Leukemia is characterized by the presence of
abnormally large numbers of immature, poorly differentiated white
blood cells in the blood; the condition often begins with bone
marrow abnormalities (1). In
children, acute lymphoblastic leukemia (ALL) is the most common
type of blood cancer. In the United States, ~4,000 patients are
diagnosed each year, accounting for ~30% of childhood cancer types
(2,3). Among these ALL cases, >80% result
from clonal proliferation of abnormal B cell progenitors (B-ALL)
(4). In recent years, cytosolic
signal transduction and molecular abnormality have been shown to
play important roles in the pathogenesis of B-ALL; the
abnormalities include gene mutations, abnormal protein interaction,
unarrested cell cycle and increased autophagy (5,6).
Knowledge on these abnormalities could help develop gene targeted
therapy. In the wake of medical improvements, increasingly more
risk factors have been discovered for B-ALL; minimizing these risk
factors could therefore lower its prevalence. Over the last few
years, the cure rate of B-ALL has substantially been raised.
Despite this improvement, relapse rate maintains at ~15% for
patients with B-ALL (7). Such
treatment failure could be associated with multiple drug resistance
or the abnormal expression of intracellular enzymes.
This present review summarized pediatric B-ALL, its
pathogenesis, risk factors, current treatment, and the monitoring
of treatment efficacy and treatment failure. Better understanding
of the underlying mechanisms of B-ALL could facilitate the
development of novel target therapies to reduce the prevalence of
B-ALL and the likelihood of relapse.
Lymphoid cells are initially developed from
pluripotent hematopoietic stem cells in the bone marrow. B cells
are primarily differentiated from common lymphoid progenitor cells
pro-B cells, pre-B cells and mature B cells. The maturation process
is normally well managed by cell signal transduction, activated
transcription factors and positive/negative selection (8). However, in the case of B-ALL,
malignancies of B precursor-stage lymphoid cells inhibit lymphoid
differentiation, leading to abnormal cell proliferation and
survival (9). The occurrence of
B-ALL is known to correlate with a series of gene mutations, which
often start at the pluripotent stem cell stage, followed by
processes of clonal expansion, differentiation, cell proliferation
and dysregulated cell apoptosis; the end result is the replacement
of normal lymphoid cells by malignant cells (10). A number of molecular pathways and
gene expressions are known to be associated with ALL, which are
elaborated below.
The ‘two hits hypothesis’ has been proposed to
explain the tumorigenesis of childhood ALL (11). Specifically, the TEL-AML1 fusion gene
is a mutation that could be present years before any clinical
symptom appears, and often the mutation has taken place earlier
in utero (12). This oncogene
can act on the hematopoietic stem cell (HSC) and produces gene
lesions; after the second genetic mutation (or environment hit),
the multistep pathway is then activated to develop ALL (13,14).
TEL-AML1 is therefore the molecular lesion that initiates the
disease; here, the leukemic cells stay at the pro-B-cell stage
(13,14). Experiments on cord blood samples are
in support of the TEL-AML1 gene mutation occurring in utero
(13,14). TEL-AML1-positive subjects have higher
risks in developing ALL. TEL-AML1 potentially induces HSC expansion
and accelerates transformation (12). Such series of events is consistent
with the ‘two hits hypothesis’ in that two gene mutations are
involved for the subsequent development of the malignancy (15).
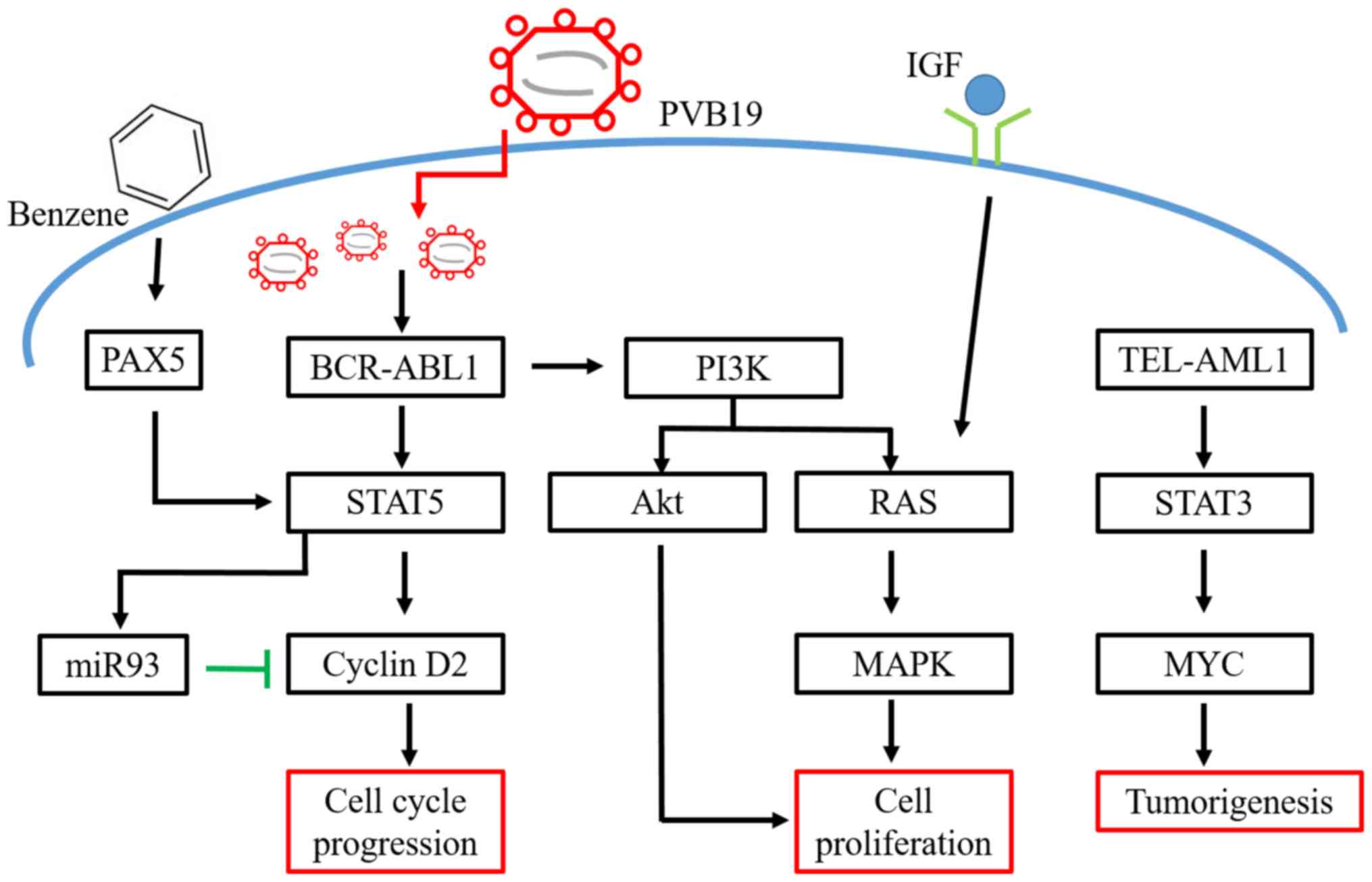
The ABL gene on chromosome 9 switches location with
the BCR gene on chromosome 22 to form the BCR-ABL fusion gene.
Chromosome 22 with the new fusion gene is referred to as the
Philadelphia chromosome (Ph) (16).
The BCR-ABL1 tyrosine kinase gene, transcribed at the Ph
chromosome, is the most common mutation in B-cell ALL. Its worst
prognosis is often associated with Ph, BCR-ABL-1 positive gene
mutation (17). BCR-ABL can promote
complex formation of GRB2, GAB2 and Son-of-Sevenless, with
subsequent activation of RAS and recruitment of PI3K (18). The activation of RAS triggers
signaling pathways of mitogen-activated protein kinase (MAPK) and
stimulates cell proliferation. In mediating cell survival and
proliferation, PI3K activates its downstream target, the
serine-threonine kinase Akt and suppresses the activity of forkhead
O transcriptional factors, degrading p27 and activating mTOR
(19,20). To stimulate cell proliferation,
BCR-ABL can regulate STAT5 activation, which also enhances cyclin
D2 expression through the downregulated expression of miR-93
(21–23).
Paired box protein Pax-5 is a B cell activator
protein, which encodes nuclear transcriptional factors. It
modulates B cell functions, including development, differentiation,
migration and proliferation (24).
Pax-5 controls B cell development from pro to mature B cells.
Abnormal expressions of Pax-5 can lead to leukemic transformation
at the early stage of tumorigenesis in B-ALL (25). The development of pro B cells is
arrested under downregulated Pax-5 expression, an evidence in
support of the critical role of Pax-5 on B cell development. Over
90% pediatric patients with B-ALL have overexpressed Pax-5
(24). Pax-5 can fuse with other
proteins, such as Janus kinase (Jak) 2, to create an active kinase
domain, leading to B cell proliferation via the Jak-STAT signaling
pathways (26).
Patients with ALL and poor prognosis or relapses
often have mutations in the RAS pathways; these mutations
frequently occur during chemotherapy and are present in clones of
relapsed leukemic cells (27). A
recent study sequenced 13 RAS pathway genes derived from 461
initially diagnosed pediatric patients with B cell precursor-ALL
and reported that 44.2% of patients displayed mutations in their
RAS pathways (28). Such RAS
mutations are also present in ~40% of relapsed pediatric patients
with ALL (27). The activation of
RAS pathways in leukemic cells impairs the efficacy of medical
therapy using drugs such as glucocorticoids or anthracycline
(29,30). HSC cells with RAS gene mutations show
uncontrolled growth (31).
Approximately 15% of pediatric patients with ALL have mutations on
both NRAS and KRAS genes. These mutations, however, show no
correlation with any other clinical symptom (32,33).
The PI3K/Akt signaling pathway is involved in cell
proliferation and cell survival. PI3K regulates the expression
levels of mTOR, Bcl-2, NFκB and other proteins that all promote
cell proliferation (34,35). The PI3K/Akt signaling pathway is
activated in various types of liquid tumors such as B cell
precursor-ALL (36) and hence it
serves an important role in pathogenesis (37). In the leukemia microenvironment,
marrow stromal cells (MSCs) promote the proliferation of leukemic
cells and strengthen their resistance to chemotherapy, through
PI3K/Akt signaling pathway (38).
MSCs secrete C-X-C motif chemokine 12 that acts on C-X-C chemokine
receptor type 4 of the leukemia blast cells and through the PI3K
and Wnt pathways exert influences on their survival and
proliferation (39). Overactivated
PI3K pathway is frequently found in B-ALL and such overactivation
is also associated with glucocorticoid resistance (40). Patients with B-ALL bearing negative
regulators of the PI3E mutation, such as phosphatase and tensin
homolog (PTEN), may have a higher chance of treatment failure and
relapse (41).
Benzene is widely present in the work environment,
such as in fumes of paint and cigarette smoke. Benzene is a known
carcinogen and parental exposure to benzene increases the risk of
childhood ALL (56). In the liver or
lung, benzene is metabolized to benzene oxide and hydroquinone,
before further converted to benzoquinone in the bone marrow, where
cytotoxicity and DNA damages typically occur (57). Prolonged DNA damages in the bone
marrow increases the risk of developing ALL (58). A recent study reported that avoiding
exposures to occupational and environmental benzene during
pregnancy lowers the risk of ALL in the offspring (59).
Currently, for childhood B-ALL, risk-directed
therapy is the standard treatment. The age of the child at
diagnosis is taken into account. Other factors considered are the
initial white blood cell count, immunophenotypic and cytogenetic
characteristics of the blast population, as well as the rapidity of
response to early treatment. Standard treatment consists of
chemotherapy that lasts 2–3 years. For many patients, complete
remission (CR) is achieved at the end of the induction phase
(60).
The success of such treatment involves a multidrug
regimen that consists of three phases (induction, consolidation and
maintenance), during which therapy or prophylaxis directed to the
central nervous system (CNS) is delivered in multiple sessions
(61).
The primary goal of induction is to reach an initial
CR and to restore normal hematopoiesis. Induction therapy involves
a weekly medication of vincristine and anthracycline for 3–4 weeks,
daily corticosteroids and asparaginase (62).
The routine use of preventive CNS therapy is a major
therapeutic advancement in the treatment of childhood ALL. CNS
treatment usually begins in the induction phase and lasts until the
end of treatment regimen. In some CNS preventive therapy protocols,
the craniospinal radiotherapy is replaced by intrathecal
chemotherapy (63).
The second phase of treatment, or consolidation, is
focused on intensive CNS therapy in combination with sustained
intensive systemic therapy. This phase of treatment starts shortly
after the patient has reached CR. The goal of consolidation
chemotherapy is to prevent leukemic regrowth, reduce residual tumor
burden and prevent the emergence of drug-resistance in other
leukemic cells (64). A combination
of several drugs is typically involved with pharmacological
mechanisms that are different from those in the induction phase;
regimens often include the following drugs: Cytarabine,
methotrexate, anthracyclines and alkylating agents (65–67).
These drugs are administered according to schedules that would
maximize drug synergy and minimize drug resistance.
After completing the consolidation (or
intensification) phase of therapy, patients will often receive a
less intensive continuation regimen (maintenance chemotherapy) that
involves daily oral 6-mercaptopurine, weekly methotrexate with
periodic vincristine, corticosteroid and intrathecal therapy.
Maintenance phase lasts for another 2–3 years. Extending the
maintenance phase thereafter has little benefits (68,69).
Approximately 15% of patients would relapse after
treatment and the overall survival of patients with relapse is
<10% (70). Thus, it is important
to develop new ALL treatment strategies that have higher CR and
lower drug toxicity. In recent years, resveratrol has been reported
to induce apoptosis, cell cycle arrest or decrease cell
proliferation through the enhanced expressions of p21 and p27
(71). The reduced expressions of
antiapoptotic proteins, myeloid cell leukemia 1 and Bcl-2 and
increased expressions of proapoptotic proteins, Bax, Bcl-2-like
protein 11 and Bad, have resulted in upregulating caspase 3
(72). In addition, everolimus is an
mTOR inhibitor, which induces autophagy and promotes cell death
(73). This drug induces apoptosis
through caspase-independent pathways and alters the mitochondrial
permeability, leading to cell dysfunction (70). Everolimus kills malignant cells
through paraptosis (70). A specific
inhibitor of mitogen activated protein kinase (MEK), selumetinib,
could treat ALL. It acts on MEK1/2 to inhibit ERK-dependent cell
proliferation (74). This drug has
been used to treat RAS-mutated ALL with good results in
vitro and in vivo (27,75).
Besides these aforementioned target therapies, other new strategies
to treat ALL are outlined below.
Autophagy is the process of breaking down
intracellular organelles for energy transfer to maintain
homeostasis compatible with cell viability. The process of
autophagy is switched on when cells are stressed or damaged to a
certain degree. Leukemic cells resistant to chemotherapy are found
to have activated autophagy (76).
Thus, targeting autophagy is a promising new therapeutic approach
to treat B-ALL. In models of ALL cell line, newly developed drugs
produce autophagy, cell cycle arrest and apoptosis. However, after
combined treatment with autophagy inhibitor, cell viability is
often downregulated. Results indicate that autophagy protects
leukemic cells against cytotoxicity caused by anti-cancer drugs
(77,78). This approach of autophagy
manipulation might be an alternative for ALL treatment.
c-Myb is the protein product of a proto-oncogene. It
has three domains: N-terminal DNA binding domain, central
transcriptional activation domain and C-terminal domain (79). The gene is carried by the avian
myeloblastosis virus and the E26 retrovirus (80). During proliferation, especially of
immature cells, the c-Myb transcription factor is overexpressed
(81). In leukemia animal models and
in vitro systems, c-Myb inhibitors delay disease onset
(2). Inhibition of c-Myb
downregulates leukemic cell proliferation, increases G0/G1 arrest
and increases sensitivity of pre-B-ALL cells to cytotoxic agents
(82,83). Experimental evidence is in support of
c-Myb being a potential target for immunotherapy of ALL (83). Furthermore, other tumor cells also
express c-Myb, making c-Myb-targeting therapy promising in clinical
practice (84).
It is important to evaluate treatment efficacy of
any ALL therapy. Apart from the traditional blood test, the levels
of microRNA and soluble interleukin receptors are used to monitor
the treatment efficacy (85,86). In a whole genome microRNA analysis on
patients with ALL, dysregulated expression of miR-128, miR-146a,
miR-155, miR-181a and miR-195 were found (87). Treatments lasting for 6 months
resulted in downregulated expressions of miR-146a, miR-155,
miR-181a and miR-195 (87). Soluble
interleukin 2 receptor (sIL-2R) has been identified as a biomarker
of the disease activity of non-Hodgkin's lymphoma and it also
reflects the tumor volume (88).
Increased expression of sIL-2R are present in other
lymphoproliferative disorders such as chronic lymphocytic leukemia
and ALL. Immature blast cells release sIL-2R before entering the
blood stream (89). Therefore,
sIL-2R is useful for monitoring the treatment efficacy of ALL.
For B-ALL patients, the frequency of relapse is
associated with multiple drug resistance (MDR) (90). The overexpression of drug efflux
pumps on leukemic cells under drug treatment is the most common
condition for MDR and consequently the intracellular drug
concentration is lowered (91).
ATP-binding cassette (ABC) transporters actively pump drugs out of
cells and their expression level is regulated by a number of
intracellular signaling pathways, including MAPK, ERK and JNK
(92,93). The ABC transporter pump is encoded by
genes including ABCB1, ABCC1 and ABCG2. Higher expression of ABCB1
gene has been reported in ALL cell lines and higher expression of
ABCB1 is known to associate with poorer prognosis and shorter
disease-free survival rates (94,95).
Another mechanism of MDR concerns glutathione (GSH) and its
associated enzymes, which are a part of the cell defense system
against chemodrug-induced reactive oxygen species stress (96,97).
Glutathione reductase is the key enzyme of the GSH redox cycle and
it reduces the sensitivity to chemodrugs in leukemic cells by
modulating redox homeostasis (98).
B-ALL is the most prevalent cancer in children.
Despite better cure rates in recent years, the disease will still
relapse for some patients. Therefore, understanding the molecular
pathways for the pathophysiology of B-ALL and its relapse
mechanisms are important (Fig. 1).
Not all patients with B-ALL have satisfactory outcomes under the
current treatments. Newly developed immune-targeting therapy would
probably enhance the efficacy of chemotherapy and improve
prognosis. Biomarkers can be used to monitor treatment course and
to provide information for drug and dose adjustments. Well-designed
clinical trials should be conducted to gain insights for
constructing more effective immune targeting therapies and
treatment protocols.
Not applicable.
The present work was supported by a grant from
Taichung Veterans General Hospital (grant no. TCVGH-NCHU1097611;
Taichung, Taiwan, R.O.C.).
Not applicable.
FLH, CYY and SJY drafted the manuscript. ECL and CLL
were involved in literature review and revising the manuscript. CYY
and SJY edited the manuscript and were involved in the conception
and design of this review article. All authors read and approved
the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Shafat MS, Gnaneswaran B, Bowles KM and
Rushworth SA: The bone marrow microenvironment-Home of the leukemic
blasts. Blood Rev. 31:277–286. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sarvaiya PJ, Schwartz JR, Hernandez CP,
Rodriguez PC and Vedeckis WV: Role of c-Myb in the survival of pre
B-cell acute lymphoblastic leukemia and leukemogenesis. Am J
Hematol. 87:969–976. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chokkalingam AP, Metayer C, Scelo G, Chang
JS, Schiffman J, Urayama KY, Ma X, Hansen HM, Feusner JH, Barcellos
LF, et al: Fetal growth and body size genes and risk of childhood
acute lymphoblastic leukemia. Cancer Causes Control. 23:1577–1585.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu YF, Wang BY, Zhang WN, Huang JY, Li
BS, Zhang M, Jiang L, Li JF, Wang MJ, Dai YJ, et al: Genomic
profiling of adult and pediatric B-cell acute lymphoblastic
leukemia. EBioMedicine. 8:173–183. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Consolaro F, Basso G, Ghaem-Magami S, Lam
EW and Viola G: FOXM1 is overexpressed in B-acute lymphoblastic
leukemia (B-ALL) and its inhibition sensitizes B-ALL cells to
chemotherapeutic drugs. Int J Oncol. 47:1230–1240. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Z, Zhu S, Zhang G and Liu S:
Inhibition of autophagy enhances the anticancer activity of
bortezomib in B-cell acute lymphoblastic leukemia cells. Am J
Cancer Res. 5:639–650. 2015.PubMed/NCBI
|
|
7
|
Tran TH, Harris MH, Nguyen JV, Blonquist
TM, Stevenson KE, Stonerock E, Asselin BL, Athale UH, Clavell LA,
Cole PD, et al: Prognostic impact of kinase-activating fusions and
IKZF1 deletions in pediatric high-risk B-lineage acute
lymphoblastic leukemia. Blood Adv. 2:529–533. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou Y, You MJ, Young KH, Lin P, Lu G,
Medeiros LJ and Bueso-Ramos CE: Advances in the molecular
pathobiology of B-lymphoblastic leukemia. Hum Pathol. 43:1347–1362.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zuckerman T and Rowe JM: Pathogenesis and
prognostication in acute lymphoblastic leukemia. F1000Prime Rep.
6:592014. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bowman RL, Busque L and Levine RL: Clonal
hematopoiesis and evolution to hematopoietic malignancies. Cell
Stem Cell. 22:157–170. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morales-Sánchez A and Fuentes-Panana EM:
Infectious etiology of childhood acute lymphoblastic leukemia,
hypotheses and evidence. In: Clinical Epidemiology of Acute
Lymphoblastic Leukemia: From the Molecules to the Clinic.
Mejia-Arangure JM: InTech Rijeka; Croatia: pp. 19–39. 2013
|
|
12
|
Schindler JW, Van Buren D, Foudi A, Krejci
O, Qin J, Orkin SH and Hock H: TEL-AML1 corrupts hematopoietic stem
cells to persist in the bone marrow and initiate leukemia. Cell
Stem Cell. 5:43–53. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ford AM, Bennett CA, Price CM, Bruin MC,
Van Wering ER and Greaves M: Fetal origins of the TEL-AML1 fusion
gene in identical twins with leukemia. Proc Natl Acad Sci USA.
95:4584–4588. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sabaawy HE, Azuma M, Embree LJ, Tsai HJ,
Starost MF and Hickstein DD: TEL-AML1 transgenic zebrafish model of
precursor B cell acute lymphoblastic leukemia. Proc Natl Acad Sci
USA. 103:15166–15171. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jan M and Majeti R: Clonal evolution of
acute leukemia genomes. Oncogene. 32:135–140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
El Fakih R, Jabbour E, Ravandi F,
Hassanein M, Anjum F, Ahmed S and Kantarjian H: Current paradigms
in the management of Philadelphia chromosome positive acute
lymphoblastic leukemia in adults. Am J Hematol. 93:286–295. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thomas DA, Faderl S, Cortes J, O'Brien S,
Giles FJ, Kornblau SM, Garcia-Manero G, Keating MJ, Andreeff M,
Jeha S, et al: Treatment of Philadelphia chromosome-positive acute
lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood.
103:4396–4407. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cilloni D and Saglio G: Molecular
pathways: BCR-ABL. Clin Cancer Res. 18:930–937. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ho WC, Pikor L, Gao Y, Elliott BE and
Greer PA: Calpain 2 regulates Akt-FoxO-p27(Kip1) protein signaling
pathway in mammary carcinoma. J Biol Chem. 287:15458–15465. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mantamadiotis T: Towards targeting
PI3K-dependent regulation of gene expression in brain cancer.
Cancers (Basel). 9(pii): E602017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schotte D, De Menezes RX, Akbari Moqadam
F, Khankahdani LM, Lange-Turenhout E, Chen C, Pieters R and Den
Boer ML: MicroRNA characterize genetic diversity and drug
resistance in pediatric acute lymphoblastic leukemia.
Haematologica. 96:703–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deininger MW, Vieira SA, Parada Y, Banerji
L, Lam EW, Peters G, Mahon FX, Köhler T, Goldman JM and Melo JV:
Direct relation between BCR-ABL tyrosine kinase activity and cyclin
D2 expression in lymphoblasts. Cancer Res. 61:8005–8013.
2001.PubMed/NCBI
|
|
23
|
Parada Y, Banerji L, Glassford J, Lea NC,
Collado M, Rivas C, Lewis JL, Gordon MY, Thomas NS and Lam EW:
BCR-ABL and interleukin 3 promote haematopoietic cell proliferation
and survival through modulation of cyclin D2 and p27Kip1
expression. J Biol Chem. 276:23572–23580. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Firtina S, Sayitoglu M, Hatirnaz O,
Erbilgin Y, Oztunc C, Cinar S, Yildiz I, Celkan T, Anak S, Unuvar
A, et al: Evaluation of PAX5 gene in the early stages of leukemic B
cells in the childhood B cell acute lymphoblastic leukemia. Leuk
Res. 36:87–92. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tiacci E, Pileri S, Orleth A, Pacini R,
Tabarrini A, Frenguelli F, Liso A, Diverio D, Lo-Coco F and Falini
B: PAX5 expression in acute leukemias: higher B-lineage specificity
than CD79a and selective association with t(8;21)-acute myelogenous
leukemia. Cancer Res. 64:7399–7404. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schinnerl D, Fortschegger K, Kauer M,
Marchante JR, Kofler R, Den Boer ML and Strehl S: The role of the
Janus-faced transcription factor PAX5-JAK2 in acute lymphoblastic
leukemia. Blood. 125:1282–1291. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Irving J, Matheson E, Minto L, Blair H,
Case M, Halsey C, Swidenbank I, Ponthan F, Kirschner-Schwabe R,
Groeneveld-Krentz S, et al: Ras pathway mutations are prevalent in
relapsed childhood acute lymphoblastic leukemia and confer
sensitivity to MEK inhibition. Blood. 124:3420–3430. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jerchel IS, Hoogkamer AQ, Ariës IM,
Steeghs EMP, Boer JM, Besselink NJM, Boeree A, van de Ven C, de
Groot-Kruseman HA, de Haas V, et al: RAS pathway mutations as a
predictive biomarker for treatment adaptation in pediatric B-cell
precursor acute lymphoblastic leukemia. Leukemia. 32:931–940. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jones CL, Gearheart CM, Fosmire S,
Delgado-Martin C, Evensen NA, Bride K, Waanders AJ, Pais F, Wang J,
Bhatla T, et al: MAPK signaling cascades mediate distinct
glucocorticoid resistance mechanisms in pediatric leukemia. Blood.
126:2202–2212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang J, Wang J, Liu Y, Sidik H, Young KH,
Lodish HF and Fleming MD: Oncogenic Kras-induced leukemogeneis:
Hematopoietic stem cells as the initial target and lineage-specific
progenitors as the potential targets for final leukemic
transformation. Blood. 113:1304–1314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shu XO, Perentesis JP, Wen W, Buckley JD,
Boyle E, Ross JA and Robison LL; Children's Oncology Group, :
Parental exposure to medications and hydrocarbons and ras mutations
in children with acute lymphoblastic leukemia: A report from the
Children's Oncology Group. Cancer Epidemiol Biomarkers Prev.
13:1230–1235. 2004.PubMed/NCBI
|
|
33
|
Al-Kzayer LF, Sakashita K, Al-Jadiry MF,
Al-Hadad SA, Ghali HH, Uyen Le TN, Liu T, Matsuda K, Abdulkadhim
JM, Al-Shujairi TA, et al: Analysis of KRAS and NRAS Gene Mutations
in Arab Asian Children With Acute Leukemia: High Frequency of RAS
Mutations in Acute Lymphoblastic Leukemia. Pediatr Blood Cancer.
62:2157–2161. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li H, Zeng J and Shen K: PI3K/AKT/mTOR
signaling pathway as a therapeutic target for ovarian cancer. Arch
Gynecol Obstet. 290:1067–1078. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu MH, Lee TH, Lee HP, Li TM, Lee IT,
Shieh PC and Tang CH: Kuei-Lu-Er-Xian-Jiao extract enhances BMP-2
production in osteoblasts. Biomedicine (Taipei). 7:22017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Toosi B, Zaker F, Alikarami F, Kazemi A
and Teremmahi Ardestanii M: VS-5584 as a PI3K/mTOR inhibitor
enhances apoptotic effects of subtoxic dose arsenic trioxide via
inhibition of NF-κB activity in B cell precursor-acute
lymphoblastic leukemia. Biomed Pharmacother. 102:428–437. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Morishita N, Tsukahara H, Chayama K,
Ishida T, Washio K, Miyamura T, Yamashita N, Oda M and Morishima T:
Activation of Akt is associated with poor prognosis and
chemotherapeutic resistance in pediatric B-precursor acute
lymphoblastic leukemia. Pediatr Blood Cancer. 59:83–89. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sanchez VE, Nichols C, Kim HN, Gang EJ and
Kim YM: Targeting PI3K signaling in acute lymphoblastic leukemia.
Int J Mol Sci. 20(pii): E4122019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang Y, Mallampati S, Sun B, Zhang J, Kim
SB, Lee JS, Gong Y, Cai Z and Sun X: Wnt pathway contributes to the
protection by bone marrow stromal cells of acute lymphoblastic
leukemia cells and is a potential therapeutic target. Cancer Lett.
333:9–17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Evangelisti C, Cappellini A, Oliveira M,
Fragoso R, Barata JT, Bertaina A, Locatelli F, Simioni C, Neri LM,
Chiarini F, et al: Phosphatidylinositol 3-kinase inhibition
potentiates glucocorticoid response in B-cell acute lymphoblastic
leukemia. J Cell Physiol. 233:1796–1811. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Silveira AB, Laranjeira AB, Rodrigues GO,
Leal PC, Cardoso BA, Barata JT, Yunes RA, Zanchin NI, Brandalise SR
and Yunes JA: PI3K inhibition synergizes with glucocorticoids but
antagonizes with methotrexate in T-cell acute lymphoblastic
leukemia. Oncotarget. 6:13105–13118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sánchez-Beato M, Sánchez-Aguilera A and
Piris MA: Cell cycle deregulation in B-cell lymphomas. Blood.
101:1220–1235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang MM and Zhu J: The regulation of
normal and leukemic hematopoietic stem cells by niches. Cancer
Microenviron. 5:295–305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang Y, Xue K, Li Z, Zheng W, Dong W, Song
J, Sun S, Ma T and Li W: c-Myc regulates the CDK1/cyclin B1
dependentG2/M cell cycle progression by histone H4 acetylation in
Raji cells. Int J Mol Med. 41:3366–3378. 2018.PubMed/NCBI
|
|
45
|
Ren Y, Bi C, Zhao X, Lwin T, Wang C, Yuan
J, Silva AS, Shah BD, Fang B, Li T, et al: PLK1 stabilizes a
MYC-dependent kinase network in aggressive B cell lymphomas. J Clin
Invest. 128:5517–5530. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Slack GW and Gascoyne RD: MYC and
aggressive B-cell lymphomas. Adv Anat Pathol. 18:219–228. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Du W, Zhou Y, Pike S and Pang Q: NPM
phosphorylation stimulates Cdk1, overrides G2/M checkpoint and
increases leukemic blasts in mice. Carcinogenesis. 31:302–310.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rahmani M, Talebi M, Hagh MF, Feizi AAH
and Solali S: Aberrant DNA methylation of key genes and Acute
Lymphoblastic Leukemia. Biomed Pharmacother. 97:1493–1500. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vasconcelos GM, Christensen BC, Houseman
EA, Xiao J, Marsit CJ, Wiencke JK, Zheng S, Karagas MR, Nelson HH,
Wrensch MR, et al: History of Parvovirus B19 infection is
associated with a DNA methylation signature in childhood acute
lymphoblastic leukemia. Epigenetics. 6:1436–1443. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Timms JA, Relton CL, Rankin J, Strathdee G
and McKay JA: DNA methylation as a potential mediator of
environmental risks in the development of childhood acute
lymphoblastic leukemia. Epigenomics. 8:519–536. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Milne E, Laurvick CL, Blair E, Bower C and
de Klerk N: Fetal growth and acute childhood leukemia: Looking
beyond birth weight. Am J Epidemiol. 166:151–159. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Groves FD, Watkins BT, Roberts DJ, Tucker
TC, Shen T and Flood TJ: Birth weight and risk of childhood acute
lymphoblastic leukemia in arizona, Illinois, and kentucky. South
Med J. 111:579–584. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Robison LL, Codd M, Gunderson P, Neglia
JP, Smithson WA and King FL: Birth weight as a risk factor for
childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol.
4:63–72. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hellström A, Ley D, Hansen-Pupp I,
Hallberg B, Ramenghi LA, Löfqvist C, Smith LE and Hard AL: Role of
insulinlike growth factor 1 in fetal development and in the early
postnatal life of premature infants. Am J Perinatol. 33:1067–1071.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Stratikopoulos E, Szabolcs M, Dragatsis I,
Klinakis A and Efstratiadis A: The hormonal action of IGF1 in
postnatal mouse growth. Proc Natl Acad Sci USA. 105:19378–19383.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Khalade A, Jaakkola MS, Pukkala E and
Jaakkola JJ: Exposure to benzene at work and the risk of leukemia:
A systematic review and meta-analysis. Environ Health. 9:312010.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xie Z, Zhang Y, Guliaev AB, Shen H, Hang
B, Singer B and Wang Z: The p-benzoquinone DNA adducts derived from
benzene are highly mutagenic. DNA Repair (Amst). 4:1399–1409. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mansell E, Zareian N, Malouf C, Kapeni C,
Brown N, Badie C, Baird D, Lane J, Ottersbach K, Blair A and Case
CP: DNA damage signalling from the placenta to foetal blood as a
potential mechanism for childhood leukaemia initiation. Sci Rep.
9:43702019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhou Y, Zhang S, Li Z, Zhu J, Bi Y, Bai Y
and Wang H: Maternal benzene exposure during pregnancy and risk of
childhood acute lymphoblastic leukemia: A meta-analysis of
epidemiologic studies. PLoS One. 9:e1104662014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cooper SL and Brown PA: Treatment of
pediatric acute lymphoblastic leukemia. Pediatr Clin North Am.
62:61–73. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Pui CH, Campana D, Pei D, Bowman WP,
Sandlund JT, Kaste SC, Ribeiro RC, Rubnitz JE, Raimondi SC, Onciu
M, et al: Treating childhood acute lymphoblastic leukemia without
cranial irradiation. N Engl J Med. 360:2730–2741. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tsurusawa M, Shimomura Y, Asami K, Kikuta
A, Watanabe A, Horikoshi Y, Matsushita T, Kanegane H, Ohta S, Iwai
A, et al: Long-term results of the Japanese childhood cancer and
leukemia study group studies 811, 841, 874 and 911 on childhood
acute lymphoblastic leukemia. Leukemia. 24:335–344. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pui CH, Pei D, Sandlund JT, Ribeiro RC,
Rubnitz JE, Raimondi SC, Onciu M, Campana D, Kun LE, Jeha S, et al:
Long-term results of St Jude total therapy studies 11, 12, 13A,
13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia.
24:371–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jabbour EJ, Faderl S and Kantarjian HM:
Adult acute lymphoblastic leukemia. Mayo Clin Proc. 80:1517–1527.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Winter SS, Holdsworth MT, Devidas M,
Raisch DW, Chauvenet A, Ravindranath Y, Ducore JM and Amylon MD:
Antimetabolite-based therapy in childhood T-cell acute
lymphoblastic leukemia: A report of POG study 9296. Pediatr Blood
Cancer. 46:179–186. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Seymour JF, Grigg AP, Szer J and Fox RM:
Cisplatin, fludarabine, and cytarabine: A novel, pharmacologically
designed salvage therapy for patients with refractory,
histologically aggressive or mantle cell non-Hodgkin's lymphoma.
Cancer. 94:585–593. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kato M and Manabe A: Treatment and biology
of pediatric acute lymphoblastic leukemia. Pediatr Int. 60:4–12.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Narayanan S and Shami PJ: Treatment of
acute lymphoblastic leukemia in adults. Crit Rev Oncol Hematol.
81:94–102. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Terwilliger T and Abdul-Hay M: Acute
lymphoblastic leukemia: A comprehensive review and 2017 update.
Blood Cancer J. 7:e5772017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Baraz R, Cisterne A, Saunders PO, Hewson
J, Thien M, Weiss J, Basnett J, Bradstock KF and Bendall LJ: mTOR
inhibition by everolimus in childhood acute lymphoblastic leukemia
induces caspase-independent cell death. PLoS One. 9:e1024942014.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Singh SK, Banerjee S, Acosta EP, Lillard
JW and Singh R: Resveratrol induces cell cycle arrest and apoptosis
with docetaxel in prostate cancer cells via a p53/p21WAF1/CIP1 and
p27KIP1 pathway. Oncotarget. 8:17216–17228. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ge J, Liu Y, Li Q, Guo X, Gu L, Ma ZG and
Zhu YP: Resveratrol induces apoptosis and autophagy in T-cell acute
lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating
p38-MAPK. Biomed Environ Sci. 26:902–911. 2013.PubMed/NCBI
|
|
73
|
Cai Y, Xia Q, Su Q, Luo R, Sun Y, Shi Y
and Jiang W: mTOR inhibitor RAD001 (Everolimus) induces apoptotic,
not autophagic cell death, in human nasopharyngeal carcinoma cells.
Int J Mol Med. 31:904–912. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ciombor KK and Bekaii-Saab T: Selumetinib
for the treatment of cancer. Expert Opin Investig Drugs.
24:111–123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kerstjens M, Driessen EM, Willekes M,
Pinhancos SS, Schneider P, Pieters R and Stam RW: MEK inhibition is
a promising therapeutic strategy for MLL-rearranged infant acute
lymphoblastic leukemia patients carrying RAS mutations. Oncotarget.
8:14835–14846. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Piya S, Andreeff M and Borthakur G:
Targeting autophagy to overcome chemoresistance in acute
myleogenous leukemia. Autophagy. 13:214–215. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Takahashi H, Inoue J, Sakaguchi K, Takagi
M, Mizutani S and Inazawa J: Autophagy is required for cell
survival under L-asparaginase-induced metabolic stress in acute
lymphoblastic leukemia cells. Oncogene. 36:4267–4276. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Takahashi H, Inoue J, Sakaguchi K, Takagi
M, Mizutani S and Inazawa J: Autophagy inhibition sensitizes acute
lymphoblastic leukemia cells to L-asparaginase. Blood. 126:3772.
2015. View Article : Google Scholar
|
|
79
|
Sakura H, Kanei-Ishii C, Nagase T,
Nakagoshi H, Gonda TJ and Ishii S: Delineation of three functional
domains of the transcriptional activator encoded by the c-myb
protooncogene. Proc Natl Acad Sci USA. 86:5758–5762. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Tanaka Y, Nomura T and Ishii S: Two
regions in c-myb proto-oncogene product negatively regulating its
DNA-binding activity. FEBS Lett. 413:162–168. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhou Y and Ness SA: Myb proteins: Angels
and demons in normal and transformed cells. Front Biosci (Landmark
Ed). 16:1109–1131. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lv M, Wang Y, Wu W, Yang S, Zhu H, Hu B,
Chen Y, Shi C, Zhang Y, Mu Q and Ouyang G: CMyc inhibitor 10058F4
increases the efficacy of dexamethasone on acute lymphoblastic
leukaemia cells. Mol Med Rep. 18:421–428. 2018.PubMed/NCBI
|
|
83
|
Liu X, Xu Y, Han L and Yi Y: Reassessing
the Potential of Myb-targeted Anti-cancer Therapy. J Cancer.
9:1259–1266. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mitra P: Transcription regulation of MYB:
A potential and novel therapeutic target in cancer. Ann Transl Med.
6:4432018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Grobbelaar C and Ford AM: The Role of
MicroRNA in paediatric acute lymphoblastic leukaemia: Challenges
for diagnosis and therapy. J Oncol. 2019:89414712019. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Nakase K, Kita K, Miwa H, Nishii K,
Shikami M, Tanaka I, Tsutani H, Ueda T, Nasu K, Kyo T, et al:
Clinical and prognostic significance of cytokine receptor
expression in adult acute lymphoblastic leukemia: Interleukin-2
receptor alpha-chain predicts a poor prognosis. Leukemia.
21:326–332. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Duyu M, Durmaz B, Gunduz C, Vergin C,
Yilmaz Karapinar D, Aksoylar S, Kavakli K, Cetingul N, Irken G,
Yaman Y, et al: Prospective evaluation of whole genome microRNA
expression profiling in childhood acute lymphoblastic leukemia.
Biomed Res Int. 2014:9675852014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yoshida N, Oda M, Kuroda Y, Katayama Y,
Okikawa Y, Masunari T, Fujiwara M, Nishisaka T, Sasaki N, Sadahira
Y, et al: Clinical significance of sIL-2R levels in B-cell
lymphomas. PLoS One. 8:e787302013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Nakase K, Kita K, Kyo T, Tsuji K and
Katayama N: High serum levels of soluble interleukin-2 receptor in
acute myeloid leukemia: Correlation with poor prognosis and CD4
expression on blast cells. Cancer Epidemiol. 36:e306–e309. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Li B, Brady SW, Ma X, Shen S, Zhang Y, Li
Y, Szlachta K, Dong L, Liu Y, Yang F, et al: Therapy-induced
mutations drive the genomic landscape of relapsed acute
lymphoblastic leukemia. Blood. 135:41–55. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Tomiyasu H, Watanabe M, Sugita K,
Goto-Koshino Y, Fujino Y, Ohno K, Sugano S and Tsujimoto H:
Regulations of ABCB1 and ABCG2 expression through MAPK pathways in
acute lymphoblastic leukemia cell lines. Anticancer Res.
33:5317–5323. 2013.PubMed/NCBI
|
|
92
|
Xie J, Jin B, Li DW, Shen B, Cong N, Zhang
TZ and Dong P: ABCG2 regulated by MAPK pathways is associated with
cancer progression in laryngeal squamous cell carcinoma. Am J
Cancer Res. 4:698–709. 2014.PubMed/NCBI
|
|
93
|
El Azreq MA, Naci D and Aoudjit F:
Collagen/β1 integrin signaling up-regulates the ABCC1/MRP-1
transporter in an ERK/MAPK-dependent manner. Mol Biol Cell.
23:3473–3484. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kourti M, Vavatsi N, Gombakis N, Sidi V,
Tzimagiorgis G, Papageorgiou T, Koliouskas D and Athanassiadou F:
Expression of multidrug resistance 1 (MDR1), multidrug
resistance-related protein 1 (MRP1), lung resistance protein (LRP),
and breast cancer resistance protein (BCRP) genes and clinical
outcome in childhood acute lymphoblastic leukemia. Int J Hematol.
86:166–173. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Baudis M, Prima V, Tung YH and Hunger SP:
ABCB1 over-expression and drug-efflux in acute lymphoblastic
leukemia cell lines with t(17;19) and E2A-HLF expression. Pediatr
Blood Cancer. 47:757–764. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhang K, Mack P and Wong KP:
Glutathione-related mechanisms in cellular resistance to anticancer
drugs. Int J Oncol. 12:871–882. 1998.PubMed/NCBI
|
|
97
|
Tsai SY, Sun NK, Lu HP, Cheng ML and Chao
CC: Involvement of reactive oxygen species in multidrug resistance
of a vincristine-selected lymphoblastoma. Cancer Sci. 98:1206–1214.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhu Z, Du S, Du Y, Ren J, Ying G and Yan
Z: Glutathione reductase mediates drug resistance in glioblastoma
cells by regulating redox homeostasis. J Neurochem. 144:93–104.
2018. View Article : Google Scholar : PubMed/NCBI
|