Introduction
Lung cancer is the one of the leading causes of
cancer-related death worldwide (1),
and non-small cell lung cancer (NSCLC) accounts for almost 80% of
these deaths (2). Cisplatin-based
chemotherapy is the main treatment used for patients with major
NSCLC (2). However, the long-term
prognosis for patients with NSCLC still remains poor, with the
5-year survival rate being only ~11% (3). Patients with NSCLC often show an
initial positive response to cisplatin treatment, but in many cases
this response is not sustained and cancer cells become resistant to
the treatment, this has become a major clinical challenge in the
treatment of NSCLC (4). Therefore,
the identification of novel therapeutic targets in NSCLC treatment
that can complement current therapy is urgently required.
The ubiquitin-proteasome system regulates cellular
protein levels with specificity and precision to optimize cellular
functions (5,6). Moreover, certain studies have
demonstrated that ubiquitination can modify protein functions,
including the regulation of subcellular localization and
protein-protein interactions (5–7). The
association between ubiquitin and tumor biology has long been
recognized, and the suppression of the proteasome has proven to be
effective in the treatment of various types of cancer, such as
myeloma and gastrointestinal cancer (6). Deubiquitinating enzymes (DUBs) can
reverse protein ubiquitination by digesting ubiquitin chains
(8).
Ubiquitin-specific protease 17 (USP17), a DUB, is
involved in the regulation of inflammation (9) and cell motility (10), the development of Th17 cells
(11) and oncogenesis (12–17). It
has been found that USP17 is highly expressed in several types of
tumor, including colon, esophageal and cervical tumors (14). In addition, USP17 regulates the Ras
pathway by altering the intracellular localization of Ras and other
small GTPases, which are crucial regulators of cellular
proliferation and migration (10).
USP17 is required for the trafficking and oncogenic signaling of
mutant epidermal growth factor receptor (EGFR) in NSCLC cells
(15). These studies verified the
role of USP17 in promoting tumor growth and metastasis. McFarlane
et al (16) reported
upregulation of USP17 in patients with NSCLC. Moreover, patients
with USP17-positive tumors had significantly shorter
recurrence-free survival times compared with those with
USP17-negative tumors, and USP17 expression was associated with the
recurrence of disease at distant sites. In addition, an in
vivo study, in which human NSCLC cells were inoculated into
nude mice, found that the suppression of USP17 in NSCLC cells
inhibited tumor growth and invasion (17). However, the biological function of
USP17 that directly regulates NSCLC progression has not been
studied fully.
The re-emergence of cancer cells is often due to the
activation of survival signals, including increased activation of
the PI3K/AKT pathway (18–20), which has been associated with NSCLC
progression. The PI3K/AKT pathway is an important pathway
downstream of EGFR. Deregulation of this pathway, due to gene
amplifications, activating oncogene mutations or the loss of PTEN,
has been observed in several types of human cancer, such as
colorectal, gastric, lung, ovarian and thyroid cancer (20–23).
In the present study, the aim was to explore the
functions and underlying molecular mechanisms of USP17 in NSCLC
cells. Moreover, the effects of inhibiting of USP17 downstream
PI3K/AKT pathway in cisplatin sensitivity of NSCLC cells were also
investigated.
Materials and methods
Cell lines
The human NSCLC A549 and H1299 cell lines were
purchased from The Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences. A549 cells were maintained in
Dulbecco's modified Eagle's medium, and H1299 cells were maintained
in RPMI-1640 medium. All media were supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Invitrogen; Thermo Fisher
Scientific, Inc.), penicillin (100 U/ml), and streptomycin (100
µg/ml) in a humidified atmosphere of 5% CO2 at 37°C. All
cells were confirmed to be free from mycoplasma contamination.
Plasmids and reagents
USP17 short hairpin (sh)RNA and USP17 overexpression
lentiviruses were purchased from Hanyin Biotech Co. Polybrene (cat.
no. 107689; Sigma-Aldrich; Merck KGaA) was used as an infection
reagent. The target sequence for USP17 shRNA-knockdown (KD) was
5′-CTCTTGAGAATGTGCCGAT-3′ (the shRNA was packaged into the
lentivirus). The negative control (NC) comprised an empty vector
without target sequences. To generate stable cell lines,
supernatant containing lentivirus (1×106 TU) was added
to A549 and H1299 cells (1×105/well), which were
subsequently screened with 1 µg/ml puromycin for 2 weeks.
Experiments were performed 72 h after infection.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
Total RNA (100 ng) was used for cDNA synthesis using the Stratagene
AffinityScript QPCR cDNA Synthesis kit (Agilent Technologies,
Inc.). The temperature protocol for the RT step was 5 min at 65°C,
60 min at 42°C and 15 min at 70°C. The cDNA samples were diluted
10-fold with nuclease-free H2O, of which 2 µl was
combined with Brilliant III Ultra-Fast SYBR® Green qPCR
Master mix (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Human RPL13A was used as an internal reference control. The primer
sequences were as follows: Human USP17 forward,
5′-GAGATTCTCCGATGTCACAGGC-3′ and reverse,
5′-TCCGTCGTGACAACTCCACCCA-3′; human RPL13A forward,
5′-CTCAAGGTGTTTGACGGCATCC-3′ and reverse,
5′-TACTTCCAGCCAACCTCGTGAG-3′. The relative expression of target
genes was determined using the 2−∆∆Cq method (24). The qPCR cycling conditions comprised
an initial denaturation step of 3 min at 95°C, followed by 45
cycles at 95°C (10 sec) and 58°C (45 sec); data were acquired at
the end of the annealing/extension phase. Melt curve analysis was
performed at the end of each run between 58–95°C and the data were
analyzed using Microsoft Excel 2013 (Microsoft Corporation).
Cell Counting Kit-8 assay (CCK-8)
Cells (5×103/well) were treated with
cisplatin (1 µM) or MK2206 (1 µM; both purchased from Selleck
Chemicals) for 0, 24, 48, 72, 96 and 120 h. The CCK-8 assay was
conducted according to the kit instructions (cat. no. CK04; Dojindo
Molecular Technologies, Inc.). Cells with or without USP17
overexpression (OE) and cells treated with MK2206 were examined.
Briefly, cells in logarithmic growth phase were trypsin-digested
and resuspended in RPMI-1640 medium. Cells were plated at equal
densities (2,000 cells/100 µl per well) in 96-well plates and
incubated at 37°C and 5% CO2 for continuous detection
over a 5-day period. At the beginning of the second day, cell
growth was terminated by the addition of 10 µl of CCK-8 solution (5
mg/ml) to the culture medium. After 2 h, the OD490 values were
determined using a microplate reader (BioTek Instruments,
Inc.).
Transwell assay
USP17-OE cells and those (2×104/well)
treated with MK2206 (1 µM) or LY294002 (5 µM; both purchased from
Selleck Chemicals) for 48 h were examined. Cells (2×104)
were detached and resuspended in serum-free medium and seeded in
the upper chamber of Matrigel-coated Transwell (precoating with
Matrigel at 4°C for 60 min) inserts with a pore size of 8 µm.
Culture medium containing 10% FBS as a chemoattractant was added to
the lower chamber. After 24 h of incubation, cells on the upper
surface of the insert were gently removed with a cotton swab.
Invasive cells (on the lower surface of the insert) were fixed with
4% paraformaldehyde (Sigma-Aldrich; Merck KGaA) at room temperature
for 15 min, stained with crystal violet (1%) at room temperature
for 60 min, and counted under a microscope (Olympus, CKX31); five
random microscopic fields were examined for each insert using
magnification, ×200.
Western blot analysis
Total protein was extracted from cells using RIPA
buffer (Beyotime Institute of Biotechnology). Protein was
quantified using a BCA assay. Protein lysates (50 µg/lane) were
separated via 8–10% SDS-PAGE and transferred onto nitrocellulose
membranes. After blocking with 5% fat-free milk at room temperature
for 30 min, the membranes were incubated with primary antibodies
(1:500) at 4°C overnight. The primary antibodies used were: Goat
anti-human USP17 (cat. no. AP5491b; Abgent, Inc.), mouse anti-actin
(cat. no. 3700P), anti-human AKT (cat. no. 4685), anti-human
phosphorylated (p)-AKT (cat. no. 4060), anti-human p-PI3K (cat. no.
4228) and anti-human PI3K (cat. no. 4257; all purchased from Cell
Signaling Technology, Inc.). Following this, membranes were
incubated with horseradish peroxidase-conjugated secondary
antibodies (1:3,000; cat. nos. 705-035-147, 705-035-150 and
705-035-152; Jackson ImmunoResearch Laboratories, Inc.) at room
temperature for 60 min. Immunoreactive proteins were visualized
using an enhanced chemiluminescence reagent (EMD Millipore).
Relative expression of p-PI3K and p-Akt was normalized to that of
actin, and then relative to the normalized total PI3K or Akt
values. Quantity One software version 4.6.9 (Bio-Rad Laboratories,
Inc.) was used to quantify the relative band intensities.
Statistical analysis
Quantitative variables were compared using one-way
ANOVA to compare differences between two or more groups, followed
by Tukey's test for post-hoc analysis. Statistical analysis was
performed using SPSS version 19.0 (IBM Corp.) and GraphPad Prism
version 5.0 (GraphPad Software, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
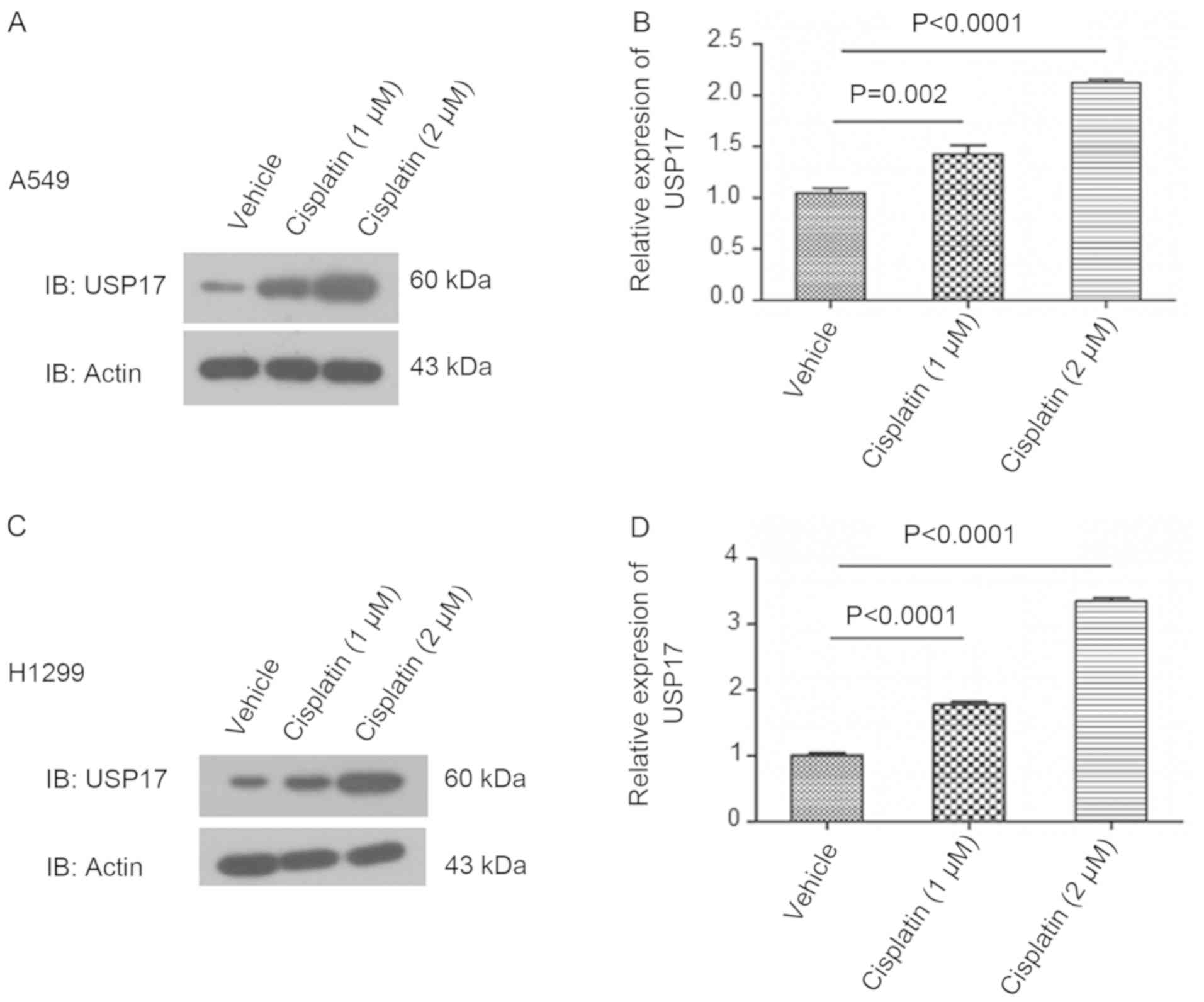
USP17 expression is upregulated in
NSCLC cells treated with cisplatin
Western blot analysis revealed increased levels of
USP17 in NSCLC cells treated with increasing concentrations of
cisplatin, in a dose-dependent manner (vehicle vs. 1 µm cisplatin
in A549, P=0.002, in H1299, P<0.0001; vehicle vs. 2 µm cisplatin
in A549, P<0.0001, in H1299, P<0.0001; Fig. 1).
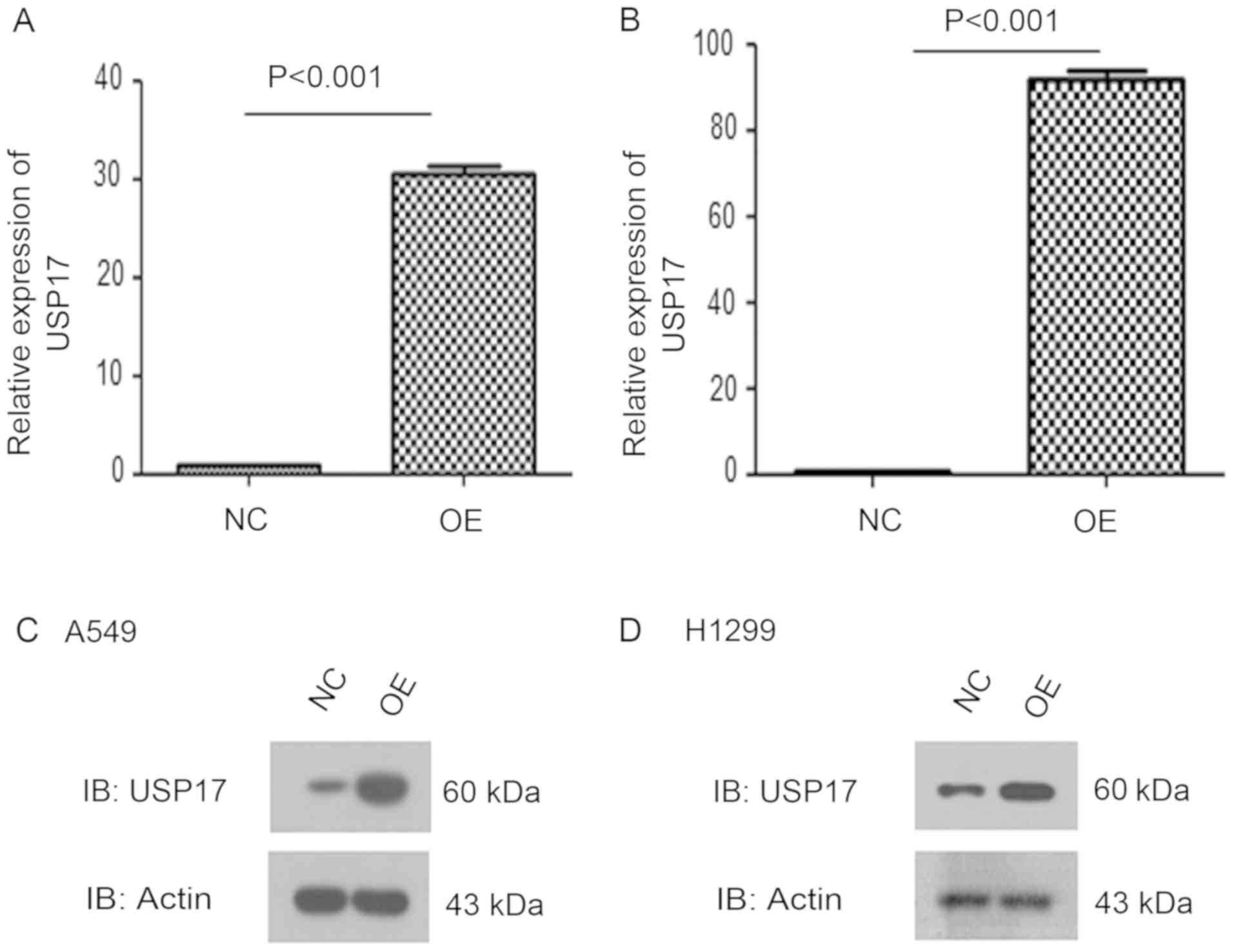
In order to address the functions of USP17 in the
response of NSCLC cells to cisplatin treatment, USP17-OE A549 and
H1299 NSCLC cells were generated. Both RT-qPCR and western blot
analysis demonstrated high expression levels of USP17 in these cell
lines, compared with those in the corresponding NC cells (NC vs.
OE, P<0.001; Fig. 2).
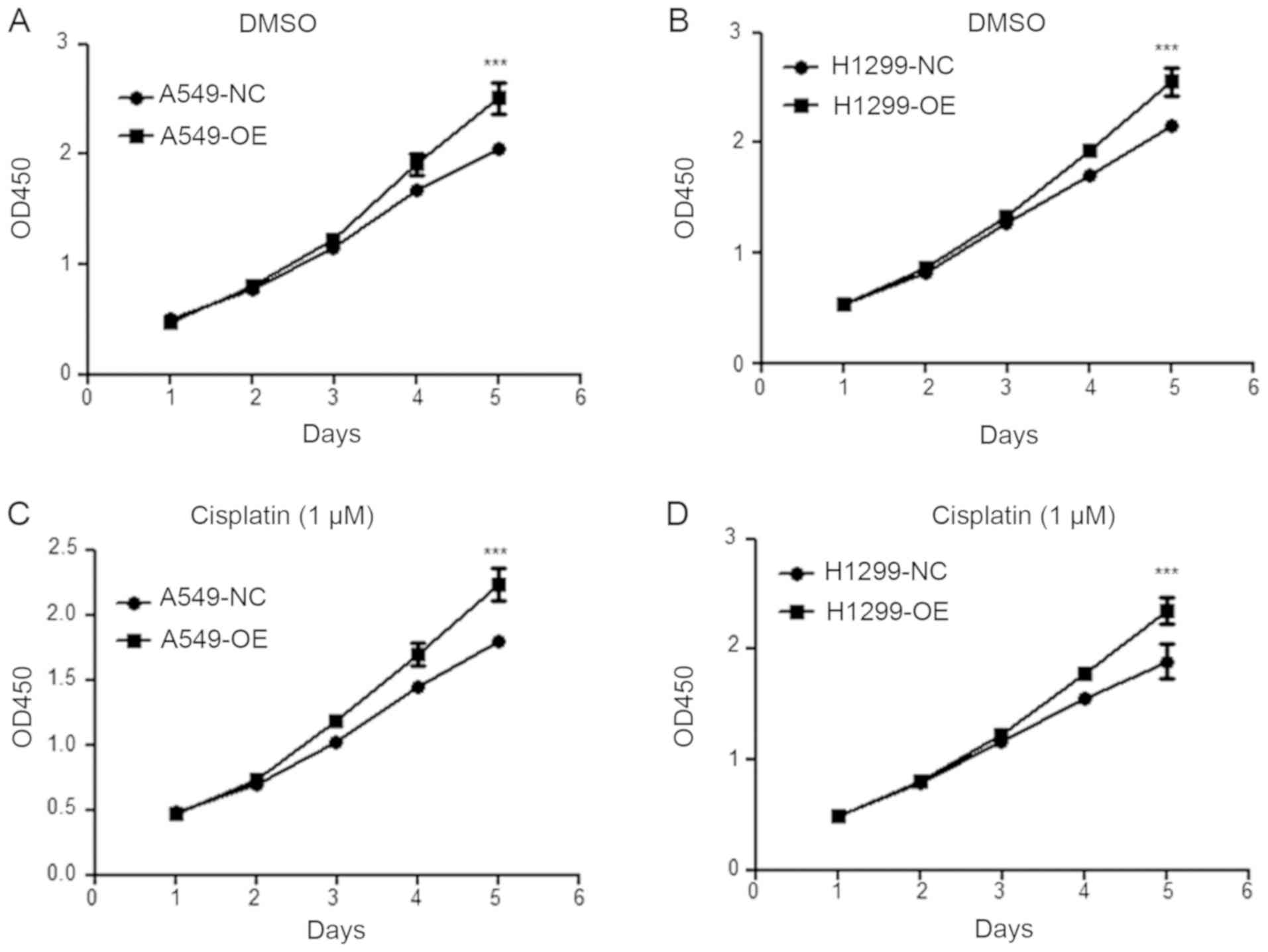
Overexpression of USP17 increases
proliferation and sustains viability in NSCLC cells
In order to investigate the biological functions of
USP17 in the transformation of NSCLC cells, a CCK-8 assay was
conducted to determine the proliferative ability of A549 (Fig. 3A) and H1299 (Fig. 3B) cells (NC vs. OE, P<0.001).
Moreover, the viability of USP17-OE cells was significantly higher
than that of the control cells when treated with cisplatin (NC vs.
OE, P<0.001; Fig. 3C and D).
These results demonstrate that the overexpression of USP17
increases proliferation and viability in NSCLC cells, independent
of cisplatin treatment.
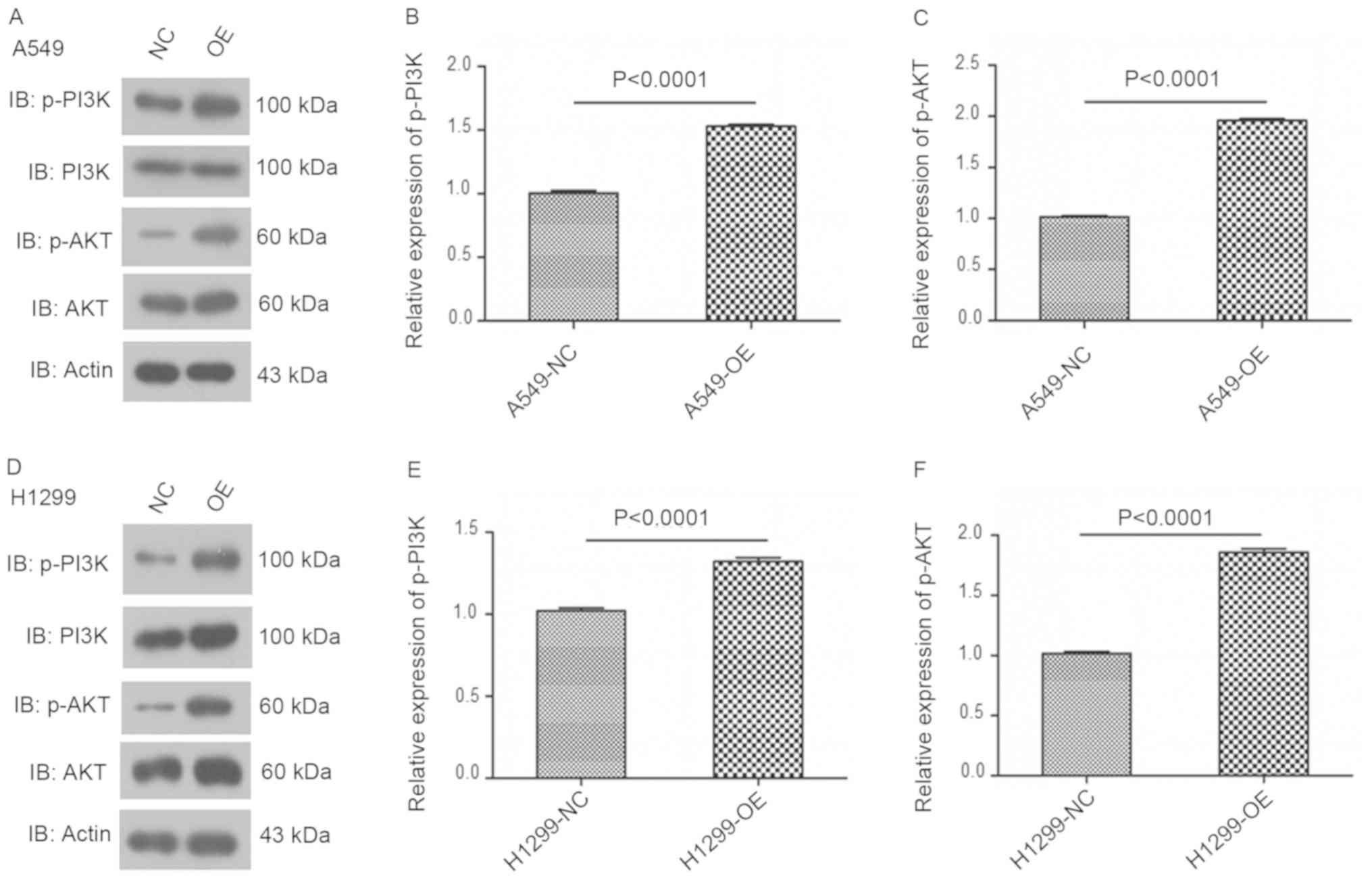
Overexpression of USP17 enhances
PI3K/AKT pathway activation in NSCLC cells
The importance of the PI3K/AKT pathway as a potent
survival mechanism in carcinogenesis and tumor metastasis is well
established (18–20). In the present study, the effect of
USP17 on the activation of the PI3K-AKT signaling pathway in NSCLC
cells was investigated. Western blot analysis demonstrated
increased p-AKT and p-PI3K levels in USP17-OE NSCLC cells, compared
with those of the control cells (NC vs. OE, P<0.0001; Fig. 4). This finding suggests the
activation of the PI3K/AKT pathway by USP17 as the underlying
mechanism of the enhanced proliferation and viability in NSCLC
cells.
Inhibition of AKT activation abolishes
USP17-mediated enhancement of proliferation and viability in
USP17-OE cells
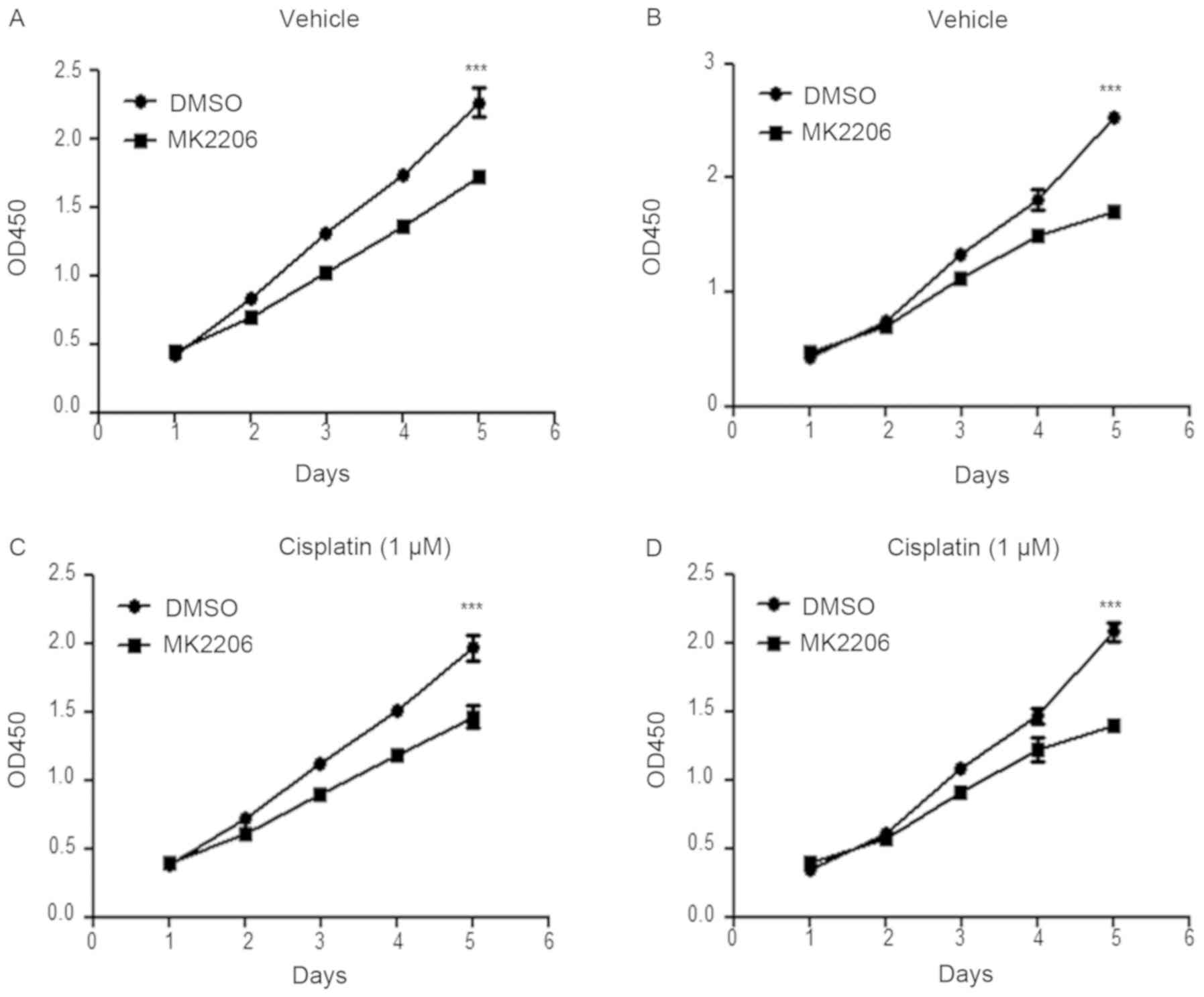
MK2206, an allosteric AKT inhibitor, has been
demonstrated to decrease cell proliferation in other types of
cancer (25–27). Following treatment with 1 µM MK2206,
the viability and proliferation of A549-USP17-OE and H1299-USP17-OE
cells were significantly decreased (DMSO vs. MK2206, P<0.001;
Fig. 5A and B), which further
supports the functional association between USP17 and AKT
activation. Moreover, the combination of the AKT inhibitor with
cisplatin significantly reduced cell viability and proliferation
(DMSO vs. MK2206, P<0.001; Fig. 5C
and D).
USP17 promotes proliferation and
invasion through PI3K/AKT activation in NSCLC
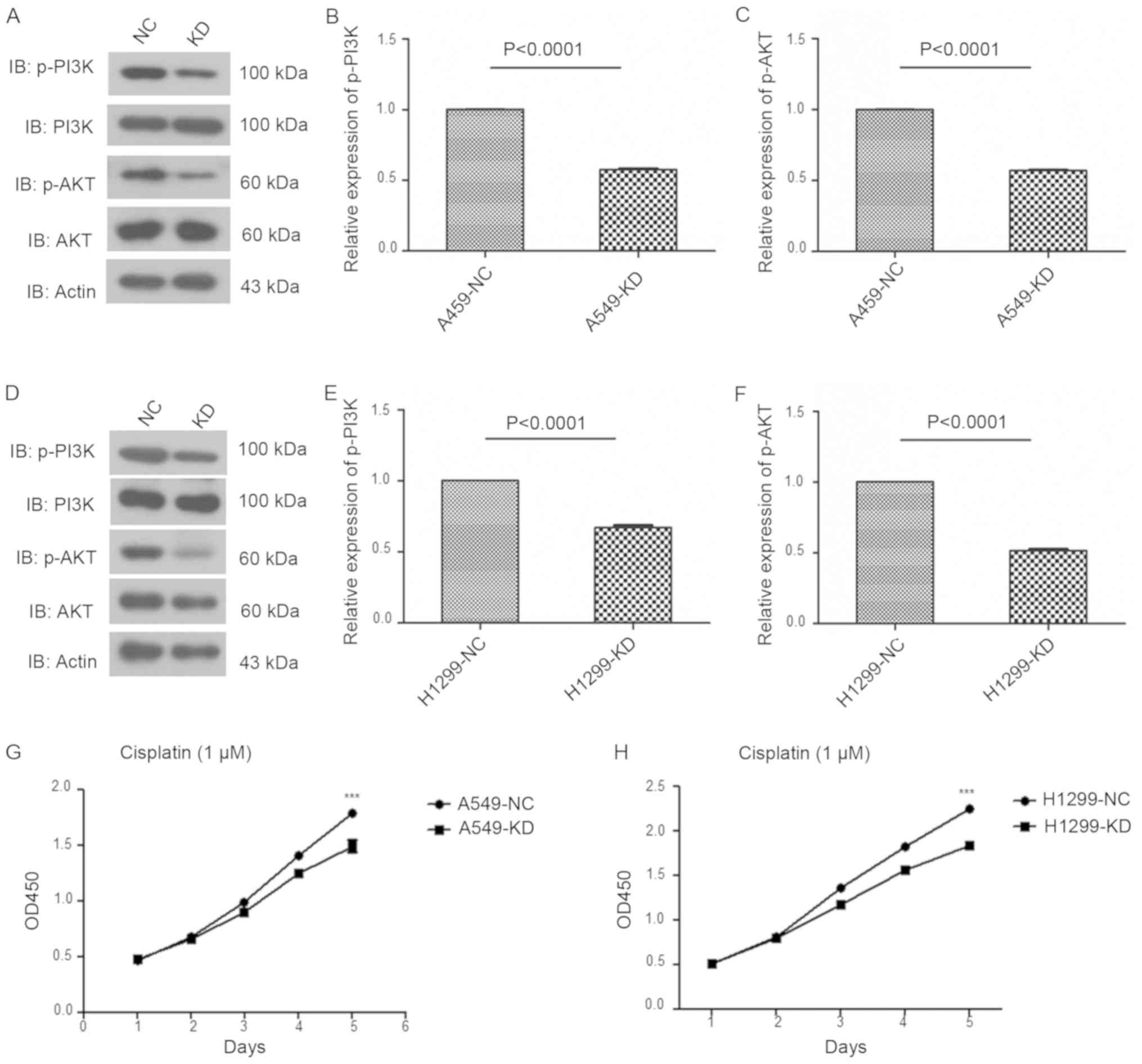
Consistent with the findings from the USP17-OE
cells, knock-down (KD) of USP17 decreased the protein expression
levels of p-AKT and p-PI3K in NSCLC cells (NC vs. KD, P<0.0001;
Fig. 6A-F). In addition, the
viability of USP17-KD cells was decreased following cisplatin
treatment (NC vs. KD, P<0.001; Fig.
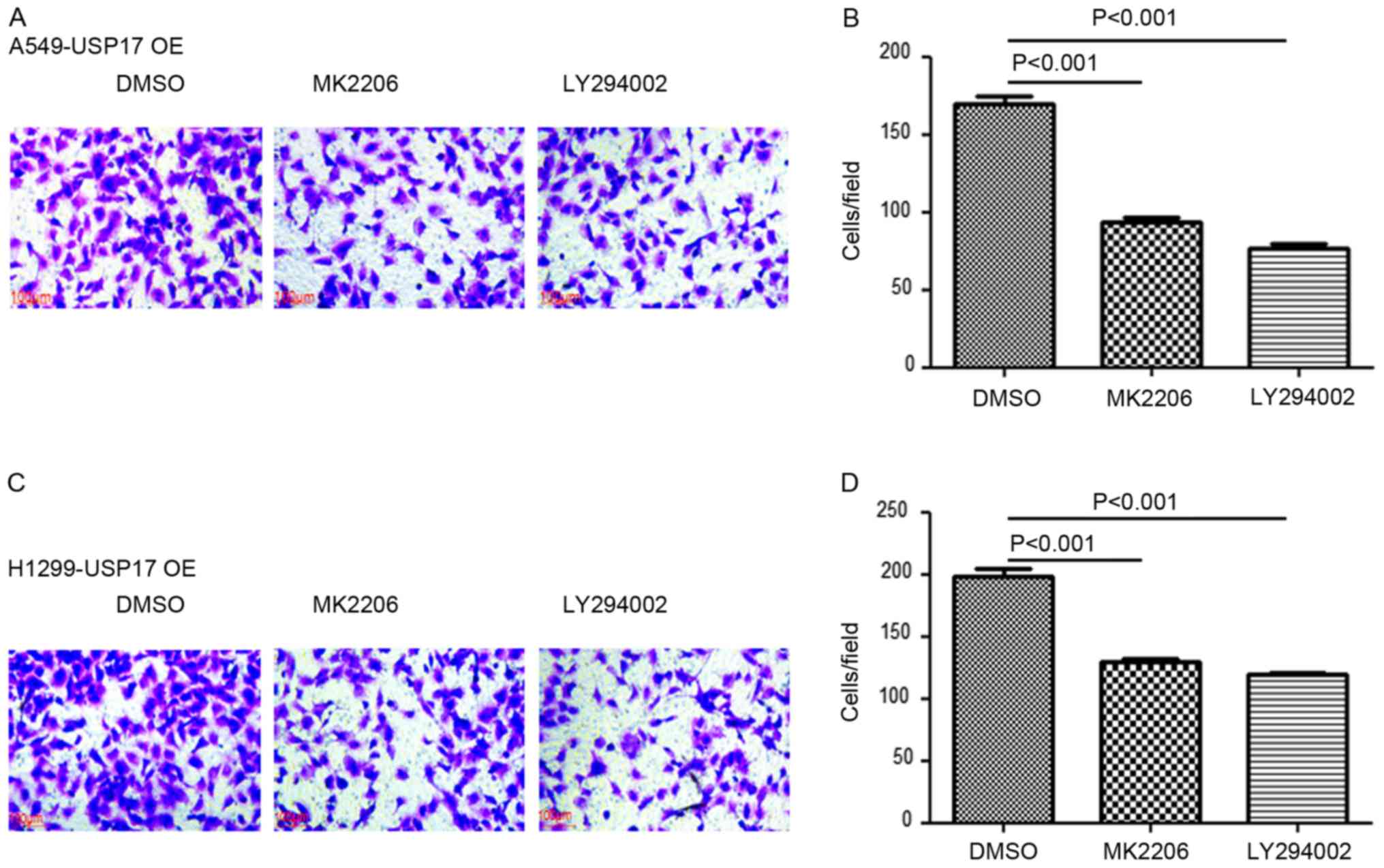
6G-H). Moreover, the invasive capacity of A549-USP17-OE and
H1299-USP17-OE cells was significantly decreased in the presence of
MK2206 and the PI3K inhibitor LY294002 (DMSO vs. MK2206,
P<0.001; DMSO vs. LY294002, P<0.001; Fig. 7).
Overall, the findings of the present study
demonstrate the promotion of NSCLC cell proliferation and viability
by USP17, via the activation of the PI3K/AKT pathway.
Discussion
The USP family is one of the five subfamilies of DUB
enzymes, which cleave polyubiquitin chains from proteins, a number
of studies have reported that USP17 has oncogenic characteristics.
Firstly, it has been shown that high levels of USP17 in lung,
colon, esophageal and cervical tumor samples promoted G1/S
transition and cellular proliferation (14–17).
Additionally, it was demonstrated that USP17 was expressed highly
in NSCLC tissues, and patients with high levels of USP17 exhibited
lower survival rates (16,17). Moreover, USP17 can be induced by
cytokines such as interleukin (IL)-4 and IL-6 (28). In the present study, it was found
that cisplatin treatment upregulated USP17 expression in a
dose-dependent manner. Moreover, increased cell proliferation was
found in USP17-OE cells compared with that of control cells, which
is consistent with previous studies (14–17).
Furthermore, it was demonstrated that the viability of USP17-OE
cells was significantly higher than that of the control cells, when
treated with cisplatin. In the present study USP17 regulated
cisplatin sensitivity in NSCLC. USP17 was initially identified as a
regulator of cell viability via signaling pathways associated with
cell death in cervical cancer (29).
It was previously reported that USP17 expression
could regulate Ras cellular localization and activation, via the
deubiquitination of Ras-converting enzyme 1, thereby inhibiting
phosphorylation of the downstream kinases, dual specificity
mitogen-activated protein kinase kinase and ERK (30). In osteosarcoma, USP17 facilitated
cell migration and invasion by deubiquitinating and stabilizing
SMAD4 (31). USP17- and
Skp1-cullin-1-F-box protein β F-box/WD repeat-containing protein
1A-regulated degradation of differentially expressed in
chondrocytes protein 1 has been shown to control the DNA-damage
response (32,33), thus indicating that the expression of
USP17 may be associated with sensitivity to chemotherapeutics,
including cisplatin.
In the present study, USP17 was found to promote the
growth of NSCLC cells via the activation of the PI3K/AKT pathway.
Aberrant activation of the PI3K/AKT pathway is often detected in
numerous types of human cancer; hence, targeting this pathway may
have therapeutic potential for the management of these tumors
(34–37). Emerging evidence indicates that the
activation of PI3K/AKT signaling by hypoxia may be a contributing
factor to drug resistance in certain types of human cancer,
including NSCLC and prostate cancer (38–40).
PI3K activation is regulated by various molecules in NSCLC, such as
DIX domain-containing protein 1, GRB2-associated-binding protein 2
and microRNAs (41–43).
There are limitations of the present study. Firstly,
the functions of USP17 in the response of NSCLC cells to cisplatin
treatment require further investigation, specifically using in
vivo animal models (44,45). Moreover, as the underlying mechanisms
of USP17 in the cisplatin response have not been investigated in
this study, it should be examined in the future.
Considerable effort has been made to identify agents
that target the activity of DUBs. However, no effective drugs have
yet entered into clinical trials. The findings from the present
study demonstrate the therapeutic potential of targeting PI3K/AKT
pathways downstream of USP17 to prevent NSCLC progression.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shanghai
Qingpu District Health and Planning Commission Research Fund (grant
no. W2017-21) and the Qingpu District Science and Technology
Commission (grant no. QKY2017-03).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SZ and HC contributed to designing research studies,
conducting experiments, acquiring data, analyzing data, providing
reagents and writing the manuscript. ZX and JY contributed to
conducting experiments and acquiring data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mao Y, Yang D and Krasna MJ: Epidemiology
of lung cancer. Surg Oncol Clin N Am. 25:439–445. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Azar FE, Azami-Aghdash S, Pournaghi-Azar
F, Mazdaki A, Rezapour A, Ebrahimi P and Yousefzadeh N:
Cost-effectiveness of lung cancer screening and treatment methods:
A systematic review of systematic reviews. BMC Health Serv Res.
17:4132017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jacobsen MM, Silverstein SC, Quinn M,
Waterston LB, Thomas CA, Benneyan JC and Han PKJ: Timeliness of
access to lung cancer diagnosis and treatment: A scoping literature
review. Lung Cancer. 112:156–164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Giaccone G: Clinical perspectives on
platinum resistance. Drugs. 59:9–37. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
D'Andrea A and Pellman D: Deubiquitinating
enzymes: A new class of biological regulators. Crit Rev Biochem Mol
Biol. 33:337–352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weathington NM and Mallampalli RK:
Emerging therapies targeting the ubiquitin proteasome system in
cancer. J Clin Invest. 124:6–12. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Welchman RL, Gordon C and Mayer RJ:
Ubiquitin and ubiquitin-like proteins as multifunctional signals.
Nat Rev Mol Cell Biol. 6:599–609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nijman SM, Luna-Vargas MP, Velds A,
Brummelkamp TR, Dirac AM, Sixma TK and Bernards R: A genomic and
functional inventory of deubiquitinating enzymes. Cell.
123:773–786. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Song H, Tao L, Chen C, Pan L, Hao J, Ni Y,
Li D, Li B and Shi G: USP17-mediated deubiquitination and
stabilization of HDAC2 in cigarette smoke extract-induced
inflammation. Int J Clin Exp Pathol. 8:10707–10715. 2015.PubMed/NCBI
|
|
10
|
de la Vega M, Kelvin AA, Dunican DJ,
McFarlane C, Burrows JF, Jaworski J, Stevenson NJ, Dib K, Rappoport
JZ, Scott CJ, et al: The deubiquitinating enzyme USP17 is essential
for GTPase subcellular localization and cell motility. Nat Commun.
2:2592011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Han L, Yang J, Wang X, Wu Q, Yin S, Li Z,
Zhang J, Xing Y, Chen Z, Tsun A, et al: The E3 deubiquitinase USP17
is a positive regulator of retinoic acid-related orphan nuclear
receptor γt (RORγt) in Th17 cells. J Biol Chem. 289:25546–25555.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Borbely G, Haldosen LA, Dahlman-Wright K
and Zhao C: Induction of USP17 by combining BET and HDAC inhibitors
in breast cancer cells. Oncotarget. 6:33623–33635. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu M, Chen H, Han C, Lan J, Xu Y, Li C,
Xue Y and Lou M: Expression and functional implications of USP17 in
glioma. Neurosci Lett. 616:125–131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McFarlane C, Kelvin AA, de la Vega M,
Govender U, Scott CJ, Burrows JF and Johnston JA: The
deubiquitinating enzyme USP17 is highly expressed in tumor
biopsies, is cell cycle regulated, and is required for G1-S
progression. Cancer Res. 70:3329–3339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McCann AP, Smyth P, Cogo F, McDaid WJ,
Jiang L, Lin J, Evergren E, Burden RE, Van Schaeybroeck S, Scott CJ
and Burrows JF: USP17 is required for trafficking and oncogenic
signaling of mutant EGFR in NSCLC cells. Cell Commun Signal.
16:772018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McFarlane C, McFarlane S, Paul I, Arthur
K, Scheaff M, Kerr K, Stevenson M, Fennell DA and Johnston JA: The
deubiquitinating enzyme USP17 is associated with non-small cell
lung cancer (NSCLC) recurrence and metastasis. Oncotarget.
4:1836–1843. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang S, Yuan J and Zheng R: Suppression
of Ubiquitin-Specific Peptidase 17 (USP17) inhibits tumorigenesis
and invasion in non-small cell lung cancer cells. Oncol Res.
24:263–269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Graff JR, Konicek BW, McNulty AM, Wang Z,
Houck K, Allen S, Paul JD, Hbaiu A, Goode RG, Sandusky GE, et al:
Increased AKT activity contributes to prostate cancer progression
by dramatically accelerating prostate tumor growth and diminishing
p27Kip1 expression. J Biol Chem. 275:24500–24505. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roy HK, Olusola BF, Clemens DL, Karolski
WJ, Ratashak A, Lynch HT and Smyrk TC: AKT proto-oncogene
overexpression is an early event during sporadic colon
carcinogenesis. Carcinogenesis. 23:201–205. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saini MK and Sanyal SN: PTEN regulates
apoptotic cell death through PI3-K/Akt/GSK3β signaling pathway in
DMH induced early colon carcinogenesis in rat. Exp Mol Pathol.
93:135–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xue G, Restuccia DF, Lan Q, Hynx D,
Dirnhofer S, Hess D, Ruegg C and Hemmings BA: Akt/PKB-mediated
phosphorylation of Twist1 promotes tumor metastasis via mediating
cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer
Discov. 2:248–259. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Yang Q, Guan H, Shi B, Ji M and Hou
P: ZNF677 suppresses Akt phosphorylation and tumorigenesis in
thyroid cancer. Cancer Res. 78:5216–5228. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang Y, Xiao G, Chen Y and Deng Y: LncRNA
MALAT1 promotes migration and invasion of non-small-cell lung
cancer by targeting miR-206 and activating Akt/mTOR signaling.
Anticancer Drugs. 29:725–735. 2018.PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Malkomes P, Lunger I, Luetticke A,
Oppermann E, Haetscher N, Serve H, Holzer K, Bechstein WO and
Rieger MA: Selective AKT Inhibition by MK-2206 represses colorectal
cancer-initiating stem cells. Ann Surg Oncol. 23:2849–2857. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wisinski KB, Tevaarwerk AJ, Burkard ME,
Rampurwala M, Eickhoff J, Bell MC, Kolesar JM, Flynn C and Liu G:
Phase I Study of an AKT Inhibitor (MK-2206) combined with lapatinib
in adult solid tumors followed by dose expansion in advanced HER2+
breast cancer. Clin Cancer Res. 22:2659–2667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Agarwal E, Chaudhuri A, Leiphrakpam PD,
Haferbier KL, Brattain MG and Chowdhury S: Akt inhibitor MK-2206
promotes anti-tumor activity and cell death by modulation of AIF
and Ezrin in colorectal cancer. BMC Cancer. 14:1452014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baek KH: Cytokine-regulated protein
degradation by the ubiquitination system. Curr Protein Pept Sci.
7:171–177. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shin JM, Yoo KJ, Kim MS, Kim D and Baek
KH: Hyaluronan- and RNA-binding deubiquitinating enzymes of USP17
family members associated with cell viability. BMC Genomics.
7:2922006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Burrows JF, Kelvin AA, McFarlane C, Burden
RE, McGrattan MJ, De la Vega M, Govender U, Quinn DJ, Dib K, Gadina
M, et al: USP17 regulates Ras activation and cell proliferation by
blocking RCE1 activity. J Biol Chem. 284:9587–9595. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song C, Liu W and Li J: USP17 is
upregulated in osteosarcoma and promotes cell proliferation,
metastasis, and epithelial-mesenchymal transition through
stabilizing SMAD4. Tumour Biol. 39:10104283177171382017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang M, He SF, Liu LL, Sun XX, Yang F, Ge
Q, Wong WK and Meng JY: Potential role of ZEB1 as a DNA repair
regulator in colorectal cancer cells revealed by cancer-associated
promoter profiling. Oncol Rep. 38:1941–1948. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim J, D'Annibale S, Magliozzi R, Low TY,
Jansen P, Shaltiel IA, Mohammed S, Heck AJ, Medema RH and
Guardavaccaro D: USP17- and SCFβTrCP-regulated degradation of DEC1
controls the DNA damage response. Mol Cell Biol. 34:4177–4185.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu J, Sun Y, Lu Y, Jiang X, Ma B, Yu L,
Zhang J, Dong X and Zhang Q: Glaucocalyxin A exerts anticancer
effect on osteosarcoma by inhibiting GLI1 nuclear translocation via
regulating PI3K/Akt pathway. Cell Death Dis. 9:7082018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koundouros N and Poulogiannis G:
Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in
Cancer. Front Oncol. 8:1602018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zheng J, Zhang M, Zhang L, Ding X, Li W
and Lu S: HSPC159 promotes proliferation and metastasis by inducing
epithelial-mesenchymal transition and activating the PI3K/Akt
pathway in breast cancer. Cancer Sci. 109:2153–2163. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhong C, Chen Y, Tao B, Peng L, Peng T,
Yang X, Xia X and Chen L: LIM and SH3 protein 1 regulates cell
growth and chemosensitivity of human glioblastoma via the PI3K/AKT
pathway. BMC Cancer. 18:7222018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gong T, Cui L, Wang H, Wang H and Han N:
Knockdown of KLF5 suppresses hypoxia-induced resistance to
cisplatin in NSCLC cells by regulating HIF-1α-dependent glycolysis
through inactivation of the PI3K/Akt/mTOR pathway. J Transl Med.
16:1642018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
O'Reilly D, Johnson P and Buchanan PJ:
Hypoxia induced cancer stem cell enrichment promotes resistance to
androgen deprivation therapy in prostate cancer. Steroids.
152:1084972019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu W, Yu WM, Zhang J, Chan RJ, Loh ML,
Zhang Z, Bunting KD and Qu CK: Inhibition of the Gab2/PI3K/mTOR
signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2)
gain-of-function mutations. Leukemia. 31:1415–1422. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang L, Cao XX, Chen Q, Zhu TF, Zhu HG and
Zheng L: DIXDC1 targets p21 and cyclin D1 via PI3K pathway
activation to promote colon cancer cell proliferation. Cancer Sci.
100:1801–1808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao W, Sun Q, Yu Z, Mao S, Jin Y, Li J,
Jiang Z, Zhang Y, Chen M, Chen P, et al: MiR-320a-3p/ELF3 axis
regulates cell metastasis and invasion in non-small cell lung
cancer via PI3K/Akt pathway. Gene. 670:31–37. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao J, Xu J and Zhang R: SRPX2 regulates
colon cancer cell metabolism by miR-192/215 via PI3K-Akt. Am J
Transl Res. 10:483–490. 2018.PubMed/NCBI
|
|
44
|
Zhang B, Liu L, Guan H, Wang H, Zhang Z
and Zhou P: HepG2 cell cycle related gene transcriptional profiles
are altered by a novel vanillin derivative BVAN08. J Med Discov.
2:170362017. View Article : Google Scholar
|
|
45
|
Deng N and Chen Y: Application of
CRISPR-Cas9 gene editing system: Non-viral delivery strategies and
improvements. J Med Discov. 3:170572018.
|