Introduction
Lung cancer is the leading cause of
cancer-associated mortality among males and the second leading
cause of cancer-associated mortality among females globally, with
an estimated 1.8 million new cases, and 1.6 million mortalities in
2012 (1). In the past 30 years, the
number lung cancer-associated deaths in China has increased by
464.84%, and ~600,000 individuals die of lung cancer each year
(2). In recent years, with the
advances in early diagnosis and clinical treatment techniques, lung
cancer treatment and diagnosis has improved (3). However, the number of patients with
lung cancer in China has grown at a rate of 0.9% per year between
2000 and 2011 (2). Due to China's
large population, such a growth rate causes a huge economic burden
on public health.
Non-small cell lung cancer (NSCLC) includes large
cell lung cancer, adenocarcinoma and squamous cell carcinoma,
accounting for ≤85% of lung cancer cases (4). For NSCLC, surgery is the best treatment
(5). However, only 25–30% of
patients are suitable for potentially curative resection (5). At present, the 5-year survival rates of
patients with stage Ia and IIIa NSCLC undergoing resection are 75
and 25%, respectively (5).
Therefore, the development of new clinical treatments and methods
of early diagnosis for lung cancer is essential.
The occurrence of lung cancer is associated with
smoking, radon, air pollution and occupational factors like
asbestos. Recently, several studies have reported that the process
of carcinogenesis is driven by the accumulation of genetic
mutation, such as K-Ras and EGFR, as well as epigenetic changes
(6–8). Epigenetic changes include DNA
methylation, histone modifications and expression of non-coding RNA
(9). Tumor development is primarily
associated with the inactivation of tumor suppressor genes and the
activation of oncogenes (8). DNA
methylation in the promoter region can prevent the activation of a
gene, leading to a downregulation of its expression (10). Previous studies have indicated that
the primary markers of lung cancer include the inactivation of
various tumor suppressor gene promoters by methylation. For
example, aberrantly methylated genes associated with homeoboxes are
frequently observed in early stage lung cancer, including SIX
homeobox (SIX), LIM homeobox (LHX), paired box (PAX) and
distal-less homeobox (DLX) (11). Hypermethylated tumor suppressor genes
commonly found in advanced non-small cell lung cancer include:
Helicase-like transcription factor (HLTF), adenovirus E1B 19 kDa
interacting protein 3 (BNIP3), member X in H2A histone family
(H2AFX), calcium voltage-gated channel subunit alpha1 G (CACNA1G),
TGFB-induced factor homebox 1 (TGIF), inhibitor of DNA binding 4
(ID4) and calcium voltage-gated channel subunit alpha1 A
(CACNA1A) (12). The present
study aimed to identify aberrantly methylated genes in tumors, and
then to develop diagnostic markers for NSCLC in plasma or other
body fluids.
Tumor necrosis factor (TNF) receptor superfamily
member 10c (TNFRSF10C) and TNFRSF10D are decoy receptors for the
TNF-related apoptosis-inducing ligand (TRAIL), a member of the TNF
family (13). TNFRSF10C and
TNFRSF10D bind to TRAIL and do not contain the death domain
necessary for apoptosis; thus, they inhibit TRAIL-induced apoptosis
by competing with the TRAIL receptors (TRAIL-R) death receptor 4
(DR4) and DR5 for binding TRAIL (13). TRAIL and its associated receptors
have been used as targets for anticancer therapeutics (14). The tolerability and safety of TRAIL
was determined in clinical phase I studies, and no dose-limiting
toxicity was observed (15,16). TRAIL-R agonists can be used as a
single agent or in combination with conventional chemotherapy drugs
for advanced NSCLC (17), colorectal
cancer (18), B-cell lymphoma
(19) and advanced or metastatic
solid tumors or non-Hodgkin's lymphoma (NHL) (20). TRAIL ligands are widely expressed in
tumor tissues and have attracted attention due to their ability to
selectively induce tumor cell apoptosis (21).
The methylation levels of TNFRSF10C and
TNFRSF10D in lung cancer have been studied in lung
adenocarcinoma (22). A previous
study explored tumor-associated genes, in which TNFRSF10C
methylation levels were higher in patients with lung adenocarcinoma
who were non-smokers compared with those who were smokers (22). However, the aforementioned study did
not compare the methylation levels of TNFRSF10C and
TNFRSF10D genes between lung cancer and normal tissues. In
the present study, the association between TNFRSF10C and
TNFRSF10D methylation levels and NSCLC was investigated by
analyzing NSCLC and distant non-tumor tissues from patients from
The First Affiliated Hospital of Fujian Medical University (Fuzhou,
China) and The Cancer Genome Atlas (TCGA) database. The aim of the
present study was to identify the contribution of methylation of
the aforementioned genes to NSCLC pathogenesis.
Materials and methods
Tissue sample collection
A total of 44 pairs of tumor tissues and distant
non-tumor tissues were collected from June 2017 to September 2018.
These samples were obtained from 44 patients with NSCLC at The
First Affiliated Hospital of Fujian Medical University (Fuzhou,
China). Patient clinicopathological information was collected,
including age, sex, family history. The average age of the patients
was 62.91±9.05 years (mean ± SD; age range, 42–87 years), including
23 female and 21 male patients. There were 32 cases of
adenocarcinoma, 12 cases of squamous cell carcinoma and 2
inflammatory pseudotumors. The research protocol was approved by
The Ethics Committee of the First Affiliated Hospital of Fujian
Medical University (Fuzhou, China; approval number, 2017-KY-068).
All subjects provided written informed consent. Samples were taken
during resection and the sampling procedure was performed under the
guidance of a pathologist to distinguish between tumor tissues and
distant non-tumor tissues (>10 cm away from the lesions).
Quantitative methylation-specific PCR
(qMSP) assay
Primer sequences were designed based on the region
of the TNFRSF10C and TNFRSF10D genes rich in CG
sites. The primer sequences were as follows: TNFRSF10C
forward, 5′-AGGGTGCGATTTAGGATTTAG-3′ and reverse,
5′-CGATAACGACGACGAACTT5′; TNFRSF10D forward,
5′-CGACGATGAAGACGACGAAT-3′ and reverse,
5′-AAACCAAACCATAACTCCTAAACC-3′. The conditions for qMSP were as
follows: Initial denaturation at 95°C for 10 min, denaturation at
95°C for 20 sec, annealing at 56°C for 20 sec and extension at 72°C
for 30 sec for 45 cycles. The melting curve program included 95°C
for 15 sec, 60°C for 60 sec and then from 60°C to 95°C at a speed
of 0.11°C/sec.
DNA was extracted from patient tissue samples
according to the instructions of the QIAamp DNA FFPE Tissue kit
(Qiagen GmbH). The extracted samples were measured for purity and
concentration using a NanoDrop 1000 spectrophotometer (Thermo
Fisher Scientific, Inc.). The DNA concentration of all samples was
>25 ng/µl, and the purity of DNA was 1.8–2.0 based on the
absorbance ratio at 260 and 280 nm. The DNA was subjected to
bisulfite modification using the EZ DNA Methylation-Gold™ kit
according to the manufacturer's instructions (Zymo Research Corp.).
The primers and the bisulfite-modified DNA were then subjected to
qMSP. The total reaction system was 10 µl, containing 5 µl SYBR
mixture, 4 µl double-distilled H2O, 0.5 µl each of the
forward and reverse primers and 0.5 µl modified DNA. To avoid
errors in sample loading, ACTB was used as an internal
reference for each sample. The primer sequences of ACTB were as
follows: Forward, 5′-TGGTGATGGAGGAGGTTTAGTAAGT-3′ and reverse,
5′-AACCAATAAAACCTACTCCTCCCTTAA-3′. A total of 100% M-Sss I (New
England BioLabs, Inc.) treated sperm DNA was used as the positive
control and nuclease-free water was used as the negative control
for each group. The qMSP product was then subjected to Qsep100 DNA
fragment analysis (BiOptic, Inc.) and visualized. Three randomly
picked qMSP products of each gene were Sanger sequenced for
validation.
ACTB is a housekeeping gene that is often
stably expressed in most healthy and tumor cells; thus, it is
commonly used as an internal reference for quantitation of mRNA
expression levels and qMSP assays (23). Completely methylated DNA referred to
human sperm DNA treated with methylated-SssI, which allowed all C
sites in the DNA to be methylated, including CG sites, and thus was
used as a positive control. The formula for quantifying methylation
levels was used to calculate the relative methylation level by the
comparison between the target gene and the ACTB gene
(24,25). For each sample, the relative
methylation value was determined using the 2−∆∆Cq
method. The sample methylation reference percentage (PMR) was
calculated using the following formula: PMR=2−∆∆Cq
×100%; ∆∆Cq=sample DNA (Cqgene-CqACTB)-fully
methylated DNA (Cqgene-CqACTB).
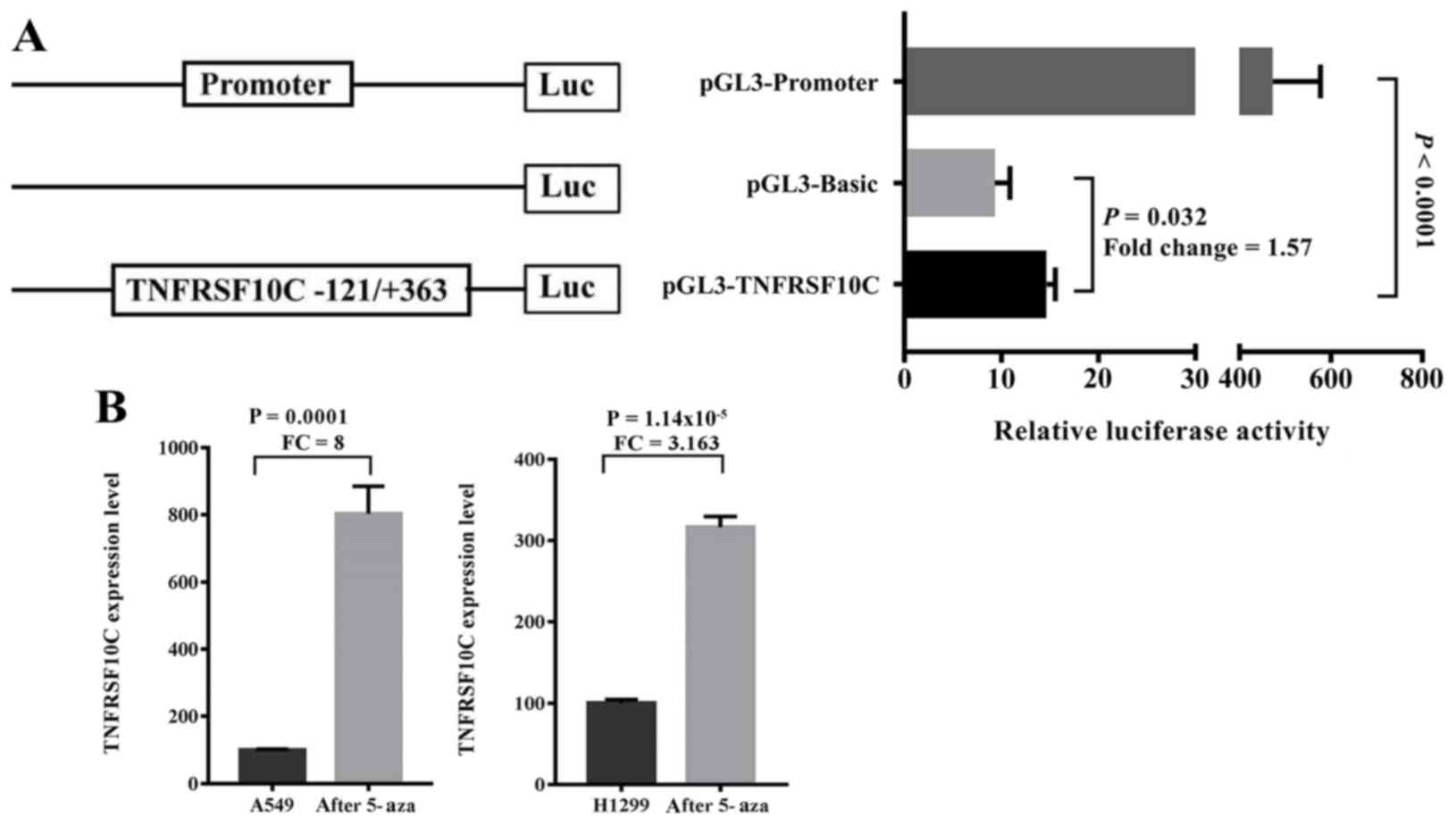
Luciferase reporter gene assay
A recombinant plasmid pGL3-TNFRSF10C containing the
selected fragment (−121 to +363 bp of TNFRSF10C, 485 bp) was
constructed. The target plasmid pGL3-TNFRSF10C was co-transfected
with the pRL-SV40 plasmid carrying the Renilla luciferase
gene to provide an internal control. Cells co-transfected with the
empty pGL3-Basic plasmid and the pRL-SV40 plasmid were used as
negative references in each experiment. The pGL3-Promoter plasmid
and the pRL-SV40 plasmid were also co-transfected as a positive
control each time. The plasmids were separately transferred into
293T cells (10% FBS, 37°C, 5% CO2; China Center for Type
Culture Collection) using FuGENE® HD Transfection
Reagent (Promega Corporation), according to the manufacturer's
protocol. The target plasmid (1 µg) and the Renilla luciferase
plasmid (0.1 µg) were transfected into 293T cells at a ratio of
10:1 using FuGENE® HD. After 24 h of transfection,
luciferase activity was detected using a dual luciferase assay kit
(Promega Corporation), according to the manufacturer's protocol.
The luciferase reaction intensity was measured at a wavelength of
~560 nm, using a SpectraMax 190 microplate reader (Molecular
Devices) by adding the Luciferase Assay Reagent II. After the
fluorescence reaction intensity was measured, the Stop&Glo
Reagent was added to the same sample to quench the aforementioned
reaction, and the Renilla luciferase reaction was
simultaneously initiated for the second measurement. Luciferase
activity was finally calculated using the Dual Luciferase Reporter
Assay system (Promega Corporation) and normalized to Renilla
luciferase activity.
Cell culture and gene expression
detection
The embryonic cells A549 and NCI-H1299 were
purchased from China Center for Type Culture Collection, and the
cells were treated with 5 µmol/l 5′-azadeoxycytidine (5-Aza; A3656,
Sigma-Aldrich; Merck KGaA) for 24 h at 37°C (25). RNA was extracted by
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
before and after 5-Aza treatment, and cDNA was obtained by using a
PrimeScript RT kit (Takara Biotechnology Co., Ltd.). The
quantitative PCR primers sequences (Sangon Biotech Co., Ltd.) were
as follows: TNFRSF10C forward, 5′-TGCACAGAGGGTGTGGATTAC-3′
and reverse, 5′-ATTCCGGAAGGTGCCTTCTTT-3′; ACTB forward,
5′-AGCACAGAGCCTCGCCTTT-3′ and reverse, 5′-AGGGTGAGGATGCCTCTCTT-3′.
The Real-time PCR system was configured using the Takara TB Green
kit (Takara Biotechnology Co., Ltd.), and the mRNA expression
levels of TNFRSF10C were detected using a StepOne Plus
real-time PCR instrument (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions for qPCR were as
follows: Initial denaturation at 95°C for 10 min, 45 cycles of
denaturation at 95°C for 20 sec, annealing at 56°C for 20 sec,
extension at 72°C for 30 sec and a final extension at 72°C for 10
min. Relative expression levels were calculated using the
2−ΔΔCq method (25) and
normalized to internal reference gene β-actin. A negative reference
was also prepared using nuclease-free water.
Bioinformatics analysis
A total of 312 pairs of lung cancer tumor tissues
and distant non-tumor tissues were obtained from TCGA database
(https://tcga.xenahubs.net) for the
analysis of differential TNFRSF10C and TNFRSF10D gene
methylation levels. The correlation between methylation and mRNA
expression levels of the TNFRSF10C gene (49 samples) were
analyzed and charted using cBioPortal (http://www.cbioportal.org). TNFRSF10C DNA
methylation and TNFRSF10C expression data from the lung
adenocarcinoma (LUAD) project were obtained by downloading the data
of 568 TCGA lung cancer samples from National Cancer Institute GDC
Data Portal (https://portal.gdc.cancer.gov/repository).
Statistical analysis
Statistical analysis was performed using SPSS
version 20.0 (IBM Corp). Nonparametric Wilcoxon signed-rank tests
were used to determine the differences in methylation between tumor
tissues and distant non-tumor tissues. The methylation level is
expressed as the median. The nonparametric Wilcoxon rank-sum test
was used to analyze the methylation differences between tumor
tissues and distant non-tumor tissues in TCGA database. The
correlation between TNFRSF10C methylation levels and mRNA
expression levels was determined using Spearman's test. Luciferase
reporter gene activities were analyzed using the Kruskal Wallis
test with Dunn's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Selected promoter fragment in the
current methylation assay
Corresponding primers for the CG-rich region of
TNFRSF10C and TNFRSF10D were selected for analysis
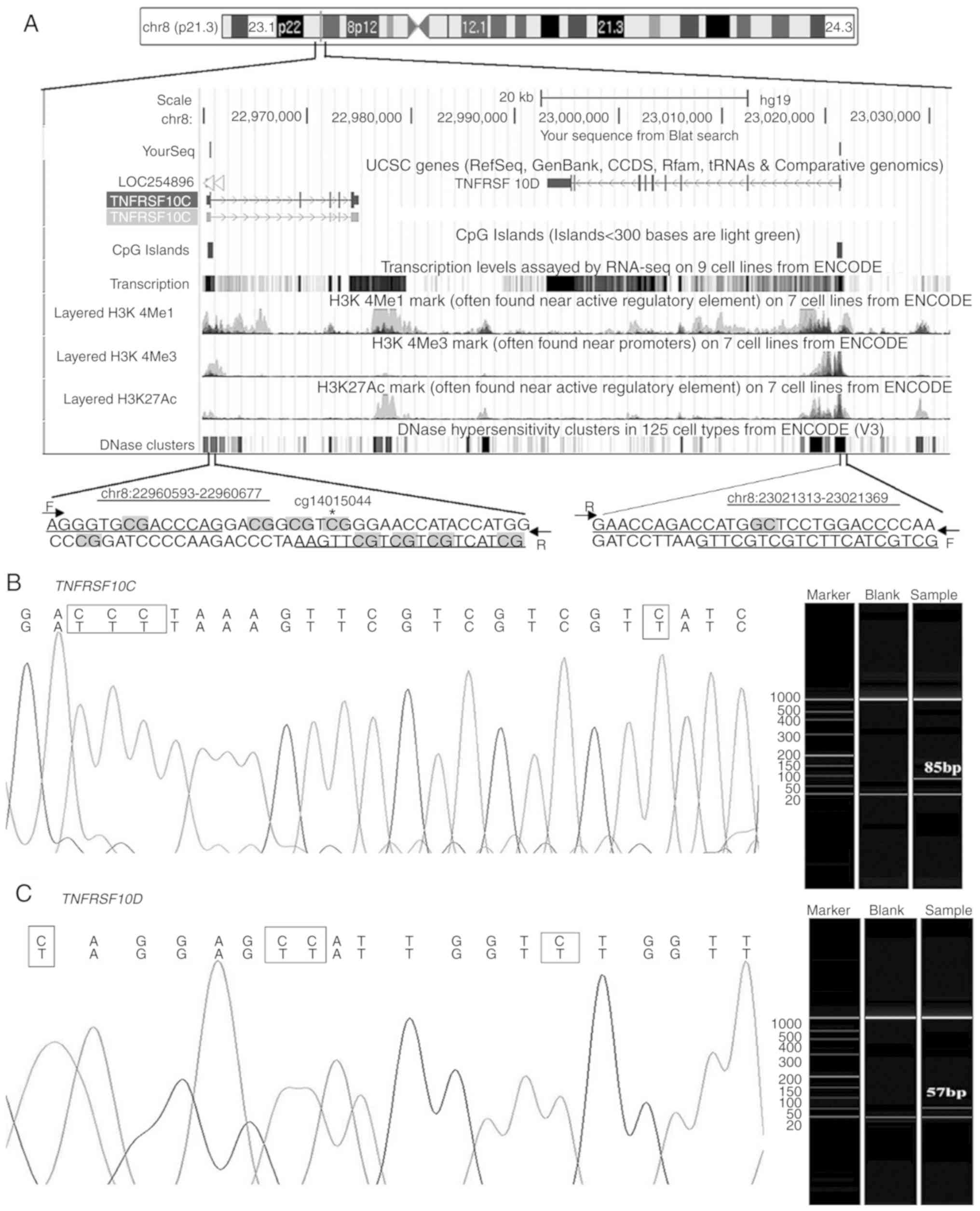
(Fig. 1A). The target fragments of
both genes were detected using capillary electrophoresis
amplification, and the amplified fragment was confirmed using
Sanger sequencing (Fig. 1B and C).
There were 7 and 15 cytosines (including 5 CG and 1 CG) in the
primer sequences of TNFRSF10C and TNFRSF10D,
respectively. The qMSP melting curves indicated that the qMSP
products of both genes were homogeneous (data not shown).
Differences in TNFRSF10C methylation
levels in tumor tissues and distant non-tumor tissues
Methylation levels of the TNFRSF10C and
TNFRSF10D genes between tumor and distant non-tumor tissues
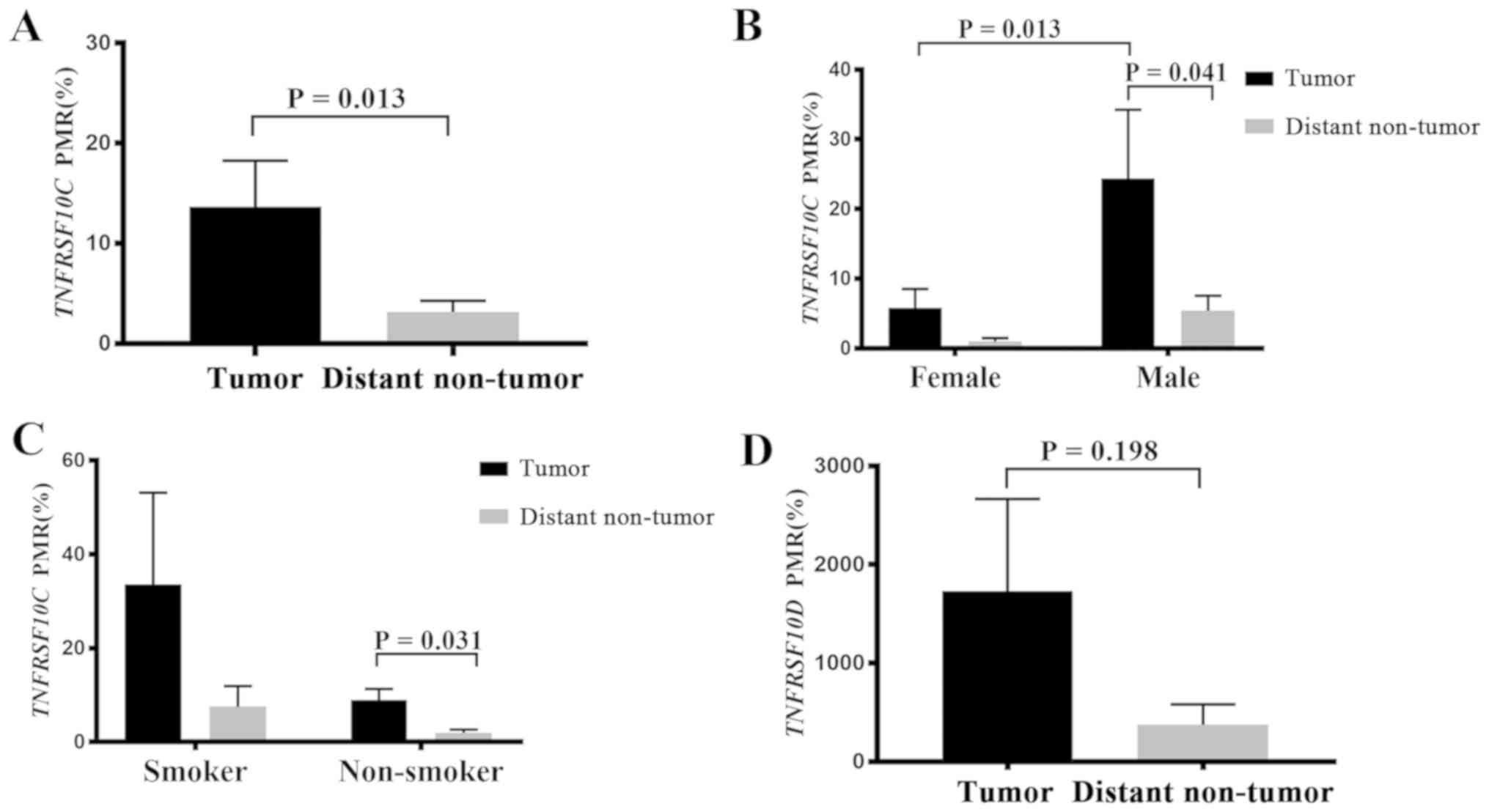
were compared in 44 patients with NSCLC. The results demonstrated
that the methylation levels of TNFRSF10C in tumor tissues
were significantly higher compared with those in distant non-tumor
tissues (P=0.013; Fig. 2A). Further
subgroup analysis of the clinicopathological data revealed a
significant association between TNFRSF10C methylation levels
and NSCLC in male patients and in non-smokers (Table I). In male patients, the methylation
levels of TNFRSF10C in tumor tissues were higher compared
with those in distant non-tumor tissues (P=0.041; Fig. 2B), and the methylation levels in
tumor tissues from male patients were significantly higher compared
with those in tumor tissues from female patients (P=0.013; Fig. 2B). In patients who were non-smokers,
TNFRSF10C methylation levels were higher in tumor tissues
compared with distant non-tumor tissues (P=0.031; Fig. 2C). However, in female patients who
were non-smokers there were no significant differences in the DNA
methylation levels of TNFRSF10C between tumor tissues and
distant non-tumor tissues (P=0.46; Table
I). In male patients who were non-smokers, the methylation
levels in tumor tissues were significantly lower compared with
those in distant non-tumor tissues (P=0.03; Table I). In male patients who were smokers,
no significant difference was observed in the TNFRSF10C DNA
methylation levels between tumor tissues and distant non-tumor
tissues (P=0.225; Table I). These
results indicated that the association of TNFRSF10C
hypomethylation with lung cancer was only observed in male patients
who were non-smokers. Due to the limited number of patients in each
subgroup, this conclusion needs to be interpreted with caution.
 | Table I.Association between tumor necrosis
factor receptor superfamily member 10c methylation levels and
clinicopathological characteristics of patients with non-small cell
lung cancer. |
Table I.
Association between tumor necrosis
factor receptor superfamily member 10c methylation levels and
clinicopathological characteristics of patients with non-small cell
lung cancer.
| Variable | n | Tumor PMR, median %
(interquartile range) | Distant non-tumor
PMR, median % (interquartile range) | P-value |
|---|
| Age |
|
|
|
|
|
≤60 | 16 |
1.430 (0,
8.360) |
1.805 (0.016,
5.475) |
0.594 |
|
>60 | 28 |
4.095 (0,
23.650) |
0.459 (0,
2.53) |
0.060 |
| Sex |
|
|
|
|
|
Female | 23 |
0.342 (0,
4.970) |
0.105 (0,
1.505) |
0.460 |
|
Male | 11 |
9.690 (1.365,
28.645) |
2.535 (0.081,
5.925) |
0.041a |
| Smoking
history |
|
|
|
|
|
Non-smoker | 34 |
2.500 (0,
10.330) |
0.639 (0,
2.272) |
0.031a |
|
Smoker | 10 |
2.730 (0,
121.05) |
5.640 (0,
8.110) |
0.225 |
| Histological
type |
|
|
|
|
|
LUSC | 30 |
1.040 (0,
9.690) |
0.490 (0,
2.142) |
0.128 |
|
LUAD | 12 |
7.250 (0,
107.300) |
5.640 (0.965,
12.290) |
0.086 |
| Cancer
location |
|
|
|
|
| Right
lung | 26 |
2.730 (0,
9.025) |
1.129 (0,
3.090) |
0.088 |
| Left
lung | 18 |
3.975 (0.535,
24.725) |
0.521 (0,
3.930) |
0.064 |
The levels of methylation of TNFRSF10D were
not significantly different in the paired samples (P=0.198;
Fig. 2D). Furthermore, subgroup
analysis of the clinicopathological data did not reveal any
significant associations between TNFRSF10D methylation
levels and NSCLC.
To ensure that the data of the inflammatory
pseudotumor did not affect the results of the experiment, the data
from the two inflammatory pseudotumors were removed from
analysis.
TNFRSF10C methylation and expression
levels
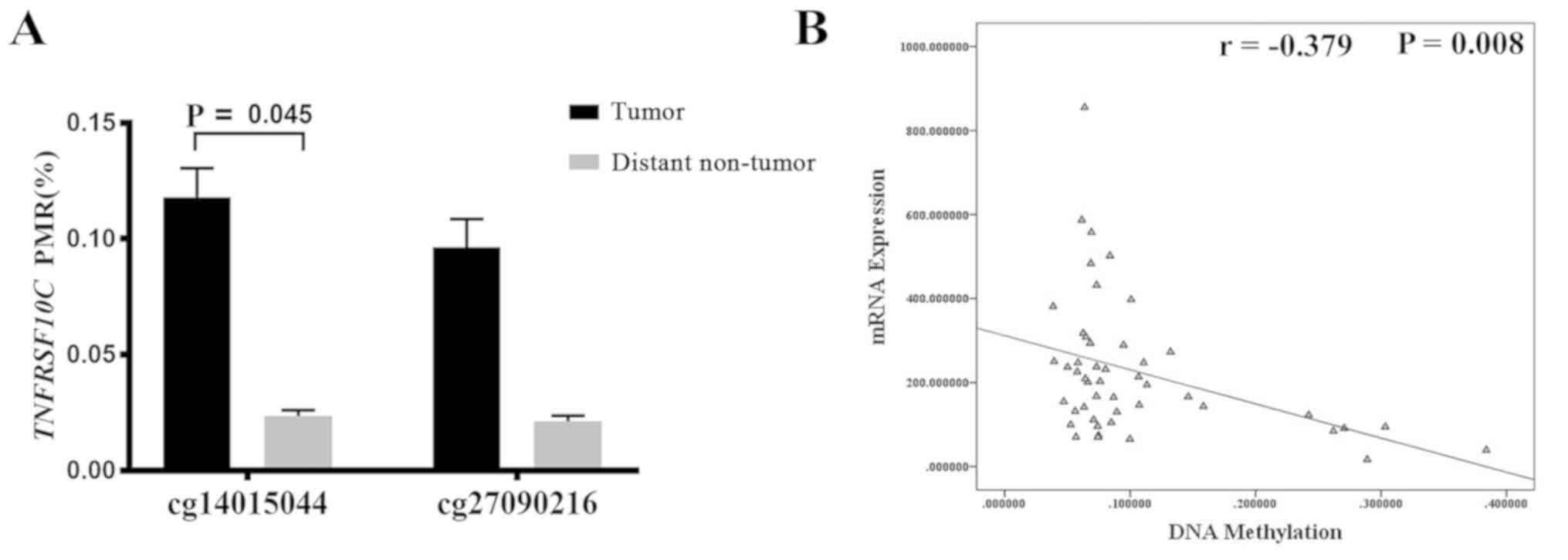
TCGA database was used to obtained methylation data
for the TNFRSF10C promoter region in lung tumor tissue
samples and to compare the methylation levels of TNFRSF10C
between tumor tissues and distant non-tumor tissues. The results
revealed that methylation levels at one of the CG sites in the
TNFRSF10C promoter region was higher in tumor tissues
compared with in distant non-tumor tissues samples (cg14015044,
P=0.045; Fig. 3A). Using lung cancer
data from the cBioPortal database, it was reported that the
methylation levels of TNFRSF10C were negatively correlated
with TNFRSF10C mRNA expression levels (r=−0.379, P=0.008;
Fig. 3B). The prognostic data from
male patients with lung cancer in TCGA database revealed that the
TNFRSF10C methylation levels had no effect on overall
patient survival time (data not shown).
TCGA data analysis also failed to identify any
significant association between TNFRSF10D and lung cancer
(data not shown). Overall, these results suggested no association
between TNFRSF10D methylation levels and lung cancer.
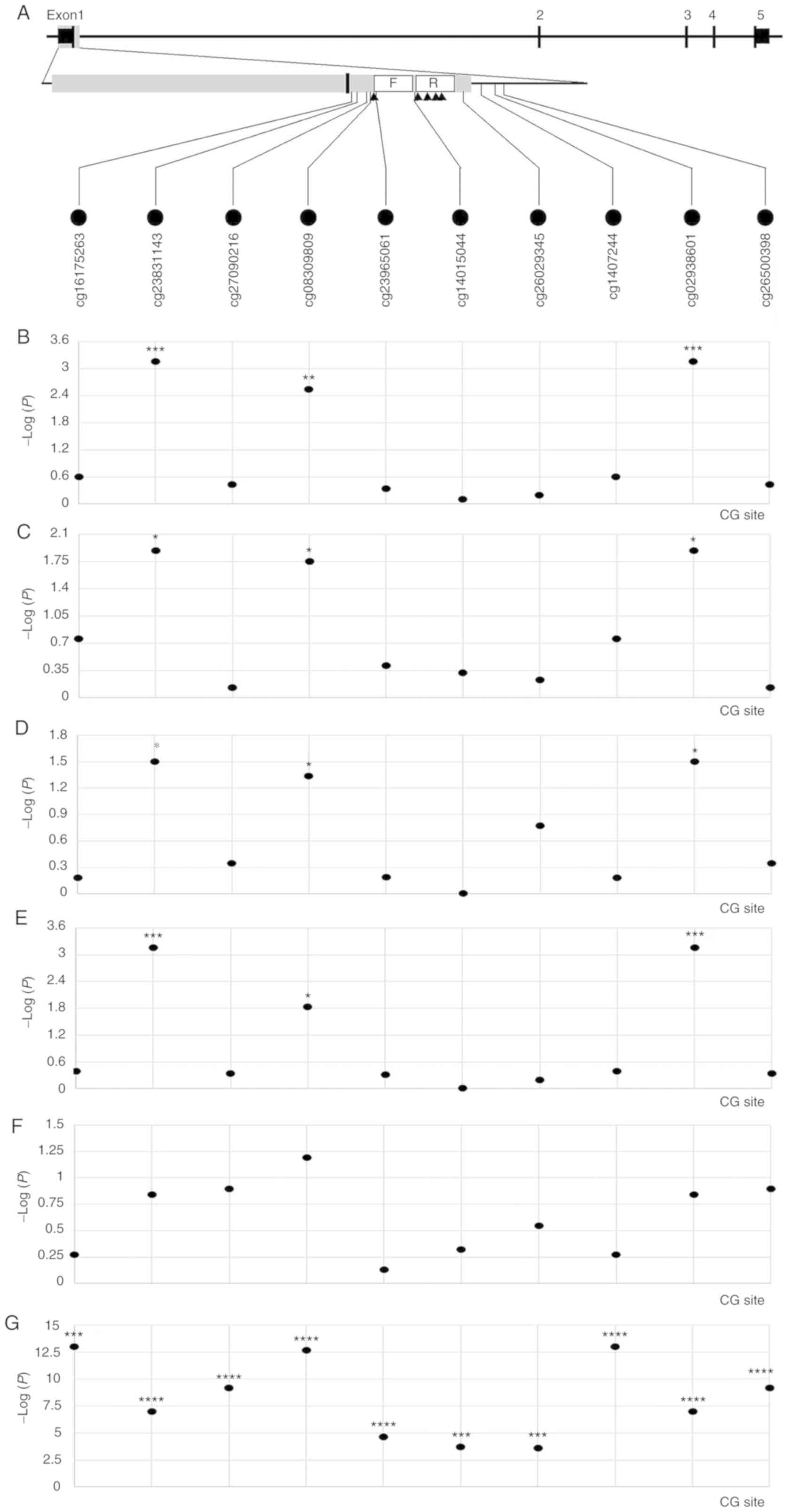
Clinicopathological, TNFRSF10C DNA
methylation and TNFRSF10C expression data from the lung
adenocarcinoma (LUAD) project were downloaded from TCGA database.
From the gene promoter region (−2,000 to +500 bp), 10 CG sites
closest to the qMSP amplicon presented in Fig. 4A were selected, and between-group
comparison and association analyses were performed. Based on the
comparison between cancer and non-cancerous tissues, it was
demonstrated that multiple CG sites, such as cg08309809, were
significantly associated with LUAD (Fig.
4B). Further subgroup analysis revealed an association between
cg08309809 and LUAD in male patients (Fig. 4C), female patients (Fig. 4D) and patients who were smokers
(Fig. 4E). There was no significant
association between cg08309809 and LUAD in the non-smoking group
(Fig. 4F); however, this may be due
to the small number of non-tumor tissues (n=2). The methylation and
expression levels of multiple CG sites in the TNFRSF10C gene
were significantly inversely correlated (r=−0.408~-0.146;
P=1.00×10−13~2.65×10−4 for different CG
sites; Fig. 4G), suggesting that
TNFRSF10C promoter methylation may be associated with gene
silencing.
Dual luciferase assay to detect
promoter activity of TNFRSF10C
A dual luciferase reporter plasmid containing the
promoter fragment of TNFRSF10C (−121 to +363 bp) was
constructed. The results demonstrated that the inserted gene
fragment significantly enhanced the transcriptional activity of the
reporter gene compared with the control group [fold-change
(FC)=1.570, P=0.032; Fig. 5A]. The
purpose of the luciferase experiment was to confirm whether the
promoter fragment regulated gene expression levels. By cloning the
fragment into the reporter vector, it was demonstrated that the
fragment enhanced the expression levels of the reporter gene,
suggesting that the fragment regulated gene expression levels.
Therefore, the methylation of this fragment may serve a role in
regulating the TNFRSF10C gene expression levels. To
determine the relationship between TNFRSF10C promoter
methylation and its transcriptional expression levels in lung
cancer cells, 5′-aza-deoxycytidine was used to treat lung cancer
cells A549 and NCI-H1299. The results demonstrated that the mRNA
expression levels of TNFRSF10C in the two lung cancer cell
lines were significantly increased following demethylation
treatment (A549, FC=8.000, P=0.0001; NCI-H1299, FC=3.163,
P=1.143×10−5; Fig.
5B).
Discussion
Decreased gene expression levels of TRAIL
decoy receptor gene TNFRSF10C caused by hypermethylation
have been observed in glioblastoma (26), prostate (27) and breast (28) cancer. The results of the present
study also demonstrated that the levels of methylation of
TNFRSF10C in NSCLC tissues were higher compared with those
in distant non-tumor tissues, which was consistent with the
aforementioned studies. The present study also demonstrated that
hypermethylation of TNFRSF10C was specific to tumor tissues
in male patients and patients who were non-smokers.
TNFRSF10C methylation levels in tumor tissues of male
patients were higher compared with those of female patients. These
results demonstrated that TNFRSF10C methylation levels were
associated with NSCLC.
Previous studies have reported that the relationship
between TNFRSF10C gene hypermethylation and downregulated
expression levels has been observed in human prostate cancer and
pancreatic cancer (27,29). In the present study, the levels of
TNFRSF10C methylation were inversely correlated with mRNA
expression levels in lung cancer tissue data from the cBioPortal
database. 5′-Aza-deoxycytidine was used to treat two lung cancer
cell lines, A549 and NCI-H1299, and the mRNA expression levels
before and after treatment were compared. The results demonstrated
that the mRNA expression levels of TNFRSF10C were
significantly increased after 5′-aza-deoxycytidine-induced
demethylation. These results indicated that the expression of
TNFRSF10C was regulated by promoter methylation.
Hypermethylation of the TNFRSF10C promoter
initiates the binding of TRAIL with tumor necrosis factors DR4 and
DR5, and thus mediates the TRAIL-R signaling pathway (30). Although TRAIL-R induces apoptosis in
a variety of cancer types, several types of tumor, such as
cholangiocarcinoma and leukemia have recently been demonstrated to
be resistant to TRAIL-R signaling (31,32). In
addition, TRAIL-R can activate the NF-κB signaling pathway and
promote the proliferation and migration of cancer cells, and thus
TRAIL-R is associated with the development of cholangiocarcinoma
and leukemia (31,32).
Although smoking can cause epigenetic changes that
induce cancer development, especially lung cancer, smoking is not
the only cause of lung cancer (33).
TRAIL-R has been demonstrated to promote the epidermal growth
factor receptor (EGFR)-KRAS signaling pathway-mediated tumor
development (34). A previous study
reported that patients who were non-smokers harbored a higher
frequency of EGFR mutations compared with smokers (35). Downregulation of TNFRSF10C
expression via methylation promotes TRAIL-R deactivation in the
EGFR-KRAS signaling pathway and promotes cancer progression and
invasion (34). The results of the
present study demonstrated that the TNFRSF10C methylation
levels were higher in tumor tissues compared with distant non-tumor
tissues in non-smokers; however, no significant differences were
observed between the groups in smokers. These results may provide
novel evidence supporting the epigenetic changes occurring in lung
cancer in patients who are non-smokers.
The incidence of lung cancer in China is high in
males at present (2). In the present
study, the methylation levels of TNFRSF10C in tumor tissues
from male patients were higher compared with distant non-tumor
tissues, and the methylation levels in tumor tissues from male
patients were significantly higher compared with those from female
patients. These findings may provide novel evidence supporting the
effects of sex on TNFRSF10C methylation levels in lung
cancer.
TRAIL-R can mediate the inhibition of tumor
development (36). TNFRSF10C and
TNFRSF10D lack a cytoplasmic domain and do not contain
transmembrane domains; thus, they are unable to participate in the
transduction of apoptotic signals (37). Hypermethylation of TNFRSF10C
promotes the binding of TRAIL to apoptotic receptors, thereby
promoting apoptosis in cancer cells and preventing tumorigenesis
(38). However, TNFRSF10D has a
truncated death domain and hence is not capable of inducing
apoptosis but protects against TRAIL-mediated apoptosis (39). The results of the present study
indicated that TNFRSF10D methylation was not associated with
lung cancer.
The present study primarily examined the methylation
levels of the TNFRSF10C and TNFRSF10D genes in lung
tumor and distant non-tumor tissues. The results demonstrated that
TNFRSF10C hypermethylation was associated with NSCLC,
especially in non-smokers and male patients. The underlying
molecular mechanisms of the hypermethylation of TNFRSF10C in
the development and progression of NSCLC need further
investigation. DNA methylation of tissue biopsy may be used as a
standard indicator of current pathological diagnosis (40,41).
However, tissue biopsy is inconvenient and traumatic. Liquid
biopsy, such as circulating tumor cells, circulating tumor DNA and
exosomes can provide more comprehensive disease information
(42,43). Furthermore, investigating the
methylation levels of TNFRSF10C and TNFRSF10D genes
of patients' blood samples, and combining data from both tissues
and blood may allow their development as novel diagnostic
biomarkers of lung cancer.
Acknowledgements
Not applicable.
Funding
This study was supported by The National Natural
Science Foundation of China (grant no. 81773055), The Science
Foundation of the Education Department of Fujian Province (grant
no. JZ160438), the Key Talents Training Program of Fujian
Provincial Health Commission (grant nos. 2017-ZQN-59 and
2017-ZQN-60), The Joint Funds for the Innovation Of Science And
Technology of Fujian province (grant no. 2017Y9113) and The K.C.
Wong Magna Fund in Ningbo University.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
YQ and LQ collected tissue samples and patient data,
analyzed the data and wrote the manuscript. MQ and MZ carried out
literature research and data interpretation. CY, JL, HL, ZZ and CC
performed the experiments. SD designed the study and critically
revised the manuscript for intellectually important content. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of the First Affiliated Hospital of Fujian Medical
University (Fuzhou, China; approval number: 2017-KY-068). All
patients provided written informed consent for participation in
this study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siege RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Inage T, Nakajima T, Yoshino I and
Yasufuku K: Early lung cancer detection. Clin Chest Med. 39:45–55.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
NSCLC Meta-analysis Collaborative Group, :
Preoperative chemotherapy for non-small-cell lung cancer: A
systematic review and meta-analysis of individual participant data.
Lancet. 383:1561–1571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Le Chevalier T: Adjuvant chemotherapy for
resectable non-small-cell lung cancer: Where is it going? Ann
Oncol. 21 (Suppl 7):vii196–vii198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ji P, Ding D, Qin N, Wang C, Zhu M, Li Y,
Dai J, Jin G, Hu Z, Shen H, et al: Systematic analyses of genetic
variants in chromatin interaction regions identified four novel
lung cancer susceptibility loci. J Cancer. 11:1075–1081. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang D, Zhao YC, Liu H, Luo S, Clarke JM,
Glass C, Su L, Shen S, Christiani DC, Gao W and Wei Q: Potentially
functional genetic variants in PLIN2, SULT2A1 and UGT1A9 genes of
the ketone pathway and survival of nonsmall cell lung cancer. Int J
Cancer. Feb 18–2020.(Epub ahead of print). View Article : Google Scholar
|
|
8
|
Jakopovic M, Thomas A, Balasubramaniam S,
Schrump D, Giaccone G and Bates SE: Targeting the epigenome in lung
cancer: Expanding approaches to epigenetic therapy. Front Oncol.
3:2612013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Berger SL, Kouzarides T, Shiekhattar R and
Shilatifard A: An operational definition of epigenetics. Genes Dev.
23:781–783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rana AK and Ankri S: Reviving the RNA
world: An insight into the appearance of RNA Methyltransferases.
Front Genet. 7:992016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rauch T, Wang Z, Zhang X, Zhong X, Wu X,
Lau SK, Kernstine KH, Riggs AD and Pfeifer GP: Homeobox gene
methylation in lung cancer studied by genome-wide analysis with a
microarray-based methylated CpG island recovery assay. Proc Natl
Acad Sci USA. 104:5527–5532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Castro M, Grau L, Puerta P, Gimenez L,
Venditti J, Quadrelli S and Sánchez-Carbayo M: Multiplexed
methylation profiles of tumor suppressor genes and clinical outcome
in lung cancer. J Transl Med. 8:862010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Emery JG, McDonnell P, Burke MB, Deen KC,
Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, et
al: Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J
Biol Chem. 273:14363–14367. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Allen JE, Krigsfeld G, Mayes PA, Patel L,
Dicker DT, Patel AS, Dolloff NG, Messaris E, Scata KA, Wang W, et
al: Dual inactivation of Akt and ERK by TIC10 signals Foxo3a
nuclear translocation, TRAIL gene induction, and potent antitumor
effects. Sci Transl Med. 5:171ra172013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. 32:1341–1350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lim B, Allen JE, Prabhu VV, Talekar MK,
Finnberg NK and El-Deiry WS: Targeting TRAIL in the treatment of
cancer: New developments. Expert Opin Ther Targets. 19:1171–1185.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Soria JC, Smit E, Khayat D, Besse B, Yang
X, Hsu CP, Reese D, Wiezorek J and Blackhall F: Phase 1b study of
dulanermin (recombinant human Apo2L/TRAIL) in combination with
paclitaxel, carboplatin, and bevacizumab in patients with advanced
non-squamous non-small-cell lung cancer. J Clin Oncol.
28:1527–1533. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wainberg ZA, Messersmith WA, Peddi PF,
Kapp AV, Ashkenazi A, Royer-Joo S, Portera CC and Kozloff MF: A
phase 1B study of dulanermin in combination with modified FOLFOX6
plus bevacizumab in patients with metastatic colorectal cancer.
Clin Colorectal Cancer. 12:248–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheah CY, Belada D, Fanale MA, Janikova A,
Czucman MS, Flinn IW, Kapp AV, Ashkenazi A, Kelley S, Bray GL, et
al: Dulanermin with rituximab in patients with relapsed indolent
B-cell lymphoma: An open-label phase 1b/2 randomised study. Lancet
Haematol. 2:e166–e174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Herbst RS, Eckhardt SG, Kurzrock R,
Ebbinghaus S, O'Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA,
Tohnya TM, Lum BL, et al: Phase I dose-escalation study of
recombinant human Apo2L/TRAIL, a dual proapoptotic receptor
agonist, in patients with advanced cancer. J Clin Oncol.
28:2839–2846. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Braithwaite AT, Marriott HM and Lawrie A:
Divergent roles for TRAIL in lung diseases. Front Med (Lausanne).
5:2122018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tessema M, Yu YY, Stidley CA, Machida EO,
Schuebel KE, Baylin SB and Belinsky SA: Concomitant promoter
methylation of multiple genes in lung adenocarcinomas from current,
former and never smokers. Carcinogenesis. 30:1132–1138. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Raff T, van der Giet M, Endemann D,
Wiederholt T and Paul M: Design and testing of beta-actin primers
for RT-PCR that do not co-amplify processed pseudogenes.
Biotechniques. 23:456–460. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kristensen LS, Mikeska T, Krypuy M and
Dobrovic A: Sensitive melting analysis after real time-methylation
specific PCR (SMART-MSP): High-throughput and probe-free
quantitative DNA methylation detection. Nucleic Acids Res.
36:e422008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vaitkienė P, Skiriutė D, Skauminas K and
Tamašauskas A: GATA4 and DcR1 methylation in glioblastomas. Diagn
Pathol. 8:72013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheng Y, Kim JW, Liu W, Dunn TA, Luo J,
Loza MJ, Kim ST, Zheng SL, Xu J, Isaacs WB and Chang BL: Genetic
and epigenetic inactivation of TNFRSF10C in human prostate cancer.
Prostate. 69:327–335. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tserga A, Michalopoulos NV, Levidou G,
Korkolopoulou P, Zografos G, Patsouris E and Saetta AA: Association
of aberrant DNA methylation with clinicopathological features in
breast cancer. Oncol Rep. 27:1630–1638. 2012.PubMed/NCBI
|
|
29
|
Cai HH, Sun YM, Miao Y, Gao WT, Peng Q,
Yao J and Zhao HL: Aberrant methylation frequency of TNFRSF10C
promoter in pancreatic cancer cell lines. Hepatobiliary Pancreat
Dis Int. 10:95–100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
von Karstedt S, Montinaro A and Walczak H:
Exploring the TRAILs less travelled: TRAIL in cancer biology and
therapy. Nat Rev Cancer. 17:352–366. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ishimura N, Isomoto H, Bronk SF and Gores
GJ: Trail induces cell migration and invasion in
apoptosis-resistant cholangiocarcinoma cells. Am J Physiol
Gastrointest Liver Physiol. 290:G129–G136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ehrhardt H, Fulda S, Schmid I, Hiscott J,
Debatin KM and Jeremias I: TRAIL induced survival and proliferation
in cancer cells resistant towards TRAIL-induced apoptosis mediated
by NF-kappaB. Oncogene. 22:3842–3852. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vaz M, Hwang SY, Kagiampakis I, Phallen J,
Patil A, O'Hagan HM, Murphy L, Zahnow CA, Gabrielson E, Velculescu
VE, et al: Chronic cigarette smoke-induced epigenomic changes
precede sensitization of bronchial epithelial cells to single-step
transformation by KRAS mutations. Cancer Cell. 32:360–376.e6. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
von Karstedt S, Conti A, Nobis M,
Montinaro A, Hartwig T, Lemke J, Legler K, Annewanter F, Campbell
AD, Taraborrelli L, et al: Cancer cell-autonomous TRAIL-R signaling
promotes KRAS-driven cancer progression, invasion, and metastasis.
Cancer Cell. 27:561–573. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chapman AM, Sun KY, Ruestow P, Cowan DM
and Madl AK: Lung cancer mutation profile of EGFR, ALK, and KRAS:
Meta-analysis and comparison of never and ever smokers. Lung
Cancer. 102:122–134. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Finnberg N, Klein-Szanto AJ and El-Deiry
WS: TRAIL-R deficiency in mice promotes susceptibility to chronic
inflammation and tumorigenesis. J Clin Invest. 118:111–123. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shivapurkar N, Toyooka S, Toyooka KO,
Reddy J, Miyajima K, Suzuki M, Shigematsu H, Takahashi T, Parikh G,
Pass HI, et al: Aberrant methylation of trail decoy receptor genes
is frequent in multiple tumor types. Int J Cancer. 109:786–792.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
van Noesel MM, van Bezouw S, Salomons GS,
Voûte PA, Pieters R, Baylin SB, Herman JG and Versteeg R:
Tumor-specific down-regulation of the tumor necrosis factor-related
apoptosis-inducing ligand decoy receptors DcR1 and DcR2 is
associated with dense promoter hypermethylation. Cancer Res.
62:2157–2161. 2002.PubMed/NCBI
|
|
39
|
Pan G, Ni J, Yu G, Wei YF and Dixit VM:
TRUNDD, a new member of the TRAIL receptor family that antagonizes
TRAIL signalling. FEBS Lett. 424:41–45. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Diaz-Lagares A, Mendez-Gonzalez J, Hervas
D, Saigi M, Pajares MJ, Garcia D, Crujerias AB, Pio R, Montuenga
LM, Zulueta J, et al: A novel epigenetic signature for early
diagnosis in lung cancer. Clin Cancer Res. 22:3361–3371. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dong S, Li W, Wang L, Hu J, Song Y, Zhang
B, Ren X, Ji S, Li J, Xu P, et al: Histone-related genes are
hypermethylated in lung cancer and hypermethylated HIST1H4F could
serve as a pan-cancer biomarker. Cancer Res. 79:6101–6112. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Constâncio V, Nunes SP, Henrique R and
Jerónimo C: DNA methylation-based testing in liquid biopsies as
detection and prognostic biomarkers for the four major cancer
types. Cells. 9(pii): E6242020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nunes SP, Diniz F, Moreira-Barbosa C,
Constâncio V, Silva AV, Oliveira J, Soares M, Paulino S, Cunha AL,
Rodrigues J, et al: Subtyping lung cancer using DNA methylation in
liquid biopsies. J Clin Med. 8(pii): E15002019. View Article : Google Scholar : PubMed/NCBI
|