on the same
allele (23). Combination therapy
comprising an allosteric inhibitor and cetuximab was also shown to
be effective in a murine model of NSCLC driven by EGFR
L858R, T790M, and/or C797S mutation (24). Furthermore, other tertiary mutations
of EGFR [L792X (11), G796D
(9), L718Q, and L844V (5)] in the EGFR tyrosine kinase domain were
reported, and novel therapies for tumors with these mutations are
required.
Other resistance mechanisms against third-generation
EGFR-TKIs involving activation of signaling pathways have been
reported. Lin et al (2) and
Ramalingam et al (3) carried
out plasma-based analyses after development of osimertinib
resistance in NSCLC. The following gene alterations were noted:
EGFR C797S mutation in 5.2–16.7%, KRAS mutation in
2.6–7.7%, BRAF mutation in 7.7%, PIK3CA mutation in
2.6%, MEK mutation in 2.6%, JAK2 mutation in 2.6%,
MET amplification in 2.6–50%, KRAS amplification in
2.6%, ERBB2 insertion in 2.6%, transformation to SCC in
9.1%, and transformation to SqCC in 4.5% of patients (2,3).
Furthermore, analysis of matched samples of pre- and
post-administration of first- or second-generation EGFR-TKIs showed
acquired resistance in addition to T790M mutation in lung
adenocarcinoma, MET amplification in 8%, ERBB2
amplification in 5%, and EGFR amplification in 16% of
patients (15). Transformation to
SCC was noted in 2.6–5.0% of patients (15), and another report demonstrated SqCC
rarely occurred (17).
The genetic alterations described above occurred in
some pretreatment EGFR mutant NSCLC tumors and patients with these
mutations can exhibit resistance to osimertinib therapy. Concurrent
gene alterations in EGFR-TKI-naïve, EGFR mutant NSCLC have
been reported. Yu et al, conducted next-generation
sequencing analysis of tissue samples and revealed EGFR
mutant NSCLC had concurrent alteration of TP53 mutation in
60%, RB1 mutation in 10%, PIK3CA mutation in 12%,
CTNNB1 mutation in 18.9%, EGFR amplification in 22%,
MDM2 mutation in 12%, CDK4 mutation in 10%,
ERBB2 amplification in 8.4%, and MET amplification in
2% of patients before EGFR-TKI treatment (15).
Gene amplification of ERBB2, MET, MDM2 or
CDK4 alone leads to poor prognosis for NSCLC regardless of
EGFR gene mutation (25–27).
Furthermore, Le et al (11)
and Blakely et al (14)
reported that cell cycle gene alteration, such as CDK4 or
CDK6, shortens PFS following osimertinib therapy. Patients
with EGFR mutant NSCLC accompanied by TP53 mutation,
ERBB2 amplification, or MET amplification before
first- or second-generation EGFR-TKI treatment exhibited a shorter
time to progression and also showed resistance to third-generation
EGFR-TKIs (12,13,28).
In this study, MDM2 gene amplification was
shown to be associated with shorter PFS for first- or
second-generation EGFR-TKIs. MDM2 is a proto-oncogene that is often
coexpressed with CDK4 in liposarcoma (29). MDM2 binds to TP53 to downregulate
transcription, leading to proteasome degradation by ubiquitination
(30).
The limitations of this study are as follows: First,
we did not evaluate EGFR mutations associated with acquired
resistance to EGFR-TKIs other than C797S (e.g., L792X, G796X and
L718Q), exon 20 insertion, mutation in driver genes KRAS,
BRAF and PIK3CA, or transformation to mesenchyme or
other pathological subtypes. Furthermore, the study cohort was
limited to NSCLC patients harboring known EGFR gene
mutations. Therefore, whether C797S mutation alone could be an
oncogenic mechanism remains unclear. Second, this study did not
evaluate single nucleotide polymorphisms or germline gene
mutations. As only seven patients had EGFR T790M mutation,
they could have germline mutations (7) rather than somatic mutations.
Finally, because our survival analysis was conducted
in a small cohort, it is possible that amplification of genes other
than MDM2, such as MDM4 and ERBB2 evaluated in
this study, could be biomarkers for EGFR-mutant NSCLC.
In conclusion, no concurrent EGFR-C797S with known
EGFR mutant NSCLC was identified in our study cohort. This result
confirms the efficacy of third-generation EGFR-TKIs; however, it is
important to remain aware of how genetic alterations can affect
EGFR-TKI responses. Therefore, further studies in a large cohort
are required to completely elucidate resistance mechanisms against
third-generation EGFR-TKIs.
Acknowledgements
The authors would like to thank Ms. Mie Otsuki, Ms.
Eiko Otomo and Ms. Yukiko Kikuta (Department of Chest Surgery,
Fukushima Medical University) for their technical supports
Funding
This work was supported by grant from AstraZeneca.
K.K. (Osaka, Japan; grant no. ESR-16-12328).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TY, SM and HS designed the study. TY, HM, HT, MW,
YO, TI, MF, NO, YM, TH, JO, MHo, MHi and YS collected and analyzed
the data. TY and HS wrote and revised the manuscript. All the
authors approved the final manuscript.
Ethics approval and consent to
participate
The current study was conducted with approval of the
ethics board at Fukushima Medical University (approval no. 2955).
Human rights and welfare of participants were protected in
accordance with the Declaration of Helsinki, and written informed
consent was obtained from participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yang JC, Ahn MJ, Kim DW, Ramalingam SS,
Sequist LV, Su WC, Kim SW, Kim JH, Planchard D, Felip E, et al:
Osimertinib in pretreated T790M-Positive advanced non-small-cell
lung cancer: AURA study phase II extension component. J Clin Oncol.
35:1288–1296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin CC, Shih JY, Yu CJ, Ho CC, Liao WY,
Lee JH, Tsai TH, Su KY, Hsieh MS, Chang YL, et al: Outcomes in
patients with non-small-cell lung cancer and acquired Thr790Met
mutation treated with osimertinib: A genomic study. Lancet Respir
Med. 6:107–116. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ramalingam SS, Yang JC, Lee CK, Kurata T,
Kim DW, John T, Nogami N, Ohe Y, Mann H, Rukazenkov Y, et al:
Osimertinib as first-line treatment of EGFR mutation-positive
advanced non-small-cell lung cancer. J Clin Oncol. 36:841–849.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Soria JC, Ohe Y, Vansteenkiste J,
Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura
F, Nogami N, Kurata T, et al: Osimertinib in untreated EGFR-mutated
advanced non-small-cell lung cancer. N Engl J Med. 378:113–125.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thress KS, Paweletz CP, Felip E, Cho BC,
Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et
al: Acquired EGFR C797S mutation mediates resistance to AZD9291 in
non-small cell lung cancer harboring EGFR T790M. Nat Med.
21:560–562. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Su KY, Chen HY, Li KC, Kuo ML, Yang JC,
Chan WK, Ho BC, Chang GC, Shih JY, Yu SL and Yang PC: Pretreatment
epidermal growth factor receptor (EGFR) T790M mutation predicts
shorter EGFR tyrosine kinase inhibitor response duration in
patients with non-small-cell lung cancer. J Clin Oncol. 30:433–440.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee JS, Hur JY, Kim HJ, Lee KY and Kim WS:
A case of concurrent de novo C797S and L858R EGFR mutation detected
in stage IA non-small cell lung cancer patient. J Thorac Oncol.
12:e179–e181. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng D, Hu M, Bai Y, Zhu X, Lu X, Wu C,
Wang J, Liu L, Wang Z, Ni J, et al: EGFR G796D mutation mediates
resistance to osimertinib. Oncotarget. 8:49671–49679.
2017.PubMed/NCBI
|
|
10
|
Ercan D, Choi HG, Yun CH, Capelletti M,
Xie T, Eck MJ, Gray NS and Jänne PA: EGFR mutations and resistance
to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res.
21:3913–3923. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Le X, Puri S, Negrao MV, Nilsson MB,
Robichaux J, Boyle T, Hicks JK, Lovinger KL, Roarty E,
Rinsurongkawong W, et al: Landscape of EGFR-Dependent and
-independent resistance mechanisms to osimertinib and continuation
therapy beyond progression in EGFR-Mutant NSCLC. Clin Cancer Res.
24:6195–6203. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takezawa K, Pirazzoli V, Arcila ME, Nebhan
CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ,
Melnick MA, et al: HER2 amplification: A potential mechanism of
acquired resistance to EGFR inhibition in EGFR-mutant lung cancers
that lack the second-site EGFRT790M mutation. Cancer Discov.
2:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ou SI, Agarwal N and Ali SM: High MET
amplification level as a resistance mechanism to osimertinib
(AZD9291) in a patient that symptomatically responded to crizotinib
treatment post-osimertinib progression. Lung Cancer. 98:59–61.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Blakely CM, Watkins TBK, Wu W, Gini B,
Chabon JJ, McCoach CE, McGranahan N, Wilson GA, Birkbak NJ, Olivas
VR, et al: Evolution and clinical impact of co-occurring genetic
alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet.
49:1693–1704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu HA, Suzawa K, Jordan E, Zehir A, Ni A,
Kim R, Kris MG, Hellmann MD, Li BT, Somwar R, et al: Concurrent
alterations in EGFR-Mutant lung cancers associated with resistance
to EGFR kinase inhibitors and characterization of MTOR as a
mediator of resistance. Clin Cancer Res. 24:3108–3118. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Okabe N, Takagi H, Mine H, Fukai S,
Minemura H and Suzuki H: Osimertinib for epidermal growth factor
receptor mutation-positive lung adenocarcinoma that transformed to
T790M-Positive squamous cell carcinoma. J Thorac Oncol.
12:e167–e169. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Watanabe K, Fukuhara T, Tsukita Y, Morita
M, Suzuki A, Tanaka N, Terasaki H, Nukiwa T and Maemondo M: EGFR
mutation analysis of circulating tumor DNA using an improved
PNA-LNA PCR clamp method. Can Respir J. 2016:52973292016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schouten JP, McElgunn CJ, Waaijer R,
Zwijnenburg D, Diepvens F and Pals G: Relative quantification of 40
nucleic acid sequences by multiplex ligation-dependent probe
amplification. Nucleic Acids Res. 30:e572002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oxnard GR, Hu Y, Mileham KF, Husain H,
Costa DB, Tracy P, Feeney N, Sholl LM, Dahlberg SE, Redig AJ, et
al: Assessment of resistance mechanisms and clinical implications
in patients with EGFR T790M-Positive lung cancer and acquired
resistance to osimertinib. JAMA Oncol. 4:1527–1534. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Niederst MJ, Hu H, Mulvey HE, Lockerman
EL, Garcia AR, Piotrowska Z, Sequist LV and Engelman JA: The
allelic context of the C797S mutation acquired upon treatment with
third-generation EGFR inhibitors impacts sensitivity to subsequent
treatment strategies. Clin Cancer Res. 21:3924–3933. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yatabe Y, Takahashi T and Mitsudomi T:
Epidermal growth factor receptor gene amplification is acquired in
association with tumor progression of EGFR-mutated lung cancer.
Cancer Res. 68:2106–2111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao J, Zou M, Lv J, Han Y and Wang G and
Wang G: Effective treatment of pulmonary adenocarcinoma harboring
triple EGFR mutations of L858R, T790M, and cis-C797S by
osimertinib, bevacizumab, and brigatinib combination therapy: A
case report. Onco Targets Ther. 11:5545–5550. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jia Y, Yun CH, Park E, Ercan D, Manuia M,
Juarez J, Xu C, Rhee K, Chen T, Zhang H, et al: Overcoming
EGFR(T790M) and EGFR(C797S) resistance with mutant-selective
allosteric inhibitors. Nature. 534:129–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sholl LM, Yeap BY, Iafrate AJ,
Holmes-Tisch AJ, Chou YP, Wu MT, Goan YG, Su L, Benedettini E, Yu
J, et al: Lung adenocarcinoma with EGFR amplification has distinct
clinicopathologic and molecular features in never-smokers. Cancer
Res. 69:8341–8348. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dworakowska D, Jassem E, Jassem J, Peters
B, Dziadziuszko R, Zylicz M, Jakóbkiewicz-Banecka J,
Kobierska-Gulida G, Szymanowska A, Skokowski J, et al: MDM2 gene
amplification: A new independent factor of adverse prognosis in
non-small cell lung cancer (NSCLC). Lung Cancer. 43:285–295. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Turke AB, Zejnullahu K, Wu YL, Song Y,
Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L,
et al: Preexistence and clonal selection of MET amplification in
EGFR mutant NSCLC. Cancer Cell. 17:77–88. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Planchard D, Loriot Y, André F, Gobert A,
Auger N, Lacroix L and Soria JC: EGFR-independent mechanisms of
acquired resistance to AZD9291 in EGFR T790M-positive NSCLC
patients. Ann Oncol. 26:2073–2078. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dei Tos AP, Doglioni C, Piccinin S, Sciot
R, Furlanetto A, Boiocchi M, Dal Cin P, Maestro R, Fletcher CD and
Tallini G: Coordinated expression and amplification of the MDM2,
CDK4, and HMGI-C genes in atypical lipomatous tumours. J Pathol.
190:531–536. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Deben C, Deschoolmeester V, Lardon F,
Rolfo C and Pauwels P: TP53 and MDM2 genetic alterations in
non-small cell lung cancer: Evaluating their prognostic and
predictive value. Crit Rev Oncol Hematol. 99:63–73. 2016.
View Article : Google Scholar : PubMed/NCBI
|